

Progressive Multifocal Leukoencephalopathy in Systemic Lupus Erythematosus: A Consequence of Patient-Intrinsic or -Extrinsic Factors?

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Blood and Bone Marrow Immunophenotyping
2.2. Genomic DNA Extraction and Exome Sequencing
2.3. Bioinformatics, Variant Calling, and Annotation
2.4. Magnetic Resonance Imaging (MRI)
3. Results
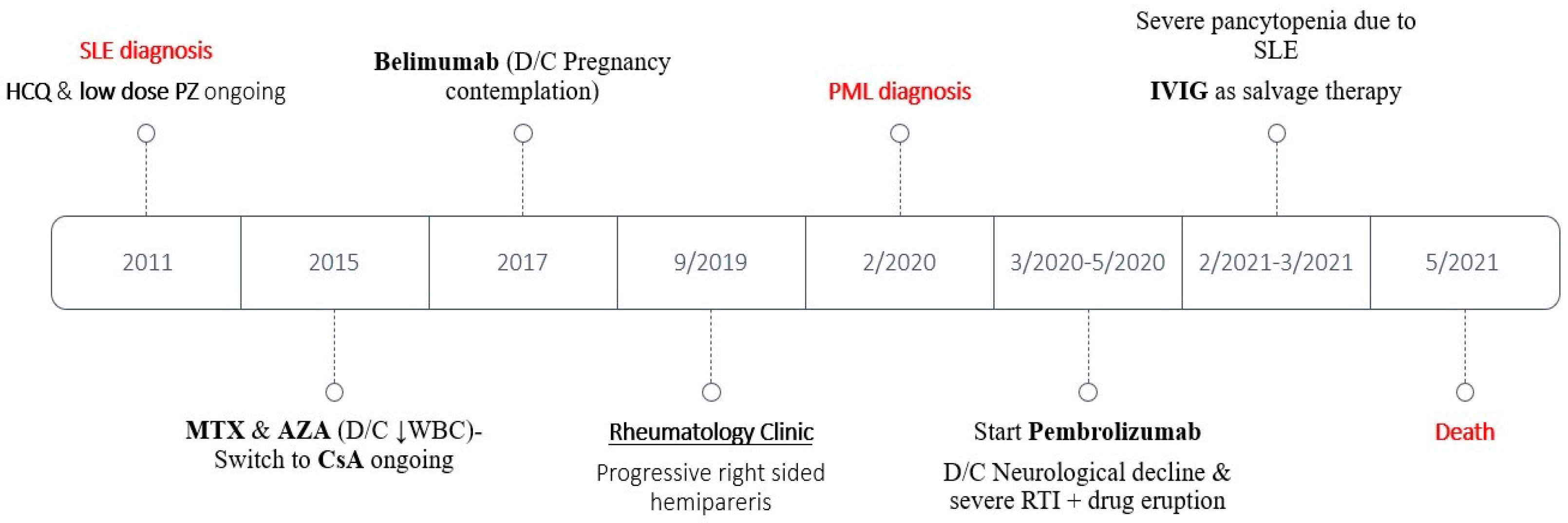
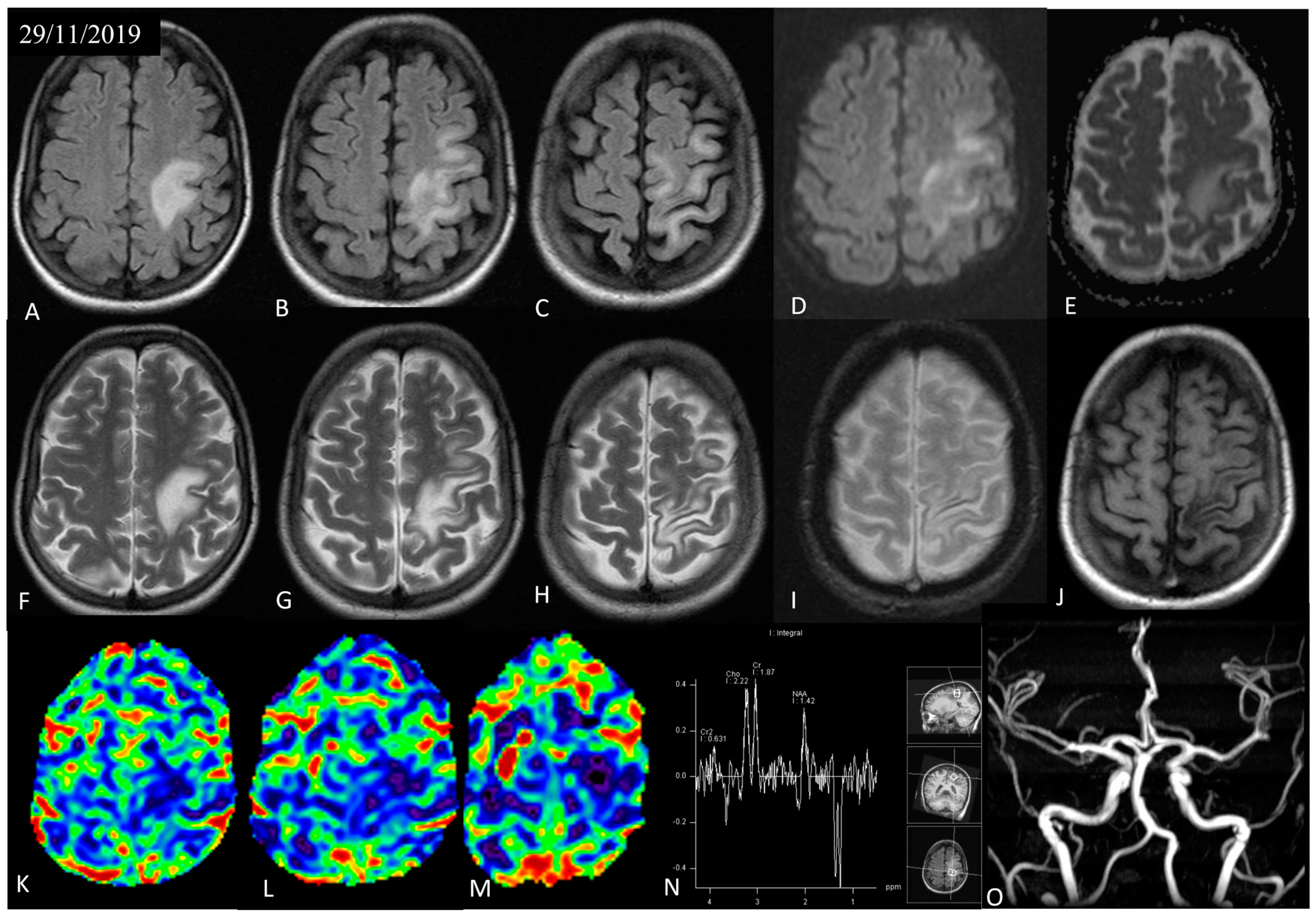
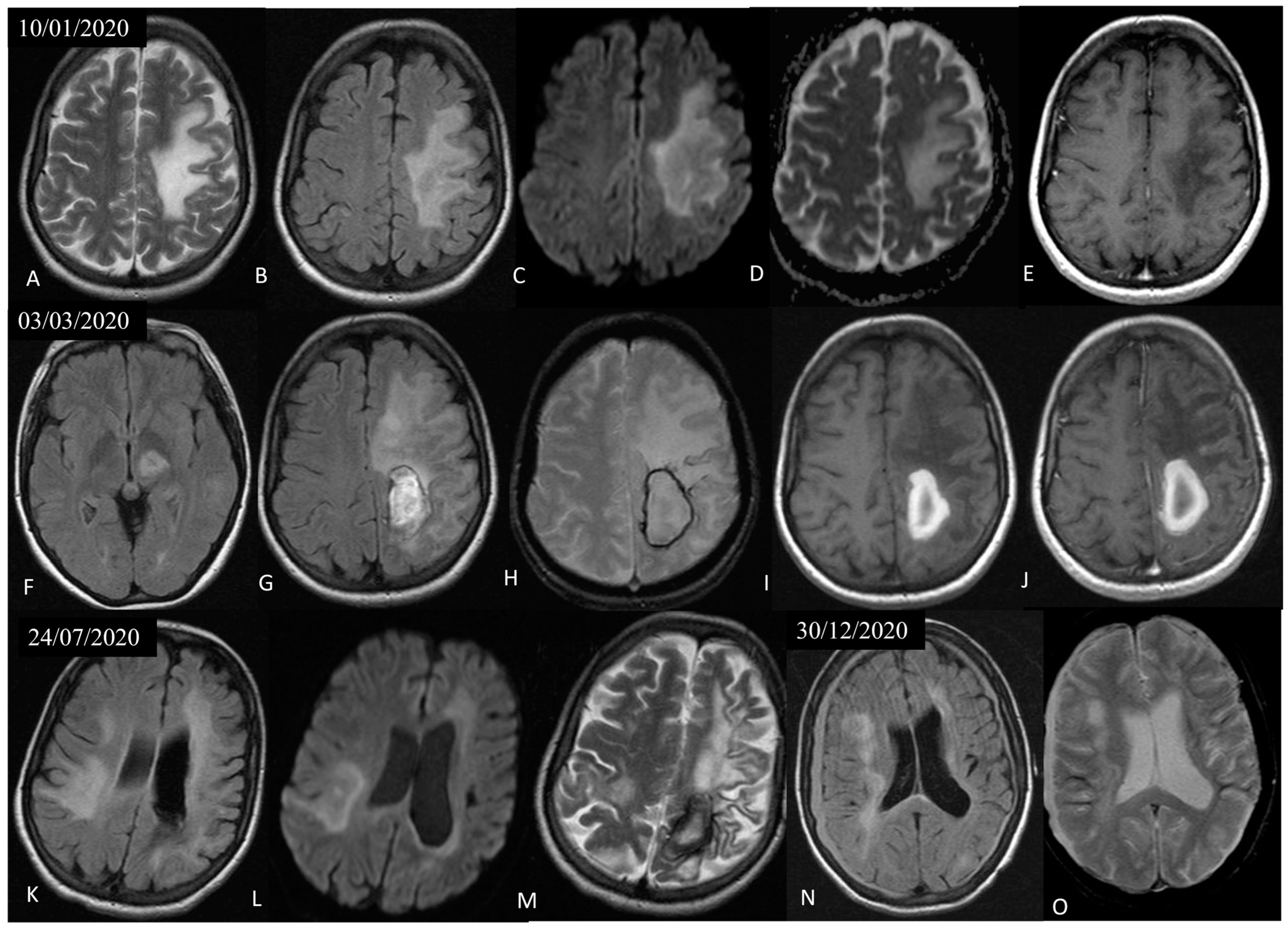
3.1. Case Presentation
3.2. Immunophenotyping Assessment
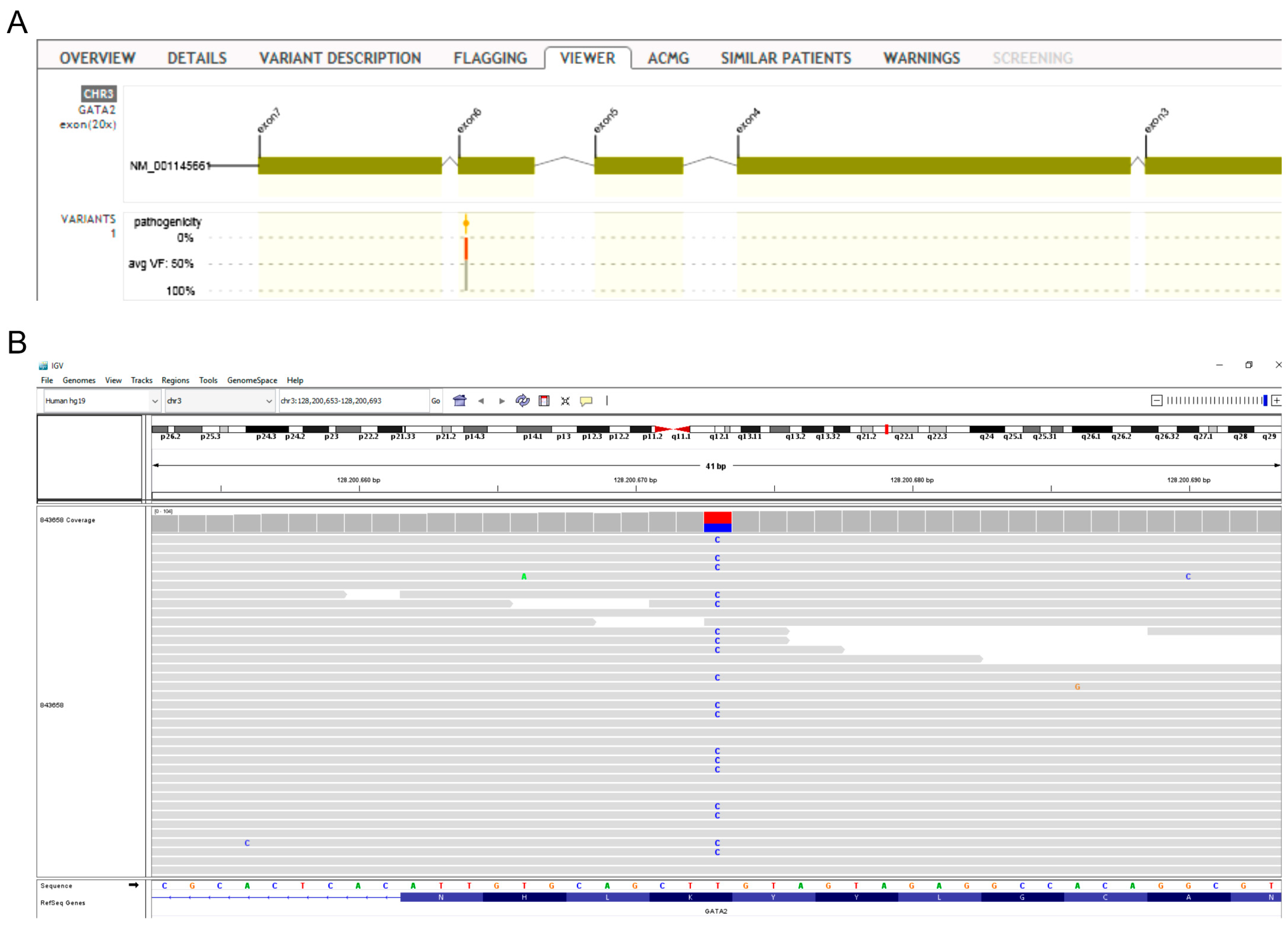
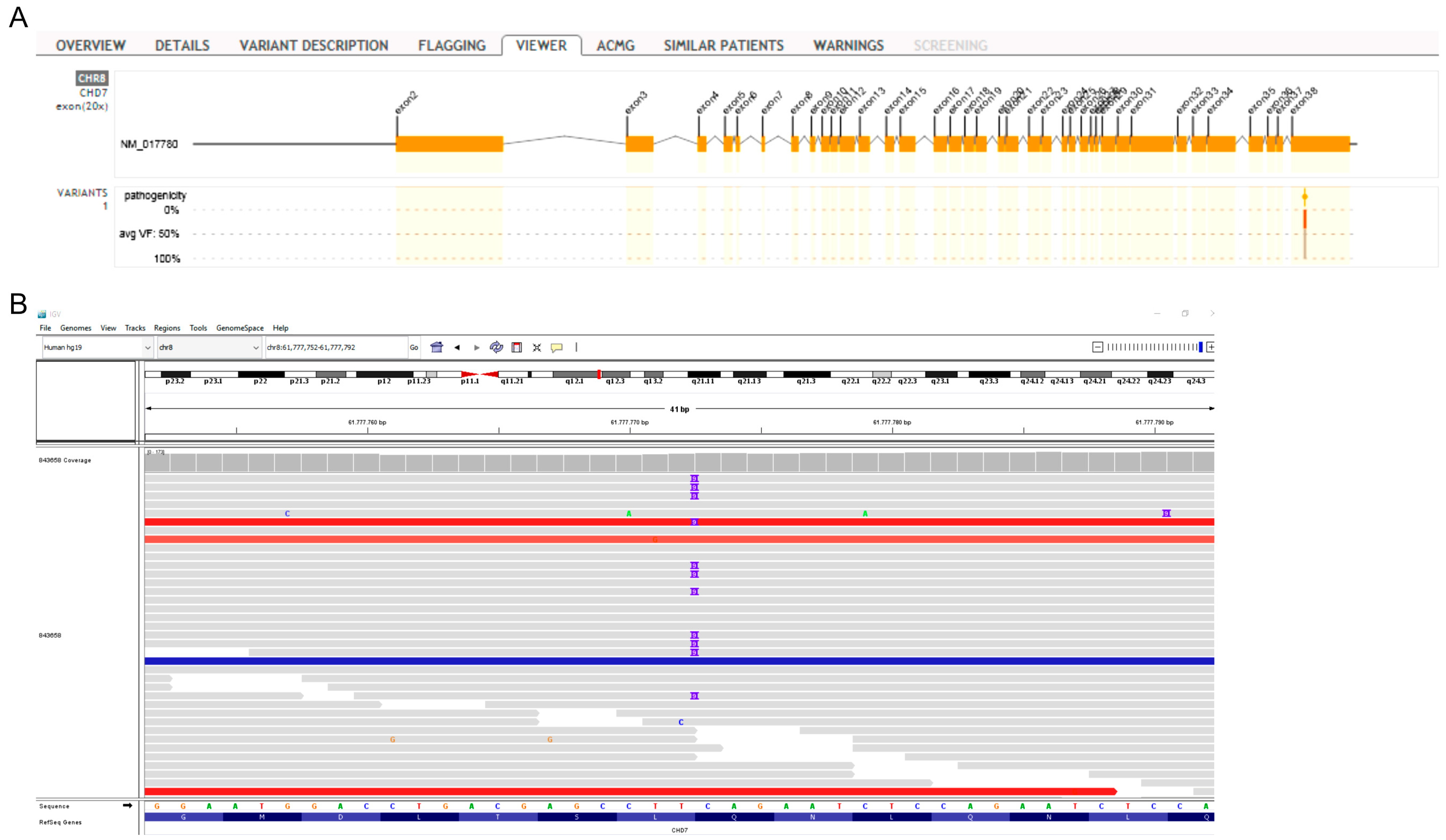
3.3. Whole Exome Sequencing Analysis
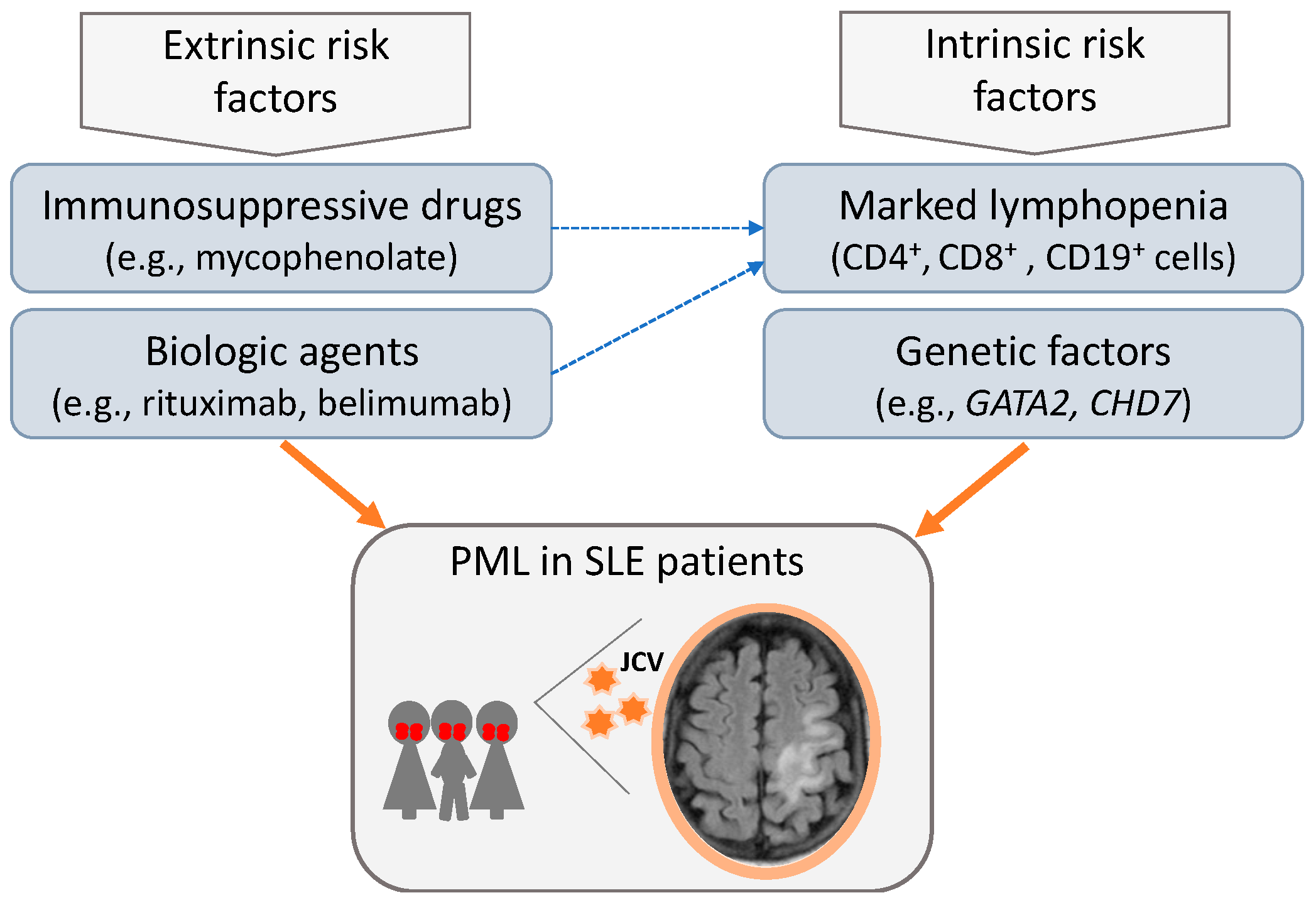
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kartau, M.; Sipila, J.O.; Auvinen, E.; Palomaki, M.; Verkkoniemi-Ahola, A. Progressive Multifocal Leukoencephalopathy: Current Insights. Degener. Neurol. Neuromuscul. Dis. 2019, 9, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Khalili, A.; Craigie, M.; Donadoni, M.; Sariyer, I.K. Host-Immune Interactions in JC Virus Reactivation and Development of Progressive Multifocal Leukoencephalopathy (PML). J. Neuroimmune Pharmacol. 2019, 14, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R.; Aksamit, A.J.; Clifford, D.B.; Davis, L.; Koralnik, I.J.; Sejvar, J.J.; Bartt, R.; Major, E.O.; Nath, A. PML diagnostic criteria: Consensus statement from the AAN Neuroinfectious Disease Section. Neurology 2013, 80, 1430–1438. [Google Scholar] [CrossRef]
- Sahraian, M.A.; Radue, E.W.; Eshaghi, A.; Besliu, S.; Minagar, A. Progressive multifocal leukoencephalopathy: A review of the neuroimaging features and differential diagnosis. Eur. J. Neurol. 2012, 19, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Venkatesan, A. Progressive Multifocal Leukoencephalopathy in HIV-Uninfected Individuals. Curr. Infect. Dis. Rep. 2016, 18, 33. [Google Scholar] [CrossRef]
- Kapoor, T.; Mahadeshwar, P.; Hui-Yuen, J.; Quinnies, K.; Tatonetti, N.; Gartshteyn, Y.; Guo, C.; Geraldino-Pardilla, L.; Askanase, A.D. Prevalence of progressive multifocal leukoencephalopathy (PML) in adults and children with systemic lupus erythematosus. Lupus Sci. Med. 2020, 7, e000388. [Google Scholar] [CrossRef]
- Henegar, C.E.; Eudy, A.M.; Kharat, V.; Hill, D.D.; Bennett, D.; Haight, B. Progressive multifocal leukoencephalopathy in patients with systemic lupus erythematosus: A systematic literature review. Lupus 2016, 25, 617–626. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kasuya, T.; Ishikawa, J.; Fujiwara, M.; Kita, Y. A case of developing progressive multifocal leukoencephalopathy while using rituximab and mycophenolate mofetil in refractory systemic lupus erythematosus. Ther. Clin. Risk Manag. 2018, 14, 1149–1153. [Google Scholar] [CrossRef]
- Molloy, E.S.; Calabrese, L.H. Progressive multifocal leukoencephalopathy in patients with rheumatic diseases: Are patients with systemic lupus erythematosus at particular risk? Autoimmun. Rev. 2008, 8, 144–146. [Google Scholar] [CrossRef]
- Brandao, M.; Damasio, J.; Marinho, A.; da Silva, A.M.; Vasconcelos, J.; Neves, E.; Almeida, I.; Farinha, F.; Vasconcelos, C. Systemic lupus erythematosus, progressive multifocal leukoencephalopathy, and T-CD4+ lymphopenia. Clin. Rev. Allergy Immunol. 2012, 43, 302–307. [Google Scholar] [CrossRef]
- Dubois, E.; Ruschil, C.; Bischof, F. Low frequencies of central memory CD4 T cells in progressive multifocal leukoencephalopathy. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e177. [Google Scholar] [CrossRef] [PubMed]
- Gomathy, S.; Panigrahi, B.; Tirlangi, P.K.; Wig, N.; Brijwal, M.; Sharma, M.C.; Garg, A.; Tripathi, M.; Mohta, S.; Doddamani, R.; et al. Progressive multifocal leukoencephalopathy in a patient with systemic lupus erythematosus and autoimmune hepatitis. Int. J. Rheum. Dis. 2022, 25, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Ibanez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol. 2002, 29, 288–291. [Google Scholar] [PubMed]
- Molloy, E.S.; Calabrese, C.M.; Calabrese, L.H. The Risk of Progressive Multifocal Leukoencephalopathy in the Biologic Era: Prevention and Management. Rheum. Dis. Clin. North. Am. 2017, 43, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Cortese, I.; Muranski, P.; Enose-Akahata, Y.; Ha, S.K.; Smith, B.; Monaco, M.; Ryschkewitsch, C.; Major, E.O.; Ohayon, J.; Schindler, M.K.; et al. Pembrolizumab Treatment for Progressive Multifocal Leukoencephalopathy. N. Engl. J. Med. 2019, 380, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef]
- Dickinson, R.E.; Griffin, H.; Bigley, V.; Reynard, L.N.; Hussain, R.; Haniffa, M.; Lakey, J.H.; Rahman, T.; Wang, X.N.; McGovern, N.; et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 2011, 118, 2656–2658. [Google Scholar] [CrossRef]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Kim, H.G.; Kurth, I.; Lan, F.; Meliciani, I.; Wenzel, W.; Eom, S.H.; Kang, G.B.; Rosenberger, G.; Tekin, M.; Ozata, M.; et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2008, 83, 511–519. [Google Scholar] [CrossRef]
- Jongmans, M.C.; Admiraal, R.J.; van der Donk, K.P.; Vissers, L.E.; Baas, A.F.; Kapusta, L.; van Hagen, J.M.; Donnai, D.; de Ravel, T.J.; Veltman, J.A.; et al. CHARGE syndrome: The phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2006, 43, 306–314. [Google Scholar] [CrossRef]
- Wong, M.T.; Scholvinck, E.H.; Lambeck, A.J.; van Ravenswaaij-Arts, C.M. CHARGE syndrome: A review of the immunological aspects. Eur. J. Hum. Genet. 2015, 23, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Nived, O.; Bengtsson, A.A.; Jonsen, A.; Sturfelt, G. Progressive multifocal leukoencephalopathy—The importance of early diagnosis illustrated in four cases. Lupus 2008, 17, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Algahtani, H.; Shirah, B.; Othman, L.; Almarri, A.K.; Alwafi, E.; Alassiri, A.H. Progressive Multifocal Leukoencephalopathy Misdiagnosed as Neuropsychiatric Systemic Lupus Erythematosus with a Catastrophic Outcome. Neurologist 2022, 27, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Law, L.Y.; Tan, I.; Prowse, M.; Sean Riminton, D.; Reddel, S.W. Progressive multifocal leukoencephalopathy in a patient with systemic lupus erythematosus: Clues to early diagnosis. J. Clin. Neurosci. 2019, 67, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Ishii, J.; Shishido-Hara, Y.; Kawamoto, M.; Fujiwara, S.; Imai, Y.; Nakamichi, K.; Kohara, N. A Punctate Magnetic Resonance Imaging Pattern in a Patient with Systemic Lupus Erythematosus Is an Early Sign of Progressive Multifocal Leukoencephalopathy: A Clinicopathological Study. Intern. Med. 2018, 57, 2727–2734. [Google Scholar] [CrossRef]
- Major, E.O.; Yousry, T.A.; Clifford, D.B. Pathogenesis of progressive multifocal leukoencephalopathy and risks associated with treatments for multiple sclerosis: A decade of lessons learned. Lancet Neurol. 2018, 17, 467–480. [Google Scholar] [CrossRef]
- Pavlovic, D.; Patel, M.A.; Patera, A.C.; Peterson, I.; Progressive Multifocal Leukoencephalopathy, C. T cell deficiencies as a common risk factor for drug associated progressive multifocal leukoencephalopathy. Immunobiology 2018, 223, 508–517. [Google Scholar] [CrossRef]
- Durali, D.; de Goer de Herve, M.G.; Gasnault, J.; Taoufik, Y. B cells and progressive multifocal leukoencephalopathy: Search for the missing link. Front. Immunol. 2015, 6, 241. [Google Scholar] [CrossRef]
- Cortese, I.; Reich, D.S.; Nath, A. Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat. Rev. Neurol. 2021, 17, 37–51. [Google Scholar] [CrossRef]
- Wei, G.; Liu, C.K.; Atwood, W.J. JC virus binds to primary human glial cells, tonsillar stromal cells, and B-lymphocytes, but not to T lymphocytes. J. Neurovirol. 2000, 6, 127–136. [Google Scholar] [CrossRef]
- Marshall, L.J.; Major, E.O. Molecular regulation of JC virus tropism: Insights into potential therapeutic targets for progressive multifocal leukoencephalopathy. J. Neuroimmune Pharmacol. 2010, 5, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Focosi, D.; Socal, M.P.; Bian, J.C.; Nabhan, C.; Hrushesky, W.J.; Bennett, A.C.; Schoen, M.W.; Berger, J.R.; Armitage, J.O.; et al. Progressive multifocal leukoencephalopathy in patients treated with rituximab: A 20-year review from the Southern Network on Adverse Reactions. Lancet Haematol. 2021, 8, e593–e604. [Google Scholar] [CrossRef] [PubMed]
- Felli, V.; Di Sibio, A.; Anselmi, M.; Gennarelli, A.; Sucapane, P.; Splendiani, A.; Catalucci, A.; Marini, C.; Gallucci, M. Progressive Multifocal Leukoencephalopathy Following Treatment with Rituximab in an HIV-Negative Patient with Non-Hodgkin Lymphoma. A Case Report and Literature Review. Neuroradiol. J. 2014, 27, 657–664. [Google Scholar] [CrossRef]
- Schroder, C.; Baerlecken, N.T.; Pannicke, U.; Dork, T.; Witte, T.; Jacobs, R.; Stoll, M.; Schwarz, K.; Grimbacher, B.; Schmidt, R.E.; et al. Evaluation of RAG1 mutations in an adult with combined immunodeficiency and progressive multifocal leukoencephalopathy. Clin. Immunol. 2017, 179, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hatchwell, E.; Smith, E.B., 3rd; Jalilzadeh, S.; Bruno, C.D.; Taoufik, Y.; Hendel-Chavez, H.; Liblau, R.; Brassat, D.; Martin-Blondel, G.; Wiendl, H.; et al. Progressive multifocal leukoencephalopathy genetic risk variants for pharmacovigilance of immunosuppressant therapies. Front. Neurol. 2022, 13, 1016377. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.Y.; Chen, Y.S.; Cheng, C.F.; Huang, S.T.; Shen, C.Y.; Hsu, P.N. Progressive multifocal leukoencephalopathy in systemic lupus erythematosus managed with pembrolizumab: A case report with literature review. Lupus 2021, 30, 1849–1855. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Marker | Peripheral Blood (% Lymphocytes) | Bone Marrow (% Lymphocytes) |
|---|---|---|
| CD3+ | 99.5 | 98.7 |
| CD3+ CD4+ | 35.0 | 51.0 |
| CD3+ CD8+ | 57.0 | 40.0 |
| CD4+ CD8+ | 3.6 | 5.2 |
| CD19+ | 0.2 | 0.8 |
| CD16+ CD56+ CD3− | 0.1 | 0.1 |
| CD16+ CD56+ CD3+ | 0.0 | 4.1 |
| CD57+ CD8+ CD3+ | 26.9 | 15.4 |
| CD57+ CD4+ CD3+ | 8.3 | 6.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emmanouilidou, E.; Kosmara, D.; Papadaki, E.; Mastorodemos, V.; Constantoulakis, P.; Repa, A.; Christopoulou, G.; Kalpadakis, C.; Avgoustidis, N.; Thomas, K.; et al. Progressive Multifocal Leukoencephalopathy in Systemic Lupus Erythematosus: A Consequence of Patient-Intrinsic or -Extrinsic Factors? J. Clin. Med. 2023, 12, 6945. https://doi.org/10.3390/jcm12216945
Emmanouilidou E, Kosmara D, Papadaki E, Mastorodemos V, Constantoulakis P, Repa A, Christopoulou G, Kalpadakis C, Avgoustidis N, Thomas K, et al. Progressive Multifocal Leukoencephalopathy in Systemic Lupus Erythematosus: A Consequence of Patient-Intrinsic or -Extrinsic Factors? Journal of Clinical Medicine. 2023; 12(21):6945. https://doi.org/10.3390/jcm12216945
Chicago/Turabian StyleEmmanouilidou, Evgenia, Despoina Kosmara, Efrosini Papadaki, Vasileios Mastorodemos, Pantelis Constantoulakis, Argyro Repa, Georgia Christopoulou, Christina Kalpadakis, Nestor Avgoustidis, Konstantinos Thomas, and et al. 2023. "Progressive Multifocal Leukoencephalopathy in Systemic Lupus Erythematosus: A Consequence of Patient-Intrinsic or -Extrinsic Factors?" Journal of Clinical Medicine 12, no. 21: 6945. https://doi.org/10.3390/jcm12216945
APA StyleEmmanouilidou, E., Kosmara, D., Papadaki, E., Mastorodemos, V., Constantoulakis, P., Repa, A., Christopoulou, G., Kalpadakis, C., Avgoustidis, N., Thomas, K., Boumpas, D., Sidiropoulos, P., & Bertsias, G. (2023). Progressive Multifocal Leukoencephalopathy in Systemic Lupus Erythematosus: A Consequence of Patient-Intrinsic or -Extrinsic Factors? Journal of Clinical Medicine, 12(21), 6945. https://doi.org/10.3390/jcm12216945