
Glaucoma as a Tauopathy—Is It the Missing Piece in the Glaucoma Puzzle?
 , ,
, , {kind=link}
Abstract
:1. Introduction
2. Ocular Structures and Glaucoma Pathophysiology
3. The Role of Tau in Neurodegenerative Disorders
4. Tau Protein’s Role in Glaucoma: A Closer Look at the Evidence
5. Oxidative Damage Is a Common Feature in Both Glaucoma and Tauopathies
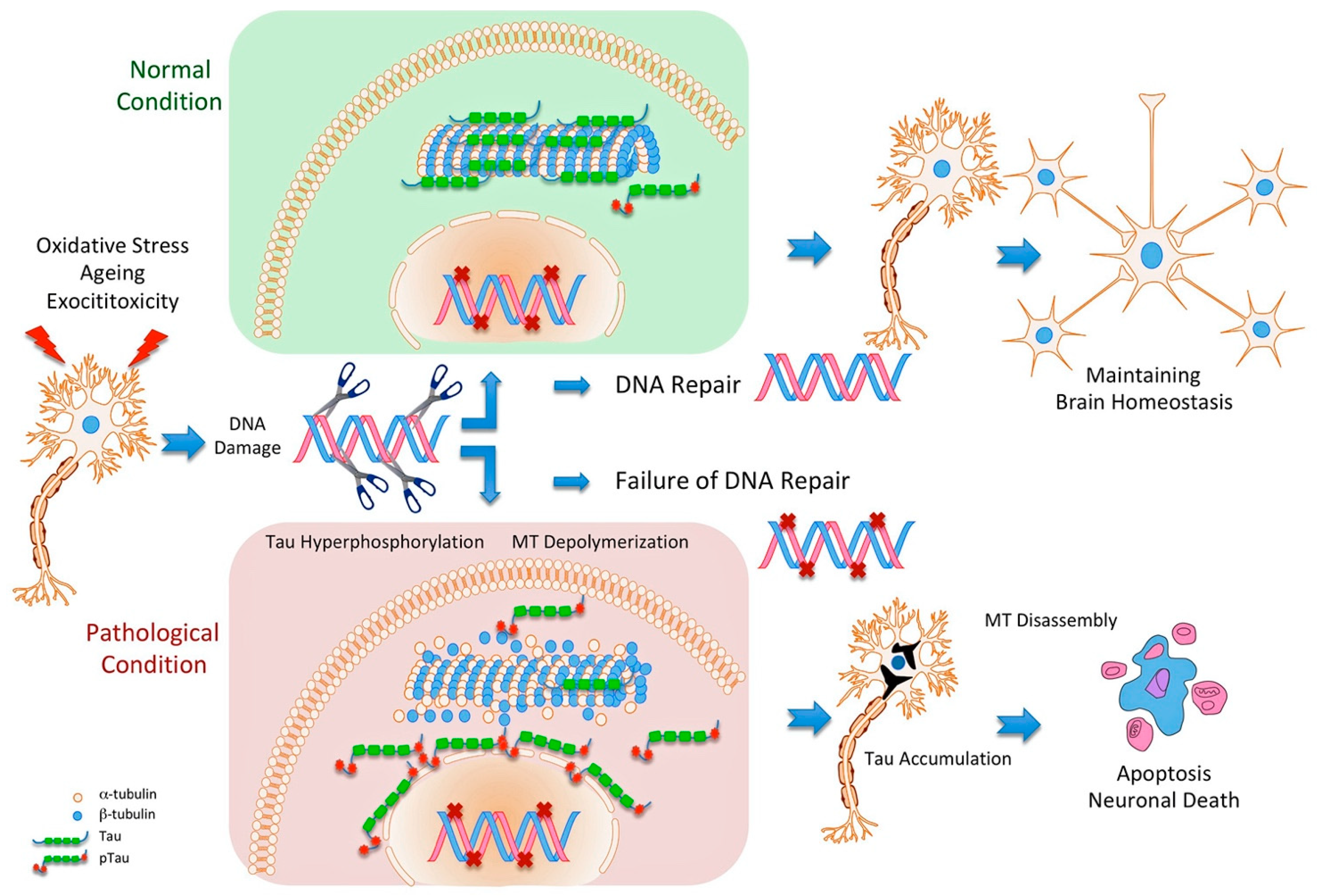
6. Oxidative DNA Damage and Tau
7. Outlook for the Future
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weinreb, R.N.; Tee Khaw, P. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Saxena, R.; Tripathi, M.; Vibha, D.; Dhiman, R. Neurodegeneration in Alzheimer’s disease and glaucoma: Overlaps and missing links. Eye 2020, 34, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Aref, A.A. Senile Dementia and Glaucoma: Evidence for a Common Link. J. Ophthalmic. Vis. Res. 2015, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Chiasseu, M.; Vargas, J.L.C.; Destroismaisons, L.; Velde, C.V.; Leclerc, N.; Di Polo, A. Tau Accumulation, Altered Phosphorylation, and Missorting Promote Neurodegeneration in Glaucoma. J. Neurosci. 2016, 36, 5785–5798. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Yoshitomi, T.; Covey, D.F.; Zorumski, C.F.; Izumi, Y. Neurosteroids and oxysterols as potential therapeutic agents for glaucoma and Alzheimer’s disease. Neuropsychiatry 2018, 8, 344–359. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta. 2010, 1802, 135–142. [Google Scholar] [CrossRef]
- Butner, K.A.; Kirschner, M.W. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef]
- Khatoon, S.; Grundke-Iqbal, I.; Iqbal, K. Brain Levels of Microtubule-Associated Protein τ Are Elevated in Alzheimer’s Disease: A Radioimmuno-Slot-Blot Assay for Nanograms of the Protein. J. Neurochem. 1992, 59, 750–753. [Google Scholar] [CrossRef]
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2017, 145, 355–368. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The Pathophysiology and Treatment of Glaucoma: A Review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Kizhatil, K.; Ryan, M.; Marchant, J.K.; Henrich, S.; John, S.W.M. Schlemm’s canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process. PLoS Biol. 2014, 12, e1001912. [Google Scholar] [CrossRef] [PubMed]
- van Zyl, T.; Yan, W.; McAdams, A.; Peng, Y.R.; Shekhar, K.; Regev, A.; Juric, D.; Sanes, J.R. Cell atlas of aqueous humor outflow pathways in eyes of humans and four model species provides insight into glaucoma pathogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 10339–10349. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, C.; dell’Omo, R.; Agnifili, L.; Bartollino, S.; Fea, A.M.; Uva, M.G.; Zeppa, L.; Mastropasqua, L. How many aqueous humor outflow pathways are there? Surv. Ophthalmol. 2020, 65, 144–170. [Google Scholar] [CrossRef]
- Quigley, H.A.; Addicks, E.M.; Green, W.R.; Maumenee, A.E. Optic Nerve Damage in Human Glaucoma: II. The Site of Injury and Susceptibility to Damage. Arch. Ophthalmol. 1981, 99, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. Available online: http://intl.iovs.org/cgi/content/full/41/11/3460 (accessed on 21 October 2023).
- Costagliola, C.; Agnifili, L.; Mastropasqua, L.; di Costanzo, A. Low-Tension Glaucoma: An Oxymoron in Ophthalmology. Prev. Chronic. Dis. 2019, 16, E10. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, K.M.; Yang, L.; Dong, Q.; Yu, J.T. Tauopathies: New perspectives and challenges. Mol. Neurodegener. 2022, 17, 28. [Google Scholar] [CrossRef]
- Götz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. 2019, 14, 239–261. [Google Scholar] [CrossRef]
- Gigant, B.; Landrieu, I.; Fauquant, C.; Barbier, P.; Huvent, I.; Wieruszeski, J.M.; Knossow, M.; Lippens, G. Mechanism of Tau-promoted microtubule assembly as probed by NMR spectroscopy. J. Am. Chem. Soc. 2014, 136, 12615–12623. [Google Scholar] [CrossRef]
- Trinczek, B.; Ebneth, A.; Mandelkow, E.M.; Mandelkow, E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 1999, 112, 2355–2367. [Google Scholar] [CrossRef] [PubMed]
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing dendritic spine pathology in Alzheimer’s disease: Problems and opportunities. Acta Neuropathol. 2015, 130, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G.; et al. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Preuss, U.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. The “jaws” model of tau-microtubule interaction examined in CHO cells. J. Cell Sci. 1997, 110, 789–800. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Catarina Silva, M.; Haggarty, S.J. Tauopathies: Deciphering Disease Mechanisms to Develop Effective Therapies. Int. J. Mol. Sci. 2020, 21, 8948. [Google Scholar] [CrossRef]
- Nisbet, R.M.; Götz, J. Amyloid-β and Tau in Alzheimer’s Disease: Novel Pathomechanisms and Non-Pharmacological Treatment Strategies. J. Alzheimers Dis. 2018, 64, S517–S527. [Google Scholar] [CrossRef]
- Jabbari, E.; Holland, N.; Chelban, V.; Jones, P.S.; Lamb, R.; Rawlinson, C.; Guo, T.; Costantini, A.A.; Tan, M.M.X.; Heslegrave, A.J.; et al. Diagnosis Across the Spectrum of Progressive Supranuclear Palsy and Corticobasal Syndrome. JAMA Neurol. 2020, 77, 377–387. [Google Scholar] [CrossRef]
- Murley, A.G.; Coyle-Gilchrist, I.; Rouse, M.A.; Simon Jones, P.; Li, W.; Wiggins, J.; Lansdall, C.; Rodríguez, P.V.; Wilcox, A.; Tsvetanov, K.A.; et al. Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain 2020, 143, 1555–1571. [Google Scholar] [CrossRef] [PubMed]
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef]
- Léger, G.C.; Banks, S.J. Neuropsychiatric symptom profile differs based on pathology in patients with clinically diagnosed behavioral variant frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 2014, 37, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Brettschneider, J.; McMillan, C.T.; Cooper, F.; Olm, C.; Arnold, S.E.; Van Deerlin, V.M.; Seeley, W.W.; Miller, B.L.; Lee, E.B.; et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 2016, 79, 272–287. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Lukic, M.J.; Irwin, D.J.; Arzberger, T.; Respondek, G.; Lee, E.B.; Coughlin, D.; Giese, A.; Grossman, M.; Kurz, C.; et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 2020, 140, 99–119. [Google Scholar] [CrossRef]
- Valentino, R.R.; Koga, S.; Walton, R.L.; Soto-Beasley, A.I.; Kouri, N.; DeTure, M.A.; Murray, M.E.; Johnson, P.W.; Petersen, R.C.; Boeve, B.F.; et al. MAPT subhaplotypes in corticobasal degeneration: Assessing associations with disease risk, severity of tau pathology, and clinical features. Acta Neuropathol. Commun. 2020, 8, 218. [Google Scholar] [CrossRef]
- Sakurai, K.; Tokumaru, A.M.; Ikeda, T.; Morimoto, S.; Inui, S.; Sumida, K.; Oba, H.; Nakagawa, M.; Matsukawa, N.; Hashizume, Y. Characteristic asymmetric limbic and anterior temporal atrophy in demented patients with pathologically confirmed argyrophilic grain disease. Neuroradiology 2019, 61, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Majtenyi, K.; Spina, S.; Murrell, J.R.; Gelpi, E.; Hoftberger, R.; Fraser, G.; Crowther, R.A.; Goedert, M.; Budka, H.; et al. White matter tauopathy with globular glial inclusions: A distinct sporadic frontotemporal lobar degeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 963–975. [Google Scholar] [CrossRef]
- Hansra, G.K.; Popov, G.; Banaczek, P.O.; Vogiatzis, M.; Jegathees, T.; Goldbury, C.S.; Cullen, K.M. The neuritic plaque in Alzheimer’s disease: Perivascular degeneration of neuronal processes. Neurobiol. Aging. 2019, 82, 88–101. [Google Scholar] [CrossRef]
- Cherry, J.D.; Kim, S.H.; Stein, T.D.; Pothast, M.J.; Nicks, R.; Meng, G.; Huber, B.R.; Mez, J.; Alosco, M.L.; Tripodis, Y.; et al. Evolution of neuronal and glial tau isoforms in chronic traumatic encephalopathy. Brain Pathol. 2020, 30, 913–925. [Google Scholar] [CrossRef]
- Humphrey, W.O.; Martindale, R.; Pendlebury, W.W.; DeWitt, J.C. Primary age-related tauopathy (PART) in the general autopsy setting: Not just a disease of the elderly. Brain Pathol. 2021, 31, 381–384. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, e20200861. [Google Scholar] [CrossRef]
- Janelidze, S.; Berron, D.; Smith, R.; Strandberg, O.; Proctor, N.K.; Dage, J.L.; Stomrud, E.; Palmqvist, S.; Mattsson-Carlgren, N.; Hansson, O. Associations of Plasma Phospho-Tau217 Levels With Tau Positron Emission Tomography in Early Alzheimer Disease. JAMA Neurol. 2021, 78, 149–156. [Google Scholar] [CrossRef]
- Chew, S.S.L.; Martins, A.; Strouthidis, N. Retinal and optic nerve changes in glaucoma: From animal study to clinical implication. Prog. Brain Res. 2015, 220, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Albarral, J.A.; Salazar, J.J.; de Hoz, R.; Marco, E.M.; Martín-Sánchez, B.; Flores-Salguero, E.; Salobrar-García, E.; López-Cuenca, I.; Barrios-Sabador, V.; Avilés-Trigueros, M.; et al. Retinal Molecular Changes Are Associated with Neuroinflammation and Loss of RGCs in an Experimental Model of Glaucoma. Int. J. Mol. Sci. 2021, 22, 2066. [Google Scholar] [CrossRef] [PubMed]
- Harwerth, R.S.; Wheat, J.L.; Fredette, M.J.; Anderson, D.R. Linking structure and function in glaucoma. Prog. Retin. Eye Res. 2010, 29, 249–271. [Google Scholar] [CrossRef]
- Doucette, L.P.; Rasnitsyn, A.; Seifi, M.; Walter, M.A. The interactions of genes, age, and environment in glaucoma pathogenesis. Surv. Ophthalmol. 2015, 60, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Sit, A.J. Intraocular pressure variations: Causes and clinical significance. Can. J. Ophthalmol. 2014, 49, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Anthony Crowther, R.; Martin, K.R.; Berg, N.; Coleman, M.; Goedert, M.; Spillantini, M.G. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: Effects on axonal viability. Neurobiol. Aging. 2011, 32, 419–433. [Google Scholar] [CrossRef]
- Yoneda, S.; Hara, H.; Hirata, A.; Fukushima, M.; Inomata, Y.; Tanihara, H. Vitreous fluid levels of β-amyloid(1-42) and tau in patients with retinal diseases. Jpn. J. Ophthalmol. 2005, 49, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.; Kasmala, L.; Proia, A.; McKinnon, S. Expression of Protein Markers of Alzheimer’s Disease in Human Glaucoma Eyes. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3269. [Google Scholar]
- Ning, A.; Cui, J.; To, E.; Ashe, K.H.; Matsubara, J. Amyloid-β Deposits Lead to Retinal Degeneration in a Mouse Model of Alzheimer Disease. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5136–5143. [Google Scholar] [CrossRef]
- Bull, N.D.; Guidi, A.; Goedert, M.; Martin, K.R.; Spillantini, M.G. Reduced Axonal Transport and Increased Excitotoxic Retinal Ganglion Cell Degeneration in Mice Transgenic for Human Mutant P301S Tau. PLoS ONE 2012, 7, e34724. [Google Scholar] [CrossRef] [PubMed]
- Schön, C.; Hoffmann, N.A.; Ochs, S.M.; Burgold, S.; Filser, S.; Steinbach, S.; Seeliger, M.W.; Arzberger, T.; Goedert, M.; Kretzschmar, H.A.; et al. Long-term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PLoS ONE 2012, 7, e53547. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; López-Cuenca, I.; Rojas, P.; Triviño, A.; Ramírez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef]
- Cronin-Golomb, A. Vision in Alzheimer’s disease. Gerontologist 1995, 35, 370–376. [Google Scholar] [CrossRef]
- Mendez, M.F.; Cherrier, M.M.; Meadows, R.S. Depth perception in Alzheimer’s disease. Percept Mot. Skills 1996, 83, 987–995. [Google Scholar] [CrossRef]
- Lee, A.G.; Martin, C.O. Neuro-ophthalmic findings in the visual variant of Alzheimer’s disease. Ophthalmology 2004, 111, 376–380. [Google Scholar] [CrossRef]
- Ho, W.L.; Leung, Y.; Tsang, A.W.T.; So, K.F.; Chiu, K.; Chang, R.C.C. Review: Tauopathy in the retina and optic nerve: Does it shadow pathological changes in the brain? Mol. Vis. 2012, 18, 2700. Available online: https://pmc/articles/PMC3501278/ (accessed on 4 July 2023). [PubMed]
- Tsai, C.S.; Ritch, R.; Schwartz, B.; Lee, S.S.; Miller, N.R.; Chi, T.; Hsieh, F.Y. Optic nerve head and nerve fiber layer in Alzheimer’s disease. Arch. Ophthalmol. 1991, 109, 199–204. [Google Scholar] [CrossRef]
- Danesh-Meyer, H.V.; Birch, H.; Ku, J.Y.F.; Carroll, S.; Gamble, G. Reduction of optic nerve fibers in patients with Alzheimer disease identified by laser imaging. Neurology 2006, 67, 1852–1854. [Google Scholar] [CrossRef]
- Iseri, P.K.; Altinaş, Ö.; Tokay, T.; Yüksel, N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J. Neuroophthalmol. 2006, 26, 18–24. [Google Scholar] [CrossRef]
- Salobrar-Garcia, E.; Hoyas, I.; Leal, M.; De Hoz, R.; Rojas, B.; Ramirez, A.I.; Salazar, J.J.; Yubero, R.; Gil, P.; Triviño, A.; et al. Analysis of Retinal Peripapillary Segmentation in Early Alzheimer’s Disease Patients. Biomed. Res. Int. 2015, 2015, 636548. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Bassi, C.J. Optic nerve damage in Alzheimer’s disease. Ophthalmology 1990, 97, 9–17. [Google Scholar] [CrossRef]
- Hinton, D.R.; Sadun, A.A.; Blanks, J.C.; Miller, C.A. Optic-nerve degeneration in Alzheimer’s disease. N. Engl. J. Med. 1986, 315, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Kesler, A.; Vakhapova, V.; Korczyn, A.D.; Naftaliev, E.; Neudorfer, M. Retinal thickness in patients with mild cognitive impairment and Alzheimer’s disease. Clin. Neurol. Neurosurg. 2011, 113, 523–526. [Google Scholar] [CrossRef]
- Kim, K.E.; Park, K.H. Macular imaging by optical coherence tomography in the diagnosis and management of glaucoma. Br. J. Ophthalmol. 2018, 102, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Cennamo, G.; Montorio, D.; Romano, M.R.; Cardone, D.M.; Minervino, C.; Reibaldi, M.; Cennamo, G. Structure-Functional Parameters in Differentiating Between Patients With Different Degrees of Glaucoma. J. Glaucoma 2016, 25, e884–e888. [Google Scholar] [CrossRef]
- Medeiros, F.A.; Zangwill, L.M.; Bowd, C.; Sample, P.A.; Weinreb, R.N. Influence of disease severity and optic disc size on the diagnostic performance of imaging instruments in glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1008–1015. [Google Scholar] [CrossRef]
- Leite, M.T.; Zangwill, L.M.; Weinreb, R.N.; Rao, H.L.; Alencar, L.M.; Sample, P.A.; Medeiros, F.A. Effect of disease severity on the performance of Cirrus spectral-domain OCT for glaucoma diagnosis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4104–4109. [Google Scholar] [CrossRef]
- Schulze, A.; Lamparter, J.; Pfeiffer, N.; Berisha, F.; Schmidtmann, I.; Hoffmann, E.M. Diagnostic ability of retinal ganglion cell complex, retinal nerve fiber layer, and optic nerve head measurements by Fourier-domain optical coherence tomography. Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.U.; Ferrari, F.; Erb, C. High Occurrence Rate of Glaucoma among Patients with Alzheimer’s Disease. Eur. Neurol. 2002, 47, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.U.; Keller, O.N.; Ferrari, F.; Maag, K.P. Association of glaucoma with neurodegenerative diseases with apoptotic cell death: Alzheimer’s disease and Parkinson’s disease. Am. J. Ophthalmol. 2002, 133, 135–137. [Google Scholar] [CrossRef]
- den Haan, J.; Verbraak, F.D.; Visser, P.J.; Bouwman, F.H. Retinal thickness in Alzheimer’s disease: A systematic review and meta-analysis. Alzheimers Dement. 2017, 6, 162–170. [Google Scholar] [CrossRef]
- Harrabi, H.; Kergoat, M.J.; Rousseau, J.; Boisjoly, H.; Schmaltz, H.; Moghadaszadeh, S.; Roy-Gagnon, M.H.; Freeman, E.E. Age-Related Eye Disease and Cognitive Function. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Su, C.W.; Lin, C.C.; Kao, C.H.; Chen, H.Y. Association Between Glaucoma and the Risk of Dementia. Medicine 2016, 95, e2833. [Google Scholar] [CrossRef]
- Varin, M.; Kergoat, M.J.; Belleville, S.; Li, G.; Rousseau, J.; Roy-Gagnon, M.H.; Moghadaszadeh, S.; Freeman, E.E. Age-Related Eye Disease and Cognitive Function: The Search for Mediators. Ophthalmology 2020, 127, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Maurano, S.T.P.; da Silva, D.J.; Ávila, M.P.; Magacho, L. Cognitive evaluation of patients with glaucoma and its comparison with individuals with Alzheimer’s disease. Int. Ophthalmol. 2018, 38, 1839–1844. [Google Scholar] [CrossRef]
- Lee, S.H.; Han, J.W.; Lee, E.J.; Kim, T.W.; Kim, H.; Kim, K.W. Cognitive Impairment and Lamina Cribrosa Thickness in Primary Open-Angle Glaucoma. Trans. Vis. Sci. Technol. 2020, 9, 17. [Google Scholar] [CrossRef]
- Glass, J.M. Visual Function and Cognitive Aging: Differential Role of Contrast Sensitivity in Verbal Versus Spatial Tasks. Psychol. Aging 2007, 22, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Bassuk, S.S.; Glass, T.A.; Berkman, L.F. Social disengagement and incident cognitive decline in community-dwelling elderly persons. Ann. Intern. Med. 1999, 131, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Mendes De Leon, C.F.; Barnes, L.L.; Schneider, J.A.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Participation in cognitively stimulating activities and risk of incident Alzheimer disease. JAMA 2002, 287, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Fong, J.; Ang, L.C.; Yücel, Y.H. Retinal tau pathology in human glaucomas. Can. J. Ophthalmol. 2008, 43, 53–60. [Google Scholar] [CrossRef]
- Oka, T.; Tamada, Y.; Nakajima, E.; Shearer, T.R.; Azuma, M. Presence of calpain-induced proteolysis in retinal degeneration and dysfunction in a rat model of acute ocular hypertension. J. Neurosci. Res. 2006, 83, 1342–1351. [Google Scholar] [CrossRef]
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Dewachter, I.; Van Leuven, F. Axonal transport, tau protein, and neurodegeneration in Alzheimer’s disease. Neuromolecular. Med. 2002, 2, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.G.; Quigley, H.A.; Zack, D.J.; Levkovitch-Verbin, H.; Kielczewski, J.; Valenta, D.; Baumrind, L.; Pease, M.E.; Klein, R.L.; Hauswirth, W.W. Gene therapy with brain-derived neurotrophic factor as a protection: Retinal ganglion cells in a rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4357–4365. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.L.; Hu, D.N.; Ritch, R.; Sharma, S.C.; Chen, C.F. Patterns of retinal ganglion cell survival after brain-derived neurotrophic factor administration in hypertensive eyes of rats. Neurosci. Lett. 2001, 305, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Ashok, A.; Singh, N.; Chaudhary, S.; Bellamkonda, V.; Kritikos, A.E.; Wise, A.S.; Rana, N.; McDonald, D.; Ayyagari, R. Retinal Degeneration and Alzheimer’s Disease: An Evolving Link. Int. J. Mol. Sci. 2020, 21, 7290. [Google Scholar] [CrossRef]
- Goldblum, D.; Kipfer-Kauer, A.; Sarra, G.M.; Wolf, S.; Frueh, B.E. Distribution of Amyloid Precursor Protein and Amyloid-β Immunoreactivity in DBA/2J Glaucomatous Mouse Retinas. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5085–5090. [Google Scholar] [CrossRef]
- Bennett, R.E.; DeVos, S.L.; Dujardin, S.; Corjuc, B.; Gor, R.; Gonzalez, J.; Roe, A.D.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Enhanced Tau Aggregation in the Presence of Amyloid β. Am. J. Pathol. 2017, 187, 1601–1612. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Dourlen, P.; Kilinc, D.; Malmanche, N.; Chapuis, J.; Lambert, J.C. The new genetic landscape of Alzheimer’s disease: From amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 2019, 138, 221–236. [Google Scholar] [CrossRef]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Vande Velde, C.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Jorgenson, E.; Hysi, P.; Khawaja, A.P.; Pendergrass, S.; Han, X.; Ong, J.S.; Hewitt, A.W.; Segrè, A.V.; Rouhana, J.M.; et al. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 2021, 12, 1258. [Google Scholar] [CrossRef]
- Zheng, C.; Liu, S.; Zhang, X.; Hu, Y.; Shang, X.; Zhu, Z.; Huang, Y.; Wu, G.; Xiao, Y.; Du, Z.; et al. Shared genetic architecture between the two neurodegenerative diseases: Alzheimer’s disease and glaucoma. Front. Aging Neurosci. 2022, 14, 880576. [Google Scholar] [CrossRef]
- Colnaghi, L.; Rondelli, D.; Muzi-Falconi, M.; Sertic, S. Tau and DNA Damage in Neurodegeneration. Brain Sci. 2020, 10, 946. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yu, Q.; Swerdlow, R.H.; Waites, C.L. Glucocorticoid-driven mitochondrial damage stimulates Tau pathology. Brain 2023, 146, 4378–4394. [Google Scholar] [CrossRef]
- Sorkhabi, R.; Ghorbanihaghjo, A.; Javadzadeh, A.; Rashtchizadeh, N.; Moharrery, M. Oxidative DNA damage and total antioxidant status in glaucoma patients. Mol. Vis. 2011, 17, 41. Available online: https://pmc/articles/PMC3021573/ (accessed on 4 September 2023). [PubMed]
- Mohanty, K.; Dada, R.; Dada, T. Oxidative DNA damage and reduced expression of DNA repair genes: Role in primary open angle glaucoma (POAG). Ophthalmic Genet. 2017, 38, 446–450. [Google Scholar] [CrossRef]
- Saccà, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA damage in the human trabecular meshwork: Clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005, 123, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Cuchra, M.; Markiewicz, L.; Mucha, B.; Pytel, D.; Szymanek, K.; Szemraj, J.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. The role of base excision repair in the development of primary open angle glaucoma in the Polish population. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2015, 778, 26–40. [Google Scholar] [CrossRef]
- Mozaffarieh, M.; Schoetzau, A.; Sauter, M.; Grieshaber, M.; Orgül, S.; Golubnitschaja, O.; Flammer, J. Comet assay analysis of single–stranded DNA breaks in circulating leukocytes of glaucoma patients. Mol. Vis. 2008, 14, 1584. Available online: https://pmc/articles/PMC2526097/ (accessed on 4 September 2023).
- Pezone, A.; Olivieri, F.; Napoli, M.V.; Procopio, A.; Avvedimento, E.V.; Gabrielli, A. Inflammation and DNA damage: Cause, effect or both. Nat. Rev. Rheumatol. 2023, 19, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Tuxworth, R.I.; Taylor, M.J.; Martin Anduaga, A.; Hussien-Ali, A.; Chatzimatthaiou, S.; Longland, J.; Thompson, A.M.; Almutiri, S.; Alifragis, P.; Kyriacou, C.P.; et al. Attenuating the DNA damage response to double-strand breaks restores function in models of CNS neurodegeneration. Brain Commun. 2019, 1, fcz005. [Google Scholar] [CrossRef]
- Yan, Z.; Liao, H.; Deng, C.; Zhong, Y.; Mayeesa, T.Z.; Zhuo, Y. DNA damage and repair in the visual center in the rhesus monkey model of glaucoma. Exp. Eye Res. 2022, 219, 109031. [Google Scholar] [CrossRef]
- Thadathil, N.; Delotterie, D.F.; Xiao, J.; Hori, R.; McDonald, M.P.; Khan, M.M. DNA Double-Strand Break Accumulation in Alzheimer’s Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol. Neurobiol. 2021, 58, 118–131. [Google Scholar] [CrossRef]
- Lyras, L.; Cairns, N.J.; Jenner, A.; Jenner, P.; Halliwell, B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 1997, 68, 2061–2069. [Google Scholar] [CrossRef]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef]
- Bartolome, F.; Carro, E.; Alquezar, C. Oxidative Stress in Tauopathies: From Cause to Therapy. Antioxidants 2022, 11, 1421. [Google Scholar] [CrossRef] [PubMed]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid. Med. Cell Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef]
- Liu, M.; Sui, D.; Dexheimer, T.; Hovde, S.; Deng, X.; Wang, K.W.; Lin, H.L.; Chien, H.T.; Kweon, H.K.; Kuo, N.S.; et al. Hyperphosphorylation Renders Tau Prone to Aggregate and to Cause Cell Death. Mol. Neurobiol. 2020, 57, 4704–4719. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Saito, Y.; Ruberu, N.N.; Sawabe, M.; Arai, T.; Tanaka, N.; Kakuta, Y.; Yamanouchi, H.; Murayama, S. Staging of Argyrophilic Grains: An Age-Associated Tauopathy. J. Neuropathol. Exp. Neurol. 2004, 63, 911–918. [Google Scholar] [CrossRef]
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 2020, 143, 2576–2593. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2021, 288, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Jana, M.; Paidi, R.K.; Majumder, M.; Raha, S.; Dasarathy, S.; Pahan, K. Tau fibrils induce glial inflammation and neuropathology via TLR2 in Alzheimer’s disease-related mouse models. J. Clin. Investig. 2023, 133, e161987. [Google Scholar] [CrossRef]
- Hernandez, M.R.; Miao, H.; Lukas, T. Astrocytes in glaucomatous optic neuropathy. Prog. Brain Res. 2008, 173, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, Y.; Leung, A.; Namekata, K.; Saitoh, S.; Bang Nguyen, H.; Takeda, A.; Danjo, Y.; Morizawa, Y.M.; Shigetomi, E.; Sano, F.; et al. Astrocytic dysfunction induced by ABCA1 deficiency causes optic neuropathy. Sci. Adv. 2022, 8, eabq1081. [Google Scholar] [CrossRef]
- Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2016, 323, 170–182. [Google Scholar] [CrossRef]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei Bs, H.S.; Zeppenfeld, D.; Xie, L.; Hongyi Kang, B.S.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Lohela, T.J.; Lilius, T.O.; Nedergaard, M. The glymphatic system: Implications for drugs for central nervous system diseases. Nat. Rev. Drug Discov. 2022, 21, 763–779. [Google Scholar] [CrossRef]
- Wostyn, P. Do normal-tension and high-tension glaucoma result from brain and ocular glymphatic system disturbances, respectively? Eye 2021, 35, 2905–2906. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; He, R.Q. Tau could protect DNA double helix structure. Biochim. Biophys. Acta Proteins Proteom. 2003, 1645, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; He, H.J.; Zhou, J.; Miao, J.Y.; Lu, J.; He, Y.G.; Pan, R.; Wei, Y.; Liu, Y.; He, R.Q. Hyperphosphorylation results in tau dysfunction in DNA folding and protection. J. Alzheimers Dis. 2013, 37, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; He, R.Q. Effect of phosphorylation and aggregation on tau binding to DNA. Protein Pept. Lett. 2002, 9, 349–357. [Google Scholar] [CrossRef]
- Benhelli-Mokrani, H.; Mansuroglu, Z.; Chauderlier, A.; Albaud, B.; Gentien, D.; Sommer, S.; Schirmer, C.; Laqueuvre, L.; Josse, T.; Buée, L.; et al. Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Res. 2018, 46, 11405–11422. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Redaelli, V.; Contiero, P.; Fabiano, S.; Tagliabue, G.; Perego, P.; Benussi, L.; Bruni, A.C.; Filippini, G.; Farinotti, M.; et al. Tau Mutations Serve as a Novel Risk Factor for Cancer. Cancer Res. 2018, 78, 3731–3739. [Google Scholar] [CrossRef] [PubMed]
- Asada-Utsugi, M.; Uemura, K.; Ayaki, T.; Uemura, M.T.; Minamiyama, S.; Hikiami, R.; Morimura, T.; Shodai, A.; Ueki, T.; Takahashi, R.; et al. Failure of DNA double-strand break repair by tau mediates Alzheimer’s disease pathology in vitro. Commun. Biol. 2022, 5, 358. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Adamec, E.; Vonsattel, J.P.; Nixon, R.A. DNA strand breaks in Alzheimer’s disease. Brain Res. 1999, 849, 67–77. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef]
- Warraich, S.T.; Yang, S.; Nicholson, G.A.; Blair, I.P. TDP-43: A DNA and RNA binding protein with roles in neurodegenerative diseases. Int. J. Biochem. Cell Biol. 2010, 42, 1606–1609. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passaro, M.L.; Matarazzo, F.; Abbadessa, G.; Pezone, A.; Porcellini, A.; Tranfa, F.; Rinaldi, M.; Costagliola, C. Glaucoma as a Tauopathy—Is It the Missing Piece in the Glaucoma Puzzle? J. Clin. Med. 2023, 12, 6900. https://doi.org/10.3390/jcm12216900
Passaro ML, Matarazzo F, Abbadessa G, Pezone A, Porcellini A, Tranfa F, Rinaldi M, Costagliola C. Glaucoma as a Tauopathy—Is It the Missing Piece in the Glaucoma Puzzle? Journal of Clinical Medicine. 2023; 12(21):6900. https://doi.org/10.3390/jcm12216900
Chicago/Turabian StylePassaro, Maria Laura, Francesco Matarazzo, Gianmarco Abbadessa, Antonio Pezone, Antonio Porcellini, Fausto Tranfa, Michele Rinaldi, and Ciro Costagliola. 2023. "Glaucoma as a Tauopathy—Is It the Missing Piece in the Glaucoma Puzzle?" Journal of Clinical Medicine 12, no. 21: 6900. https://doi.org/10.3390/jcm12216900
APA StylePassaro, M. L., Matarazzo, F., Abbadessa, G., Pezone, A., Porcellini, A., Tranfa, F., Rinaldi, M., & Costagliola, C. (2023). Glaucoma as a Tauopathy—Is It the Missing Piece in the Glaucoma Puzzle? Journal of Clinical Medicine, 12(21), 6900. https://doi.org/10.3390/jcm12216900