
Artificial Intelligence-Assisted Transcriptomic Analysis to Advance Cancer Immunotherapy
Abstract
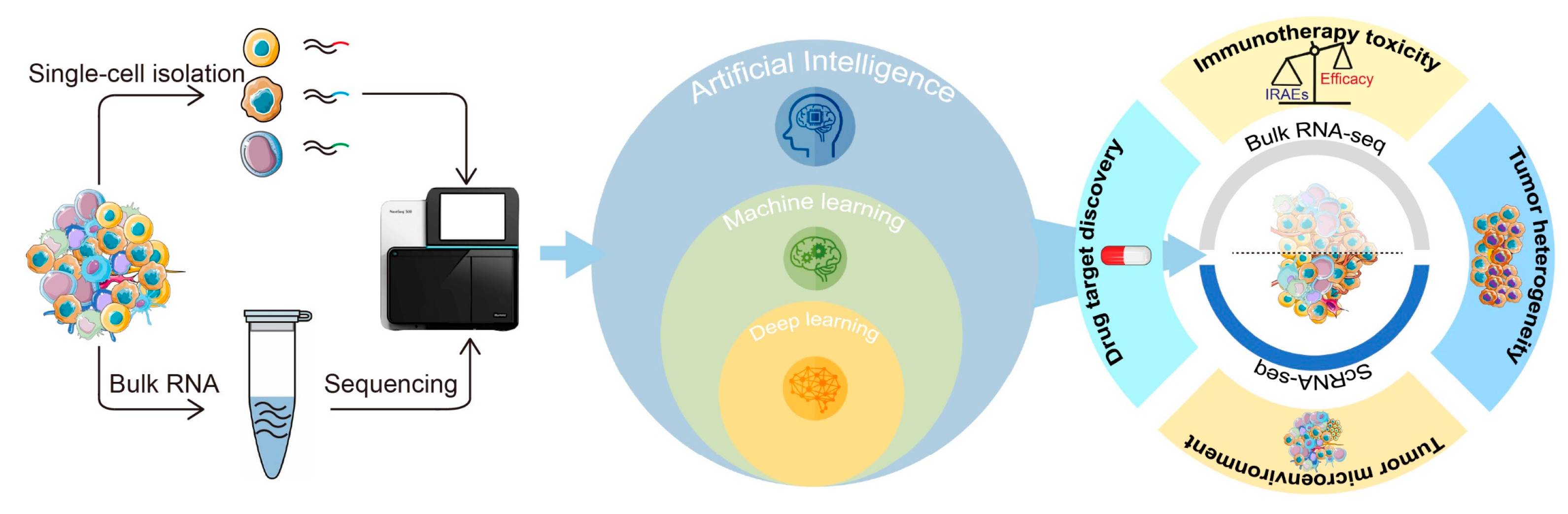
:1. Introduction
2. Artificial Intelligence Fosters the Application of Transcriptomics in Cancer Research
3. New Insights into Cancer Immunotherapy Based on AI-Assisted Transcriptomic Analysis
3.1. Tumor Heterogeneity
3.2. Tumor Microenvironment
3.3. Immunotherapy Toxicity
3.4. Drug Resistance and New Target Discovery
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suresh, K.; Naidoo, J.; Lin, C.T.; Danoff, S. Immune Checkpoint Immunotherapy for Non-Small Cell Lung Cancer. Chest 2018, 154, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Kanani, A.; Veen, T.; Søreide, K. Neoadjuvant immunotherapy in primary and metastatic colorectal cancer. Br. J. Surg. 2021, 108, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yu, J.; Meng, X. A good start of immunotherapy in esophageal cancer. Cancer Med. 2019, 8, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Friedman, C.; Proverbs-Singh, T.A.; Postow, M.A. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors. JAMA Oncol. 2016, 2, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Liu, J.; Fan, Z.; Zhao, W.; Zhou, X. Machine Intelligence in Single-Cell Data Analysis: Advances and New Challenges. Front. Genet. 2021, 12, 655536. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Liu, X.; Huang, J. Artificial intelligence for prediction of response to cancer immunotherapy. Semin. Cancer Biol. 2022, 87, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, L.; Xu, Y.; Yang, J. Machine learning meets omics: Applications and perspectives. Briefings Bioinform. 2021, 23, bbab460. [Google Scholar] [CrossRef]
- Houy, N.; Le Grand, F. Optimizing immune cell therapies with artificial intelligence. J. Theor. Biol. 2018, 461, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Shameer, K.; Johnson, K.; Glicksberg, B.; Dudley, J.T.; Sengupta, P.P. The whole is greater than the sum of its parts: Combining classical statistical and machine intelligence methods in medicine. Heart 2018, 104, 1228. [Google Scholar] [CrossRef]
- Luca, B.A.; Steen, C.B.; Matusiak, M.; Azizi, A.; Varma, S.; Zhu, C.; Przybyl, J.; Espín-Pérez, A.; Diehn, M.; Alizadeh, A.A.; et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell 2021, 184, 5482–5496.e28. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Ma, A.; Wang, Q.-E.; Liu, B.; Li, L.; Xu, D.; Ma, Q. Deep transfer learning of cancer drug responses by integrating bulk and single-cell RNA-seq data. Nat. Commun. 2022, 13, 6494. [Google Scholar] [CrossRef]
- Gao, S.; Han, L.; Luo, D.; Liu, G.; Xiao, Z.; Shan, G.; Zhang, Y.; Zhou, W. Modeling drug mechanism of action with large scale gene-expression profiles using GPAR, an artificial intelligence platform. BMC Bioinform. 2021, 22, 17. [Google Scholar] [CrossRef]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A Global View of Gene Activity and Alternative Splicing by Deep Sequencing of the Human Transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Leshchyk, A.; Johnson, N.T.; Korkin, D. A hybrid deep clustering approach for robust cell type profiling using single-cell RNA-seq data. Rna 2020, 26, 1303–1319. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Duan, X.; Gao, F.; Wang, W.; Liu, L.; Wang, X. Dissecting cancer heterogeneity based on dimension reduction of transcriptomic profiles using extreme learning machines. PLoS ONE 2018, 13, e0203824. [Google Scholar] [CrossRef]
- Amodio, M.; van Dijk, D.; Srinivasan, K.; Chen, W.S.; Mohsen, H.; Moon, K.R.; Campbell, A.; Zhao, Y.; Wang, X.; Venkataswamy, M.; et al. Exploring single-cell data with deep multitasking neural networks. Nat. Methods 2019, 16, 1139–1145. [Google Scholar] [CrossRef]
- Shen, Y.; Chu, Q.; Timko, M.P.; Fan, L. scDetect: A rank-based ensemble learning algorithm for cell type identification of single-cell RNA sequencing in cancer. Bioinformatics 2021, 37, 4115–4122. [Google Scholar] [CrossRef]
- Wang, D.; Hou, S.; Zhang, L.; Wang, X.; Liu, B.; Zhang, Z. iMAP: Integration of multiple single-cell datasets by adversarial paired transfer networks. Genome Biol. 2021, 22, 63. [Google Scholar] [CrossRef]
- Borcherding, N.; Voigt, A.P.; Liu, V.; Link, B.K.; Zhang, W.; Jabbari, A. Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated with Disease Progression. Clin. Cancer Res. 2019, 25, 2996–3005. [Google Scholar] [CrossRef]
- Del Giudice, M.; Peirone, S.; Perrone, S.; Priante, F.; Varese, F.; Tirtei, E.; Fagioli, F.; Cereda, M. Artificial Intelligence in Bulk and Single-Cell RNA-Sequencing Data to Foster Precision Oncology. Int. J. Mol. Sci. 2021, 22, 4563. [Google Scholar] [CrossRef]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. Iscience 2022, 25, 103798. [Google Scholar] [CrossRef]
- Michot, J.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef]
- Suphavilai, C.; Chia, S.; Sharma, A.; Tu, L.; Da Silva, R.P.; Mongia, A.; DasGupta, R.; Nagarajan, N. Predicting heterogeneity in clone-specific therapeutic vulnerabilities using single-cell transcriptomic signatures. Genome Med. 2021, 13, 189. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.-Y.; Shin, H.-T.; Sohn, K.-A.; Shin, S.-Y.; Park, W.-Y.; Joung, J.-G. Assessment of intratumoral heterogeneity with mutations and gene expression profiles. PLoS ONE 2019, 14, e0219682. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Gu, S.S.; Wong, C.J.; Yang, L.; Ouardaoui, N.; Li, D.; Zhang, W.; Brown, M.; Liu, X.S. Machine learning on syngeneic mouse tumor profiles to model clinical immunotherapy response. Sci. Adv. 2022, 8, eabm8564. [Google Scholar] [CrossRef]
- Barnes, B.M.; Nelson, L.; Tighe, A.; Burghel, G.J.; Lin, I.-H.; Desai, S.; McGrail, J.C.; Morgan, R.D.; Taylor, S.S. Distinct transcriptional programs stratify ovarian cancer cell lines into the five major histological subtypes. Genome Med. 2021, 13, 140. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Z.; Zhou, K.; Wang, C.; Jiang, L.; Zhang, L.; Yang, Y.; Luo, W.; Qiao, W.; Wang, G.; et al. Deciphering cell lineage specification of human lung adenocarcinoma with single-cell RNA sequencing. Nat. Commun. 2021, 12, 6500. [Google Scholar] [CrossRef]
- Zhao, H.; Gao, Y.; Miao, J.; Chen, S.; Li, J.; Li, Z.; Yin, C.; Yue, W. Single-cell RNA-seq highlights a specific carcinoembryonic cluster in ovarian cancer. Cell Death Dis. 2021, 12, 1082. [Google Scholar] [CrossRef]
- van Galen, P.; Hovestadt, V.; Wadsworth, M.H., II.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Fan, J.; Lee, H.-O.; Lee, S.; Ryu, D.-E.; Lee, S.; Xue, C.; Kim, S.J.; Kim, K.; Barkas, N.; Park, P.J.; et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 2018, 28, 1217–1227. [Google Scholar] [CrossRef]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Deng, Y.; Tan, Y.; Zhou, D.; Bai, Y.; Cao, T.; Zhong, C.; Huang, W.; Ou, Y.; Guo, L.; Liu, Q.; et al. Single-Cell RNA-Sequencing Atlas Reveals the Tumor Microenvironment of Metastatic High-Grade Serous Ovarian Carcinoma. Front. Immunol. 2022, 13, 923194. [Google Scholar] [CrossRef] [PubMed]
- Thankamony, A.; Subbalakshmi, A.; Jolly, M.; Nair, R. Lineage Plasticity in Cancer: The Tale of a Skin-Walker. Cancers 2021, 13, 3602. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, R.; Yang, Z.; Li, W.; Yang, J.; Wang, Z.; Bai, H.; Cui, Y.; Tian, Y.; Wu, Z.; et al. Molecular profiling of human non-small cell lung cancer by single-cell RNA-seq. Genome Med. 2022, 14, 87. [Google Scholar] [CrossRef]
- Ono, H.; Arai, Y.; Furukawa, E.; Narushima, D.; Matsuura, T.; Nakamura, H.; Shiokawa, D.; Nagai, M.; Imai, T.; Mimori, K.; et al. Single-cell DNA and RNA sequencing reveals the dynamics of intra-tumor heterogeneity in a colorectal cancer model. BMC Biol. 2021, 19, 207. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, L.; Khatib, S.A.; Chang, C.-W.; Heinrich, S.; Dominguez, D.A.; Forgues, M.; Candia, J.; Hernandez, M.O.; Kelly, M.; et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 75, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Yang, T.; Xing, H.; Wang, Y.; Gao, L.; Guo, X.; Xing, B.; Wang, Y.; Ma, W. Machine learning revealed stemness features and a novel stemness-based classification with appealing implications in discriminating the prognosis, immunotherapy and temozolomide responses of 906 glioblastoma patients. Briefings Bioinform. 2021, 22, bbab032. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Menden, K.; Marouf, M.; Oller, S.; Dalmia, A.; Magruder, D.S.; Kloiber, K.; Heutink, P.; Bonn, S. Deep learning–based cell composition analysis from tissue expression profiles. Sci. Adv. 2020, 6, eaba2619. [Google Scholar] [CrossRef]
- Dai, Y.; Qiang, W.; Lin, K.; Gui, Y.; Lan, X.; Wang, D. An immune-related gene signature for predicting survival and immunotherapy efficacy in hepatocellular carcinoma. Cancer Immunol. Immunother. 2020, 70, 967–979. [Google Scholar] [CrossRef]
- Cai, W.-Y.; Dong, Z.-N.; Fu, X.-T.; Lin, L.-Y.; Wang, L.; Ye, G.-D.; Luo, Q.-C.; Chen, Y.-C. Identification of a Tumor Microenvironment-relevant Gene set-based Prognostic Signature and Related Therapy Targets in Gastric Cancer. Theranostics 2020, 10, 8633–8647. [Google Scholar] [CrossRef]
- Liang, Q.; Wu, J.; Zhao, X.; Shen, S.; Zhu, C.; Liu, T.; Cui, X.; Chen, L.; Wei, C.; Cheng, P.; et al. Establishment of tumor inflammasome clusters with distinct immunogenomic landscape aids immunotherapy. Theranostics 2021, 11, 9884–9903. [Google Scholar] [CrossRef]
- Lu, Z.; Chen, H.; Jiao, X.; Zhou, W.; Han, W.; Li, S.; Liu, C.; Gong, J.; Li, J.; Zhang, X.; et al. Prediction of immune checkpoint inhibition with immune oncology-related gene expression in gastrointestinal cancer using a machine learning classifier. J. Immunother. Cancer 2020, 8, e000631. [Google Scholar] [CrossRef]
- Chen, X.; Chen, H.; Yao, H.; Zhao, K.; Zhang, Y.; He, D.; Zhu, Y.; Cheng, Y.; Liu, R.; Xu, R.; et al. Turning up the heat on non-immunoreactive tumors: Pyroptosis influences the tumor immune microenvironment in bladder cancer. Oncogene 2021, 40, 6381–6393. [Google Scholar] [CrossRef] [PubMed]
- Thedinga, K.; Herwig, R. A gradient tree boosting and network propagation derived pan-cancer survival network of the tumor microenvironment. Iscience 2021, 25, 103617. [Google Scholar] [CrossRef] [PubMed]
- Schizas, D.; Charalampakis, N.; Kole, C.; Mylonas, K.S.; Katsaros, I.; Zhao, M.; Ajani, J.A.; Psyrri, A.; Karamouzis, M.V.; Liakakos, T. Immunotherapy for esophageal cancer: A 2019 update. Immunotherapy 2020, 12, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, M.; Liang, J.; Hu, Z.; Huang, Y.; Li, M.; Pang, Y.; Lu, T.; Sui, Q.; Zhan, C.; et al. Dissecting the single-cell transcriptome network underlying esophagus non-malignant tissues and esophageal squamous cell carcinoma. Ebiomedicine 2021, 69, 103459. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, F.; Chen, F.; Chen, Y.; Ge, D.; Zhang, S.; Lu, C. Transcriptomics based multi-dimensional characterization and drug screen in esophageal squamous cell carcinoma. Ebiomedicine 2021, 70, 103510. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Mei, Y.; Xiao, W.; Hu, H.; Lu, G.; Chen, L.; Sun, Z.; Lü, M.; Ma, W.; Jiang, T.; Gao, Y.; et al. Single-cell analyses reveal suppressive tumor microenvironment of human colorectal cancer. Clin. Transl. Med. 2021, 11, e422. [Google Scholar] [CrossRef]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.-J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’Gorman, W.E.; Au-Yeung, A.; et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020, 579, 274–278. [Google Scholar] [CrossRef]
- Caushi, J.X.; Zhang, J.; Ji, Z.; Vaghasia, A.; Zhang, B.; Hsiue, E.H.-C.; Mog, B.J.; Hou, W.; Justesen, S.; Blosser, R.; et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 2021, 596, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Lu, S.; Hua, J.; Xu, J.; Wei, M.; Liang, C.; Meng, Q.; Liu, J.; Zhang, B.; Wang, W.; Yu, X.; et al. Turning towards nonimmunoreactive tumors: Evaluation of cancer-associated fibroblasts enables prediction of the immune microenvironment and treatment sensitivity in pancreatic cancer. Comput. Struct. Biotechnol. J. 2022, 20, 3911–3923. [Google Scholar] [CrossRef]
- Yang, X.; An, Z.; Hu, Z.; Xi, J.; Dai, C.; Zhu, Y. Expression Analysis of Ligand-Receptor Pairs Identifies Cell-to-Cell Crosstalk between Macrophages and Tumor Cells in Lung Adenocarcinoma. J. Immunol. Res. 2022, 2022, 9589895. [Google Scholar] [CrossRef] [PubMed]
- Steen, C.B.; Luca, B.A.; Esfahani, M.S.; Azizi, A.; Sworder, B.J.; Nabet, B.Y.; Kurtz, D.M.; Liu, C.L.; Khameneh, F.; Advani, R.H.; et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell 2021, 39, 1422–1437.e10. [Google Scholar] [CrossRef]
- Chuah, S.; Lee, J.; Song, Y.; Kim, H.-D.; Wasser, M.; Kaya, N.A.; Bang, K.; Lee, Y.J.; Jeon, S.H.; Suthen, S.; et al. Uncoupling immune trajectories of response and adverse events from anti-PD-1 immunotherapy in hepatocellular carcinoma. J. Hepatol. 2022, 77, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Marschner, D.; Falk, M.; Javorniczky, N.R.; Hanke-Müller, K.; Rawluk, J.; Schmitt-Graeff, A.; Simonetta, F.; Haring, E.; Dicks, S.; Ku, M.; et al. MicroRNA-146a regulates immune-related adverse events caused by immune checkpoint inhibitors. J. Clin. Investig. 2020, 5, e132334. [Google Scholar] [CrossRef] [PubMed]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef]
- Bai, X.; Wang, X.; Ma, G.; Song, J.; Liu, X.; Wu, X.; Zhao, Y.; Liu, X.; Liu, Z.; Zhang, W.; et al. Improvement of PD-1 Blockade Efficacy and Elimination of Immune-Related Gastrointestinal Adverse Effect by mTOR Inhibitor. Front. Immunol. 2021, 12, 793831. [Google Scholar] [CrossRef] [PubMed]
- Berner, F.; Bomze, D.; Lichtensteiger, C.; Walter, V.; Niederer, R.; Ali, O.H.; Wyss, N.; Bauer, J.; Freudenmann, L.K.; Marcu, A.; et al. Autoreactive napsin A–specific T cells are enriched in lung tumors and inflammatory lung lesions during immune checkpoint blockade. Sci. Immunol. 2022, 7, eabn9644. [Google Scholar] [CrossRef]
- Zhu, H.; Galdos, F.X.; Lee, D.; Waliany, S.; Huang, Y.V.; Ryan, J.; Dang, K.; Neal, J.W.; Wakelee, H.A.; Reddy, S.A.; et al. Identification of Pathogenic Immune Cell Subsets Associated With Checkpoint Inhibitor–Induced Myocarditis. Circulation 2022, 146, 316–335. [Google Scholar] [CrossRef]
- Bronson, S.; Stirling, E.; Westwood, B.; Triozzi, P.; Soto-Pantoja, D. 806 PD-1 blockade affects inflammation and metabolic flexibility to potentially mediate cardiac immune-related adverse events. J. Immunother. Cancer 2021, 9, A843. [Google Scholar] [CrossRef]
- Llewellyn, H.P.; Arat, S.; Gao, J.; Wen, J.; Xia, S.; Kalabat, D.; Oziolor, E.; Virgen-Slane, R.; Affolter, T.; Ji, C. T cells and monocyte-derived myeloid cells mediate immunotherapy-related hepatitis in a mouse model. J. Hepatol. 2021, 75, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Chu, Y.; Misoi, M.; Suarez-Almazor, M.E.; Tayar, J.H.; Lu, H.; Buni, M.; Kramer, J.; Rodriguez, E.; Hussain, Z.; et al. Distinct molecular and immune hallmarks of inflammatory arthritis induced by immune checkpoint inhibitors for cancer therapy. Nat. Commun. 2022, 13, 1970. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Hur, J.Y.; Cho, J.; Ku, B.M.; Koh, J.; Koh, J.Y.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; et al. Immune-related adverse events are clustered into distinct subtypes by T-cell profiling before and early after anti-PD-1 treatment. Oncoimmunology 2020, 9, 1722023. [Google Scholar] [CrossRef]
- Grigoriou, M.; Banos, A.; Hatzioannou, A.; Kloetgen, A.; Kouzis, P.; Aggouraki, D.; Zakopoulou, R.; Bamias, G.; Kassi, E.; Mavroudis, D.; et al. Regulatory T-cell Transcriptomic Re-programming Characterizes Adverse Events by Checkpoint Inhibitors in Solid Tumors. Cancer Immunol. Res. 2021, 9, 726–734. [Google Scholar] [CrossRef]
- Das, R.; Bar, N.; Ferreira, M.; Newman, A.M.; Zhang, L.; Bailur, J.K.; Bacchiocchi, A.; Kluger, H.; Wei, W.; Halaban, R.; et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J. Clin. Investig. 2018, 128, 715–720. [Google Scholar] [CrossRef]
- Siwicki, M.; Gort-Freitas, N.A.; Messemaker, M.; Bill, R.; Gungabeesoon, J.; Engblom, C.; Zilionis, R.; Garris, C.; Gerhard, G.M.; Kohl, A.; et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci. Immunol. 2021, 6, eabi7083. [Google Scholar] [CrossRef]
- Bareche, Y.; Kelly, D.; Abbas-Aghababazadeh, F.; Nakano, M.; Esfahani, P.; Tkachuk, D.; Mohammad, H.; Samstein, R.; Lee, C.-H.; Morris, L.; et al. Leveraging big data of immune checkpoint blockade response identifies novel potential targets. Ann. Oncol. 2022, 33, 1304–1317. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Cooley, L.S.; Rudewicz, J.; Souleyreau, W.; Emanuelli, A.; Alvarez-Arenas, A.; Clarke, K.; Falciani, F.; Dufies, M.; Lambrechts, D.; Modave, E.; et al. Experimental and computational modeling for signature and biomarker discovery of renal cell carcinoma progression. Mol. Cancer 2021, 20, 136. [Google Scholar] [CrossRef]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, A.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Bulanova, D.; Oikkonen, J.; Häkkinen, A.; Zhang, K.; Zheng, S.; Wang, W.; Erkan, E.P.; Carpén, O.; Joutsiniemi, T.; et al. Network-guided identification of cancer-selective combinatorial therapies in ovarian cancer. Briefings Bioinform. 2021, 22, bbab272. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Rockstroh, A.; Lehman, M.; Ratther, E.; Jain, A.; Anand, A.; Gupta, A.; Bhattacharya, N.; Poonia, S.; Rai, P.; et al. Gene expression based inference of cancer drug sensitivity. Nat. Commun. 2022, 13, 5680. [Google Scholar] [CrossRef] [PubMed]
- Onieva, J.L.; Xiao, Q.; Berciano-Guerrero, M.; Laborda-Illanes, A.; de Andrea, C.; Chaves, P.; Piñeiro, P.; Garrido-Aranda, A.; Gallego, E.; Sojo, B.; et al. High IGKC-Expressing Intratumoral Plasma Cells Predict Response to Immune Checkpoint Blockade. Int. J. Mol. Sci. 2022, 23, 9124. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, N.; Wang, T.; Zhang, E.; Wang, X.; Zheng, J. Biomarkers of the Response to Immune Checkpoint Inhibitors in Metastatic Urothelial Carcinoma. Front. Immunol. 2020, 11, 1900. [Google Scholar] [CrossRef] [PubMed]
- Somani, V.K.; Zhang, D.; Dodhiawala, P.B.; Lander, V.E.; Liu, X.; Kang, L.-I.; Chen, H.-P.; Knolhoff, B.L.; Li, L.; Grierson, P.M.; et al. IRAK4 Signaling Drives Resistance to Checkpoint Immunotherapy in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2022, 162, 2047–2062. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Corvino, D.; Nowlan, B.; Aguilera, A.R.; Ng, S.S.; Braun, M.; Cillo, A.R.; Bald, T.; Smyth, M.J.; Engwerda, C.R. NKG7 Is Required for Optimal Antitumor T-cell Immunity. Cancer Immunol. Res. 2022, 10, 154–161. [Google Scholar] [CrossRef]
- Lim, Y.W.; Coles, G.L.; Sandhu, S.K.; Johnson, D.S.; Adler, A.S.; Stone, E.L. Single-cell transcriptomics reveals the effect of PD-L1/TGF-β blockade on the tumor microenvironment. BMC Biol. 2021, 19, 107. [Google Scholar] [CrossRef]
- He, D.; Wang, D.; Lu, P.; Yang, N.; Xue, Z.; Zhu, X.; Zhang, P.; Fan, G. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene 2020, 40, 355–368. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef]
- Gao, J.; Wu, Z.; Zhao, M.; Zhang, R.; Li, M.; Sun, D.; Cheng, H.; Qi, X.; Shen, Y.; Xu, Q.; et al. Allosteric inhibition reveals SHP2-mediated tumor immunosuppression in colon cancer by single-cell transcriptomics. Acta Pharm. Sin. B 2021, 12, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Z.; Zhu, Y.; Fu, J.; Zhao, X.; Zhang, Y.; Wang, S.; Wu, J.; Wang, K.; Wu, R.; et al. Single-Cell Transcriptome Analysis Uncovers Intratumoral Heterogeneity and Underlying Mechanisms for Drug Resistance in Hepatobiliary Tumor Organoids. Adv. Sci. 2021, 8, e2003897. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Phoon, Y.P.; Karlinsey, K.; Tian, Y.F.; Thapaliya, S.; Thongkum, A.; Qu, L.; Matz, A.J.; Cameron, M.; Cameron, C.; et al. A high OXPHOS CD8 T cell subset is predictive of immunotherapy resistance in melanoma patients. J. Exp. Med. 2021, 219, e20202084. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, K.; Portell, A.J.; Ivanova, E.V.; Lizotte, P.H.; Mahadevan, N.R.; Greene, J.R.; Vajdi, A.; Gurjao, C.; Teceno, T.; Taus, L.J.; et al. Dynamic single-cell RNA sequencing identifies immunotherapy persister cells following PD-1 blockade. J. Clin. Investig. 2021, 131, e135038. [Google Scholar] [CrossRef]
- Samanta, D.; Huang, T.Y.-T.; Shah, R.; Yang, Y.; Pan, F.; Semenza, G.L. BIRC2 Expression Impairs Anti-Cancer Immunity and Immunotherapy Efficacy. Cell Rep. 2020, 32, 108073. [Google Scholar] [CrossRef]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Input Data | AI Model | Tumor | Function | Application | Ref. |
|---|---|---|---|---|---|
| Bulk RNA-seq data | ML: Innovative one-class logistic regression | Pan-cancer | Indicating prognosis, molecular subtypes, and immunotherapy efficacy | ITH | [52] |
| ScRNA-seq data | ML: RF | Acute myeloid leukemia | Distinguishing cell types in the acute myeloid leukemia ecosystem | ITH | [40] |
| ScRNA-seq data | ML: Consensus clustering | Hepatocellular carcinoma and Intrahepatic cholangiocarcinoma | Determining tumor cell states | ITH | [51] |
| Spatial RNA-seq data and images | DL: CNN | Breast cancer | Linking gene expression with visual features in cell morphology | ITH | [91] |
| Bulk RNA-seq and ICB response data | ML: NMF | Syngeneic cancer cell lines and mouse model across 13 cancer types | Reliably stratifying patients regarding ICB response | ITH | [34] |
| Bulk RNA-seq, scRNA-seq and drug response data | ML: RF | Ovarian cancer | Guiding identification of safe and effective combinatorial treatment regimens | Drug resistance | [92] |
| Bulk RNA-seq and mutations data | ML: SVM | 12 cancer types in TCGA | Discriminating tumors with heterogeneous cell populations | ITH | [33] |
| ScRNA-seq data | DL: Denoising autoencoder | Malignant melanoma | Addressing a substantial amount of noise | ITH | [23] |
| ScRNA-seq data | ML: k-TSP | Various types of cancer | Classifying a single cell type and minimizing the batch effects | ITH | [26] |
| Bulk RNA-seq data | DL: ELM | Gastric cancer, ovarian cancer, and so on | Identifying cancer molecular subtypes that are much more clinically relevant | ITH | [24] |
| Bulk RNA-seq and scRNA-seq data | DL: DNN | 550 cancer cell lines | Inferring treatment response in cancers | Drug resistance | [93] |
| ScRNA-seq data | DL: GAN, deep autoencoders | Colorectal cancer | Identifying the batch-specific cell types | ITH, TME | [27] |
| Bulk RNA-seq data | ML: NMF | Ovarian cancer | Supporting the classification of new model systems of ovarian cancer subtypes | ITH | [35] |
| Bulk RNA-seq and scRNA-seq data | DL: DaNN | Various types of cancer | Predicting drug responses in scRNA-seq | ITH, drug resistance | [16] |
| Bulk RNA-seq, scRNA-seq and spatial RNA-seq data | ML: EcoTyper | 16 types of carcinoma | Providing a framework for large-scale profiling of cellular ecosystems in any tissue | TME, ITH | [15] |
| Bulk RNA-seq data | ML: SVM | 33 tumor types from TCGA | Confirming the classification of inflammasome | TME | [60] |
| Bulk RNA-seq data | ML: linear SVM | Gastrointestinal cancer | Predicting immunotherapy response in patients | TME | [61] |
| Bulk RNA-seq data | ML: XGBoost, RWR | 25 tumor types from TCGA | Predicting the TME and immune states of patients | TME | [63] |
| Bulk RNA-seq and scRNA-seq data | ML: XGBoost | Lung adenocarcinoma | Predicting the prognosis of ligand–receptor gene pairs | TME | [73] |
| Bulk RNA-seq data | ML: RF, LASSO | Pancreatic cancer | Predicting the abundance of immune cell infiltration and the response of immunotherapy | TME | [72] |
| Bulk RNA-seq and scRNA-seq data | ML: PCA, RF | 12 tumor types, melanoma | Predicting response to ICIs | Drug resistance | [88,94] |
| Bulk RNA-seq data | ML: LASSO | Urothelial carcinoma | Estimating ICI response and prognosis of patients | Drug resistance | [95] |
| Bulk RNA-seq data | ML: RSF, PCA | Renal cell carcinoma | Predicting tumor progression and relapse | Target discovery | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gui, Y.; He, X.; Yu, J.; Jing, J. Artificial Intelligence-Assisted Transcriptomic Analysis to Advance Cancer Immunotherapy. J. Clin. Med. 2023, 12, 1279. https://doi.org/10.3390/jcm12041279
Gui Y, He X, Yu J, Jing J. Artificial Intelligence-Assisted Transcriptomic Analysis to Advance Cancer Immunotherapy. Journal of Clinical Medicine. 2023; 12(4):1279. https://doi.org/10.3390/jcm12041279
Chicago/Turabian StyleGui, Yu, Xiujing He, Jing Yu, and Jing Jing. 2023. "Artificial Intelligence-Assisted Transcriptomic Analysis to Advance Cancer Immunotherapy" Journal of Clinical Medicine 12, no. 4: 1279. https://doi.org/10.3390/jcm12041279
APA StyleGui, Y., He, X., Yu, J., & Jing, J. (2023). Artificial Intelligence-Assisted Transcriptomic Analysis to Advance Cancer Immunotherapy. Journal of Clinical Medicine, 12(4), 1279. https://doi.org/10.3390/jcm12041279