
Immunothrombosis: Molecular Aspects and New Therapeutic Perspectives


Abstract
:1. Introduction
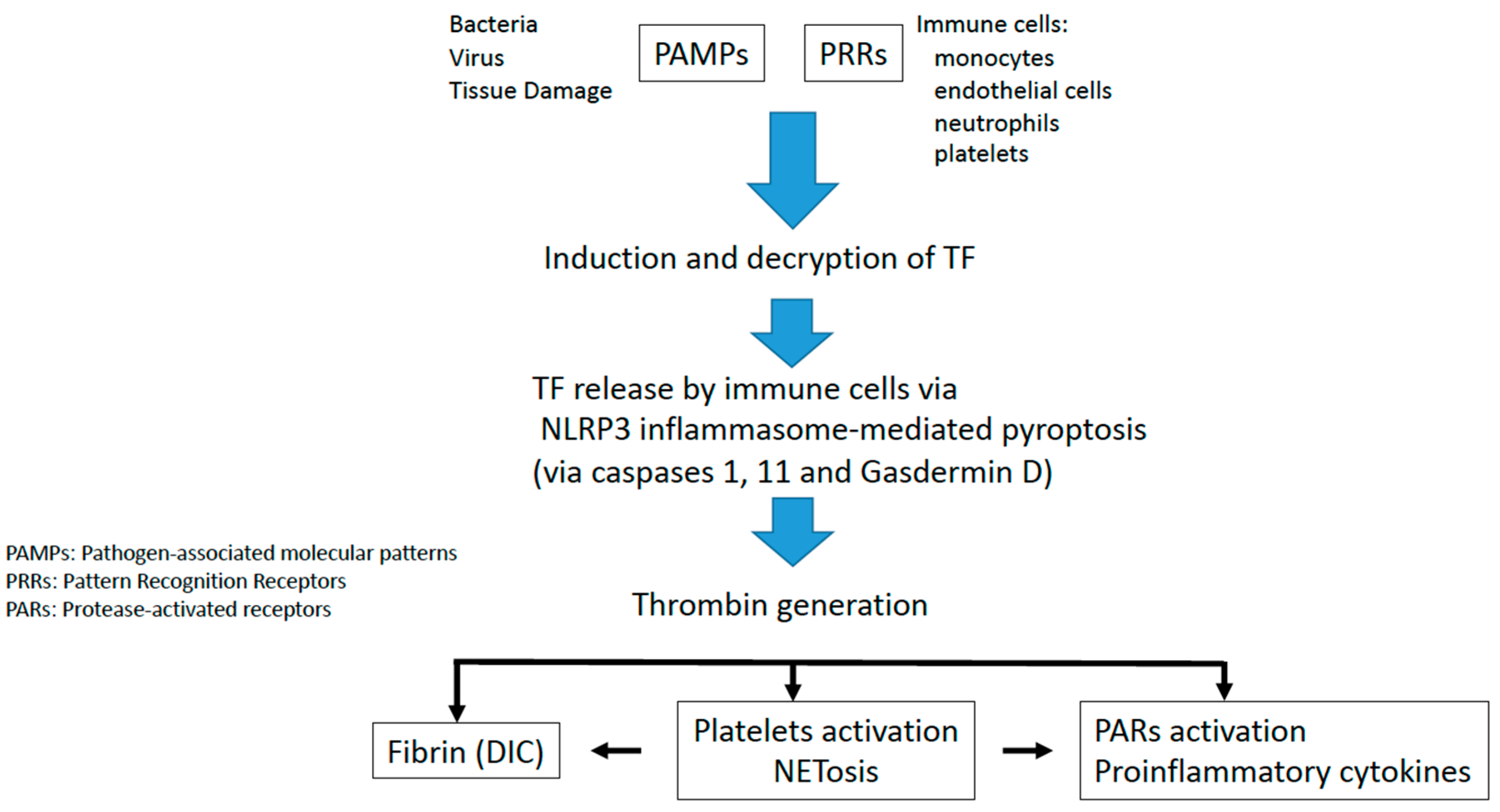
2. Tissue Factor Initiates Immunothrombosis
3. Inflammasome Activation
4. Role of NETs in Immunothrombosis
5. Role of the Von Willebrand Factor in Immunothrombosis
6. Infectious Disease-Induced Immunothrombosis
7. Thromboinflammatory Conditions
7.1. Arterial Diseases and Immunothrombosis
7.2. Autoimmune Diseases
8. Immunothrombosis as a New Therapeutical Target
- (a)
- Targeting coagulation
- (b)
- Targeting NETs
- (c)
- Targeting inflammation
- (d)
- Targeting Complement

{kind=link}
| Target | Drugs |
|---|---|
| Coagulation | |
| Anticoagulant | Low molecular weight heparin [66], fondaparinux [76]. |
| Antiplatelets | Aspirin [77], ticagrelor [78]. |
| NETS | Colchicine [73,74], heparin [79], aspirin [77], ticagrelor [78], DNASes [26]. |
| Inflammation | |
| JAK-STAT pathway inhibitors | Baricitinib, ruxolitinib, tafacitinib [80]. |
| STING inhibitors | Nitrofurans, acrylamides, indole ureas [68]. |
| Inflammasome inhibitors (NLRP3) | MCC950 [71], colchicine [73,74]. |
| Gasdermin D inhibitors | Dimetil fumarate [70,71]. |
| HMGB1 inhibitors | Peptide p5779, m2G7, metformin, thrombomodulin [72]. |
| Complement | Eculizumab [75]. |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Delvaeye, M.; Conway, E.M. Coagulation and innate immune responses: Can we view them separately? Blood 2009, 114, 2367–2374. [Google Scholar] [CrossRef]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef]
- Páramo, J.A.; Lecumberri, R. New mechanisms in vein thrombosis: Immunothrombosis. Med. Clin. 2019, 153, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2012, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Tissue Factor. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef]
- Wu, C.; Lu, W.; Zhang, Y.; Zhang, G.; Shi, X.; Hisada, Y.; Grover, X.; Zhang, X.; Li, L.; Xiang, B.; et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 2019, 50, 1401–1411.e4. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 4, 85. [Google Scholar] [CrossRef]
- Chen, V.M.; Hogg, P.J. Encryption and decryption of tissue factor. J. Thromb. Haemost. 2013, 11 (Suppl. S1), 277–284. [Google Scholar] [CrossRef]
- Wang, J.; Pendurthi, U.R.; Vijaya Mohan Rao, L. Sphingomyelin encrypts tissue factor: ATP-induced activation of A-SMase leads to tissue factor decryption and microvesicle shedding. Blood Adv. 2017, 1, 849. [Google Scholar] [CrossRef]
- Geddings, J.E.; Hisada, Y.; Boulaftali, Y.; Getz, T.M.; Whelihan, M.; Fuentes, R.; Dee, R.; Cooley, B.C.; Key, N.S.; Wolberg, A.S.; et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J. Thromb. Haemost. 2016, 14, 153–166. [Google Scholar] [CrossRef]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 1–24. [Google Scholar]
- Páramo, J.A. Neutrófilos como instigadores de trombosis: Más allá de la protección antimicrobiana. Rev. Clin. Esp. 2020, 220, 583–586. [Google Scholar] [CrossRef]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Jayarangaiah, A.; Kariyanna, P.T.; Chen, X.; Jayarangaiah, A.; Kumar, A. COVID-19-Associated Coagulopathy: An Exacerbated Immunothrombosis Response. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620943293. [Google Scholar] [CrossRef]
- Ryan, T.A.J.; O’Neill, L.A.J. Innate immune signaling and immunothrombosis: New insights and therapeutic opportunities. Eur. J. Immunol. 2022, 52, 1024–1034. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983–996.e6. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, J.; Zhang, G.; Wu, C.; Abdel-Latif, A.; Smyth, S.S.; Shiroishi, T.; Mackman, N.; Wei, Y.; Tao, M.; et al. Inflammasome activation promotes venous thrombosis through pyroptosis. Blood Adv. 2021, 5, 2619–2623. [Google Scholar] [CrossRef] [PubMed]
- Páramo, J.A. Microvascular thrombosis and clinical implications. Med. Clin. 2021, 156, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.J.; Preston, R.J.S.; O’Neill, L.A.J. Immunothrombosis and the molecular control of tissue factor by pyroptosis: Prospects for new anticoagulants. Biochem. J. 2022, 479, 731–750. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Montealegre Sanchez, G.A.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wang, H.; Tang, Y.; Fan, Z.; Xiao, X.; Chen, F. High mobility group box 1 protein induces tissue factor expression in vascular endothelial cells via activation of NF-κB and Egr-1. Thromb. Haemost. 2009, 102, 352. [Google Scholar]
- van Bruggen, S.; Martinod, K. The coming of age of neutrophil extracellular traps in thrombosis: Where are we now and where are we headed? Immunol. Rev. 2022, 1–23. [Google Scholar] [CrossRef]
- Hidalgo, A.; Libby, P.; Soehnlein, O.; Aramburu, I.V.; Papayannopoulos, V.; Silvestre-Roig, C. Neutrophil extracellular traps: From physiology to pathology. Cardiovasc. Res. 2022, 118, 2737–2753. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Yang, J.; Wu, Z.; Long, Q.; Huang, J.; Hong, T.; Liu, W.; Lin, J. Insights Into Immunothrombosis: The Interplay Among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front. Immunol. 2020, 11, 3116. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kühne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu-Bercu, A.; Grassi, L.; Frontini, M.; Salles-Crawley, I.I.; Woollard, K.J.; Crawley, J.T.B. Activated αiibβ3 on platelets mediates flow-dependent netosis via slc44a2. Elife 2020, 9, e53353. [Google Scholar] [CrossRef]
- McKenna, E.; Wubben, R.; Isaza-Correa, J.M.; Melo, A.M.; Mhaonaigh, A.U.; Conlon, N.; O’Donnell, J.S.; Cheallaigh, C.N.; Hurley, T.; Stevenson, N.J.; et al. Neutrophils in COVID-19: Not Innocent Bystanders. Front. Immunol. 2022, 13, 2548. [Google Scholar] [CrossRef]
- Brehm, M.A. Von Willebrand factor processing. Hamostaseologie 2017, 37, 59–72. [Google Scholar] [CrossRef]
- Bernardo, A.; Ball, C.; Nolasco, L.; Choi, H.; Moake, J.L.; Dong, J.F. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J. Thromb. Haemost. 2005, 3, 562–570. [Google Scholar] [CrossRef]
- Ono, T.; Mimuro, J.; Madoiwa, S.; Soejima, K.; Kashiwakura, Y.; Ishiwata, A.; Takano, K.; Ohmori, T.; Sakata, Y. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: Its correlation with development of renal failure. Blood 2006, 107, 528–534. [Google Scholar] [CrossRef]
- Staessens, S.; Denorme, F.; Francois, O.; Desender, L.; Dewaele, T.; Vanacker, P.; Deckmyn, H.; Vanhoorelbeke, K.; Andersson, T.; De Meyer, S.F. Structural analysis of ischemic stroke thrombi: Histological indications for therapy resistance. Haematologica 2020, 105, 498–507. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; DE Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef]
- South, K.; Lane, D.A. ADAMTS-13 and von Willebrand factor: A dynamic duo. J. Thromb. Haemost. 2018, 16, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Ramakrishna, K.; Dhamoon, A.S. Sepsis: The evolution in definition, pathophysiology, and management. SAGE Open Med. 2019, 7, 2050312119835043. [Google Scholar] [CrossRef] [PubMed]
- Klavina, P.A.; Leon, G.; Curtis, A.M.; Preston, R.J.S. Dysregulated haemostasis in thrombo-inflammatory disease. Clin. Sci. 2022, 136, 1809–1829. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, J.I.; Trenor, C.C.; Furie, B.C.; Furie, B. Tissue factor-bearing microparticles and thrombus formation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 728–733. [Google Scholar] [CrossRef]
- Denorme, F.; Vanhoorelbeke, K.; de Meyer, S.F. von Willebrand Factor and Platelet Glycoprotein Ib: A Thromboinflammatory Axis in Stroke. Front. Immunol. 2019, 10, 2884. [Google Scholar] [CrossRef]
- Rautiainen, L.; Cirko, A.; Pavare, J.; Balmaks, R.; Grope, I.; Katirlo, I.; Gersone, G.; Tretjakovs, P.; Gardovska, D. Assessment of ADAMTS-13 level in hospitalized children with serious bacterial infections as a possible prognostic marker. Medicina 2019, 55, 503. [Google Scholar] [CrossRef]
- Shorr, A.F.; Bernard, G.R.; Dhainaut, J.-F.; Russell, J.R.; Macias, W.L.; Nelson, D.R.; Sundin, D.P. Protein C concentrations in severe sepsis: An early directional change in plasma levels predicts outcome. Crit. Care 2006, 10, R92. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Young, H.A.; Jahrling, P.B.; Davis, K.J.; Kagan, E.; Hensley, L.E. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: Overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 2003, 188, 1618–1629. [Google Scholar] [CrossRef]
- Funderburg, N.; Mayne, E.; Sieg, S.F.; Asaad, R.; Jiang, W.; Kalinowska, M.; Luciano, A.A.; Stevens, W.; Rodriguez, B.; Brenchley, J.M.; et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: Relationship to in vivo coagulation and immune activation. Blood 2010, 115, 161–167. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Páramo, J.A. Inflammatory response in relation to COVID-19 and other prothrombotic phenotypes. Reumatol. Clínica 2022, 18, 1–4. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Loo, J.; Spittle, D.A.; Newnham, M. COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax 2021, 76, 412–420. [Google Scholar] [CrossRef]
- Wagner, D.D.; Heger, L.A. Thromboinflammation: From Atherosclerosis to COVID-19. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1103–1112. [Google Scholar] [CrossRef]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef]
- Alkim, H.; Ayaz, S.; Alkim, C.; Ulker, A.; Sahin, B. Continuous active state of coagulation system in patients with nonthrombotic inflammatory bowel disease. Clin. Appl. Thromb. Hemost. 2011, 17, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Kume, K.; Yamasaki, M.; Tashiro, M.; Yoshikawa, I.; Otsuki, M. Activations of coagulation and fibrinolysis secondary to bowel inflammation in patients with ulcerative colitis. Intern. Med. 2007, 46, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Moschonas, I.; Tselepis, A. Platelet-derived microparticles induce the formation of neutrophil extracellular traps. Atherosclerosis 2018, 275, e106. [Google Scholar] [CrossRef]
- Vetrano, S.; Ploplis, V.A.; Sala, E.; Sandoval-Cooper, M.; Donahue, D.L.; Correale, C.; Arena, V.; Spinelli, A.; Repici, A.; Malesci, A.; et al. Unexpected role of anticoagulant protein C in controlling epithelial barrier integrity and intestinal inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 19830–19835. [Google Scholar] [CrossRef]
- Amoroso, A.; Mitterhofer, A.P.; del Porto, F.; Garzia, P.; Ferri, G.M.; Galluzzo, S.; Vadacca, M.; Caccavo, D.; Afeltra, A. Antibodies to anionic phospholipids and anti-β2-GPI: Association with thrombosis and thrombocytopenia in systemic lupus erythematosus. Hum. Immunol. 2003, 64, 265–273. [Google Scholar] [CrossRef]
- Gustafsson, J.; Gunnarsson, I.; Börjesson, O.; Pettersson, S.; Möller, S.; Fei, G.-Z.; Elvin, K.; Simard, J.F.; Hansson, L.-O.; E Lundberg, I.; et al. Predictors of the first cardiovascular event in patients with systemic lupus erythematosus—A prospective cohort study. Arthritis Res. Ther. 2009, 11, R186. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Inflammatory cytokines in systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 2011, 432595. [Google Scholar] [CrossRef]
- Müller-Calleja, N.; Hollerbach, A.; Royce, J.; Ritter, S.; Pedrosa, D.; Madhusudhan, T.; Teifel, S.; Meineck, M.; Hauser, F.; Canisius, A.; et al. Lipid presentation by the protein C receptor links coagulation with autoimmunity. Science 2021, 371, eabc0956. [Google Scholar] [CrossRef]
- Mackman, N.; Bergmeier, W.; Stouffer, G.A.; Weitz, J.I. Therapeutic strategies for thrombosis: New targets and approaches. Nat. Rev. Drug Discov. 2020, 19, 333–352. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, X.; Li, Z.; He, Z.; Yang, X.; Cheng, X.; Peng, Y.; Xue, Q.; Bai, Y.; Zhang, R.; et al. Heparin prevents caspase-11-dependent septic lethality independent of anticoagulant properties. Immunity 2021, 54, 454–467.e6. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; van der Goot, F.G.; Turcatti, G.; Behrendt, B.; Ablasser, A.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, D.C.; Travis, O.K.; Tramel, R.W.; Borges-Rodriguez, M.; Baik, C.H.; Greer, M.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE 2020, 15, e0234039. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Silvis, M.J.; Fiolet, A.T.; Opstal, T.S.; Dekker, M.; Suquilanda, D.; Zivkovic, M.; Duyvendak, M.; The, S.H.; Timmers, L.; Bax, W.A.; et al. Colchicine reduces extracellular vesicle NLRP3 inflammasome protein levels in chronic coronary disease: A LoDoCo2 biomarker substudy. Atherosclerosis 2021, 334, 93–100. [Google Scholar] [CrossRef]
- Campbell, C.M.; Kahwash, R. Will Complement Inhibition Be the New Target in Treating COVID-19–Related Systemic Thrombosis? Circulation 2020, 141, 1739–1741. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Okamoto, K.; Ohike, T.; Tajirika, T.; Aihara, K.; Watanabe, S.; Kayhanian, H. Enoxaparin and fondaparinux attenuates endothelial damage in endotoxemic rats. J. Trauma Acute Care Surg. 2012, 72, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2013, 345, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Mitsios, A.; Chrysanthopoulou, A.; Arampatzioglou, A.; Angelidou, I.; Vidali, V.; Ritis, K.; Skendros, P.; Stakos, D. Ticagrelor exerts immune-modulatory effect by attenuating neutrophil extracellular traps. Int. J. Mol. Sci. 2020, 21, 3625. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Baldini, C.; Moriconi, F.R.; Galimberti, S.; Libby, P.; De Caterina, R. The JAK-STAT pathway: An emerging target for cardiovascular disease in rheumatoid arthritis and myeloproliferative neoplasms. Eur. Heart J. 2021, 42, 4389–4400. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcos-Jubilar, M.; Lecumberri, R.; Páramo, J.A. Immunothrombosis: Molecular Aspects and New Therapeutic Perspectives. J. Clin. Med. 2023, 12, 1399. https://doi.org/10.3390/jcm12041399
Marcos-Jubilar M, Lecumberri R, Páramo JA. Immunothrombosis: Molecular Aspects and New Therapeutic Perspectives. Journal of Clinical Medicine. 2023; 12(4):1399. https://doi.org/10.3390/jcm12041399
Chicago/Turabian StyleMarcos-Jubilar, María, Ramón Lecumberri, and José A. Páramo. 2023. "Immunothrombosis: Molecular Aspects and New Therapeutic Perspectives" Journal of Clinical Medicine 12, no. 4: 1399. https://doi.org/10.3390/jcm12041399
APA StyleMarcos-Jubilar, M., Lecumberri, R., & Páramo, J. A. (2023). Immunothrombosis: Molecular Aspects and New Therapeutic Perspectives. Journal of Clinical Medicine, 12(4), 1399. https://doi.org/10.3390/jcm12041399