
Peptides Derived from a Plant Protease Inhibitor of the Coagulation Contact System Decrease Arterial Thrombus Formation in a Murine Model, without Impairing Hemostatic Parameters

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Plant Material
2.3. Peptides
2.4. Animals
2.5. Plasma and Platelets
2.6. Inhibitory Activity
2.7. Coagulation Assays
2.8. Platelet Aggregation
2.9. Photochemical-Induced Arterial Thrombosis Model
2.10. Platelet Behavior in Intravital Microscopy
2.11. Bleeding Time
2.12. Statistical Analysis
3. Results
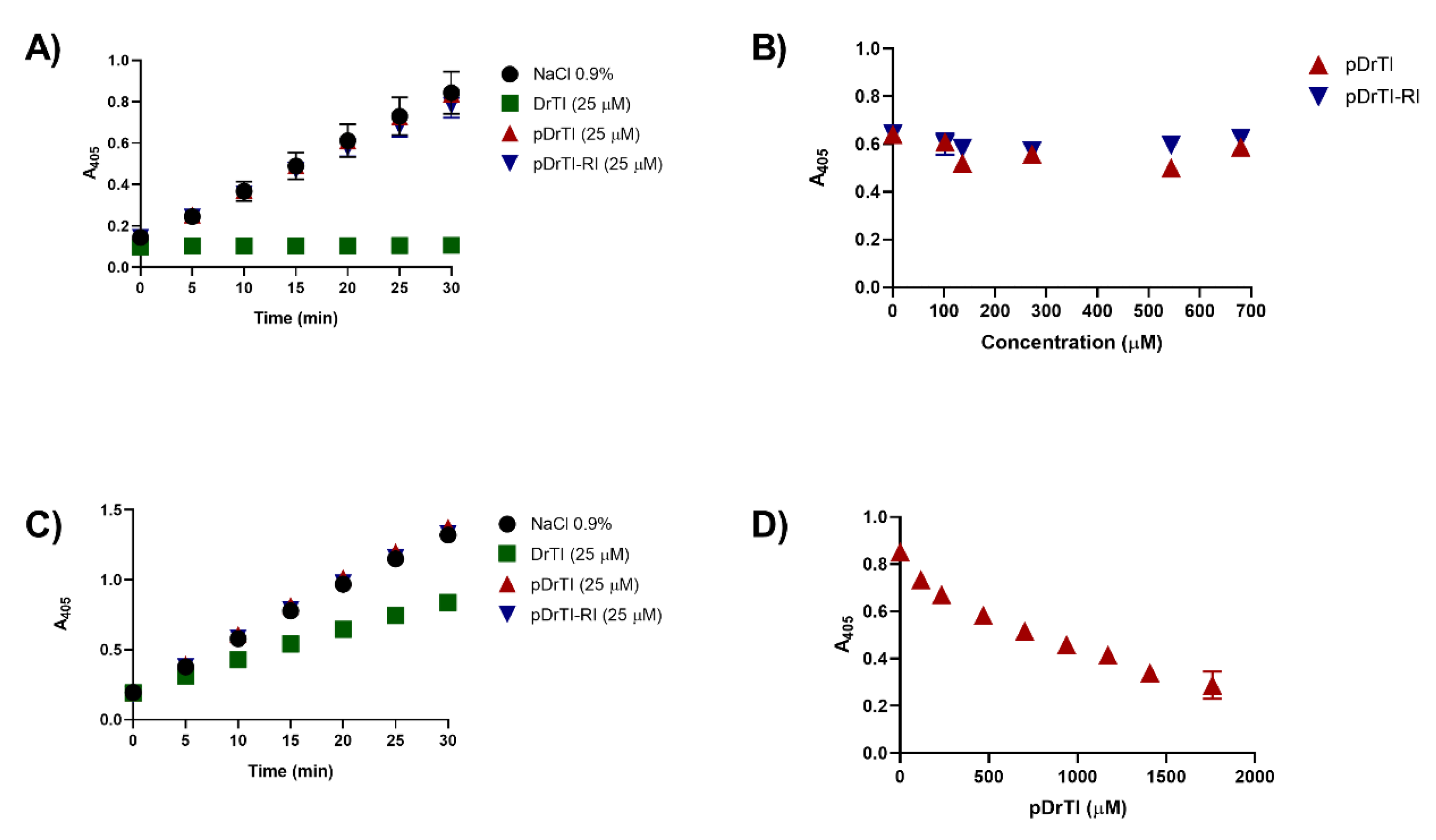
3.1. Inhibitory Activity
3.2. Coagulation Assays
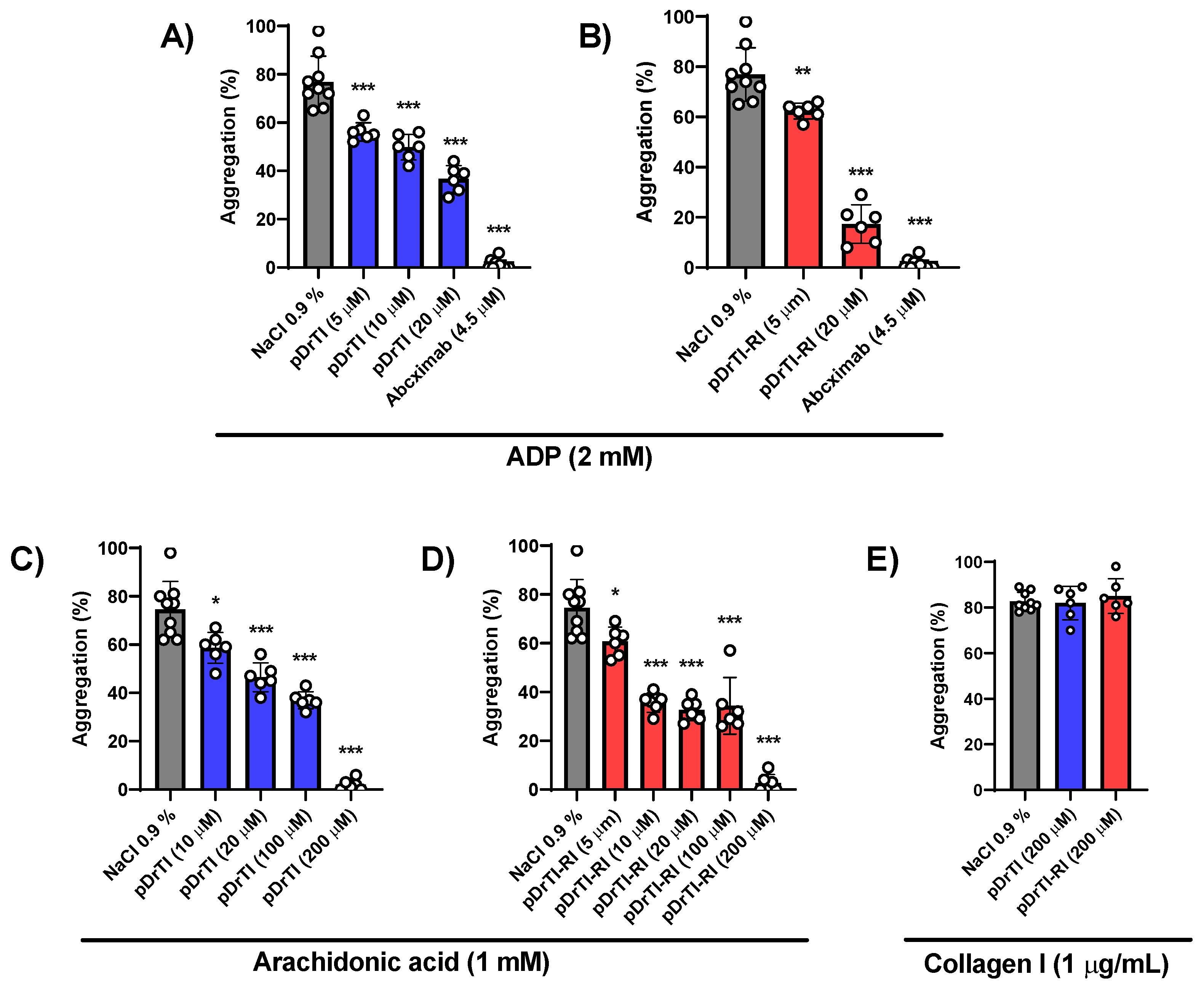
3.3. Platelet Aggregation
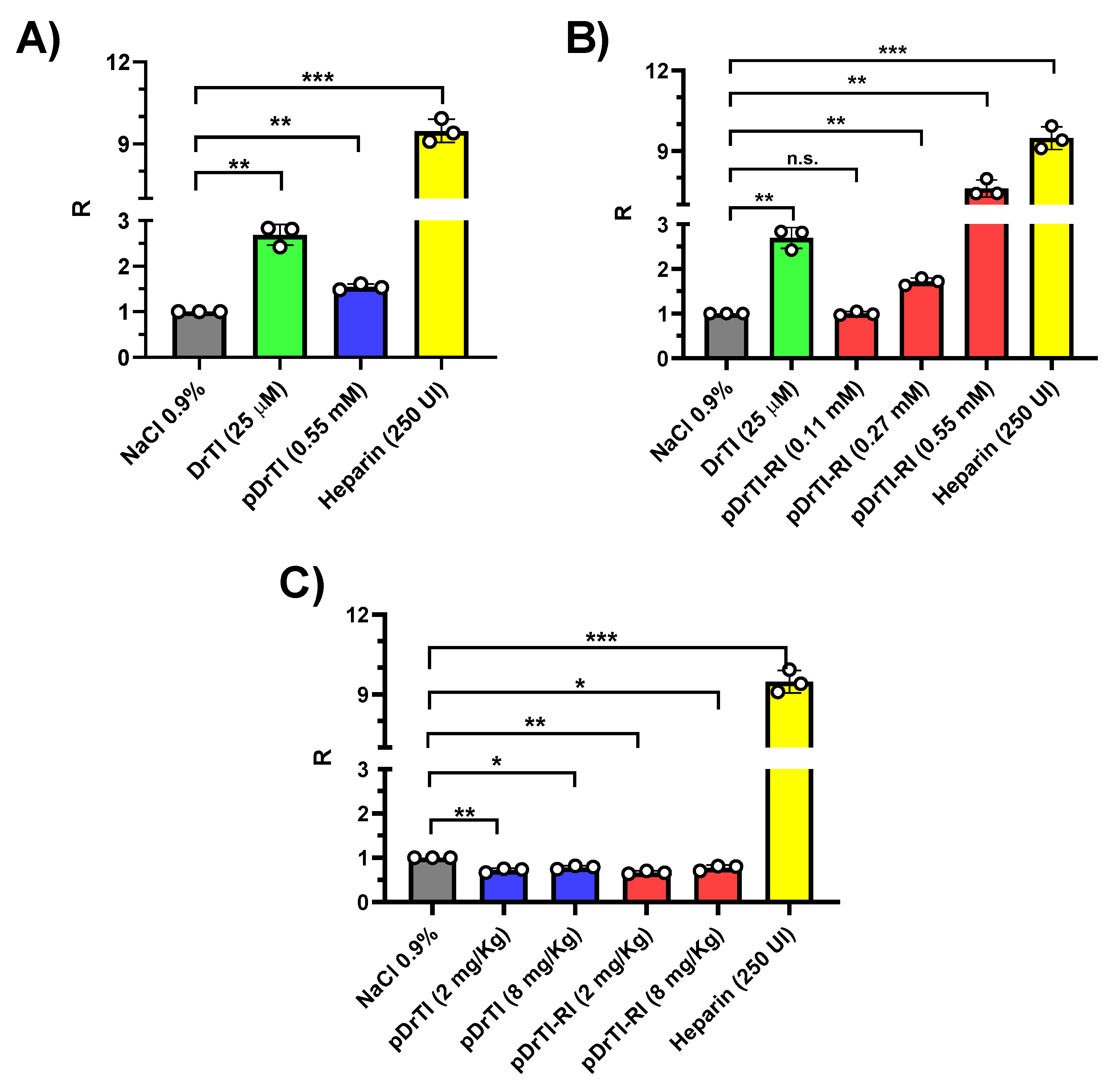
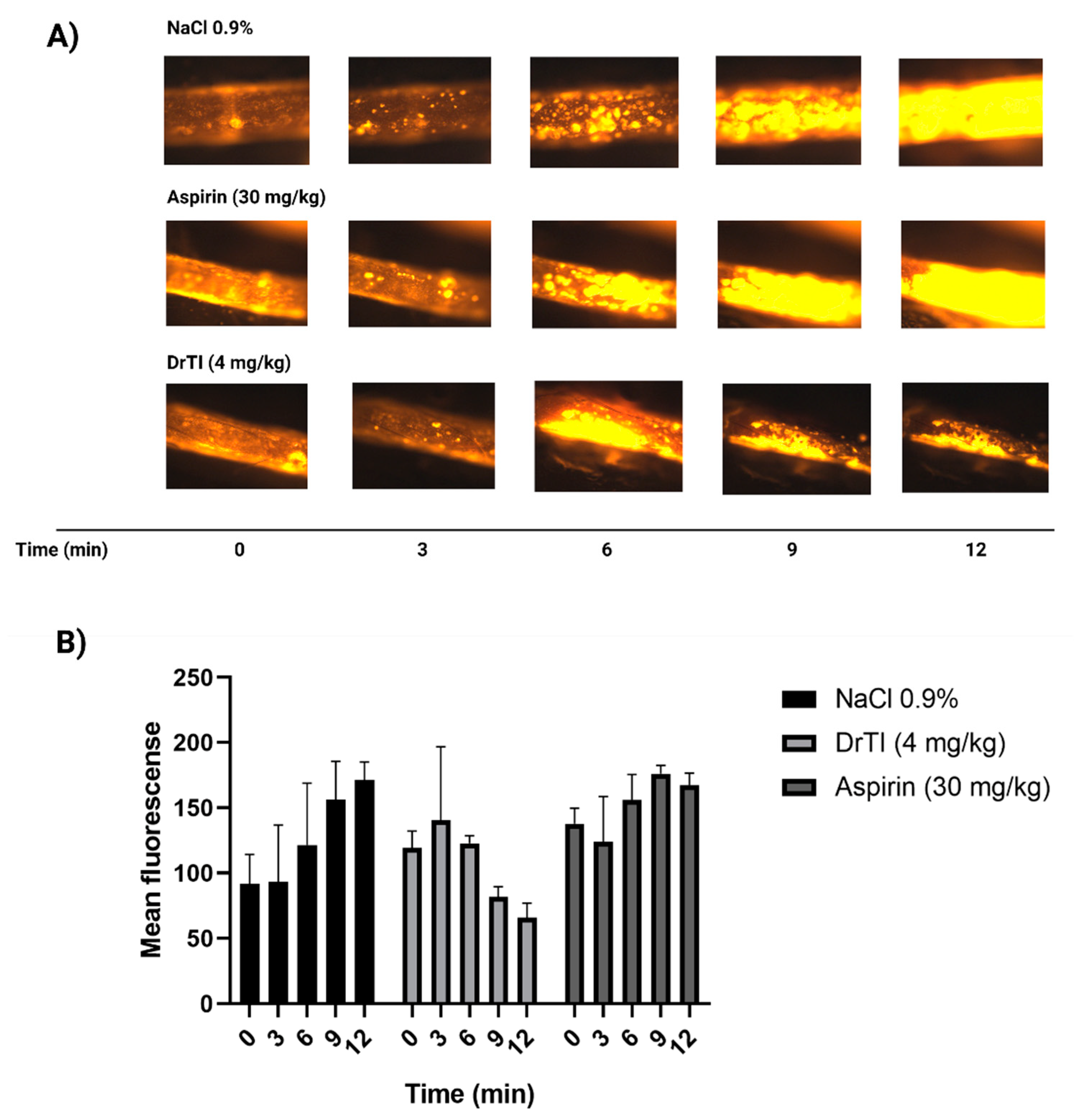
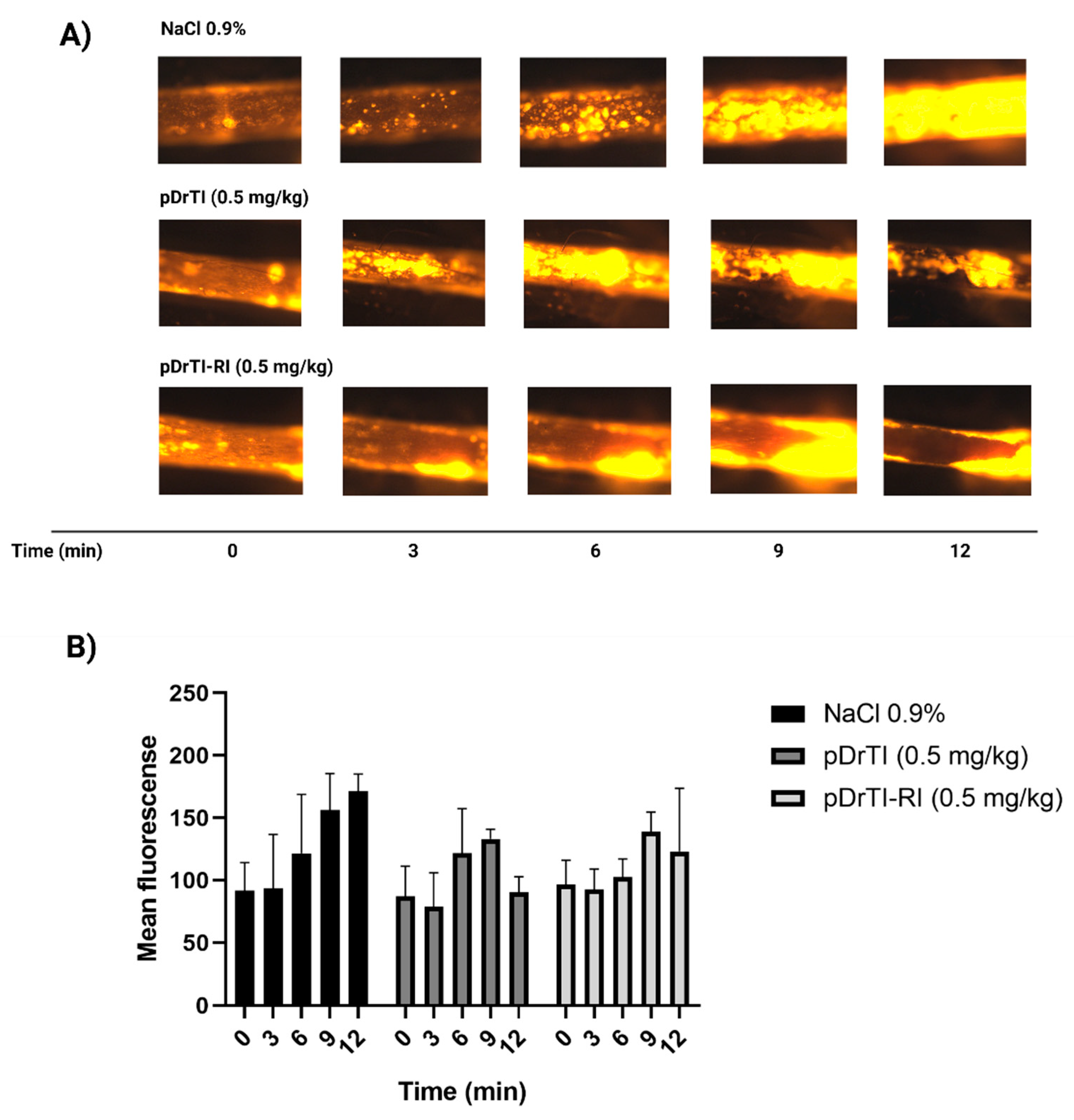
3.4. Photochemical-Induced Arterial Thrombosis Model
3.5. Platelet Behavior in Intravital Microscopy
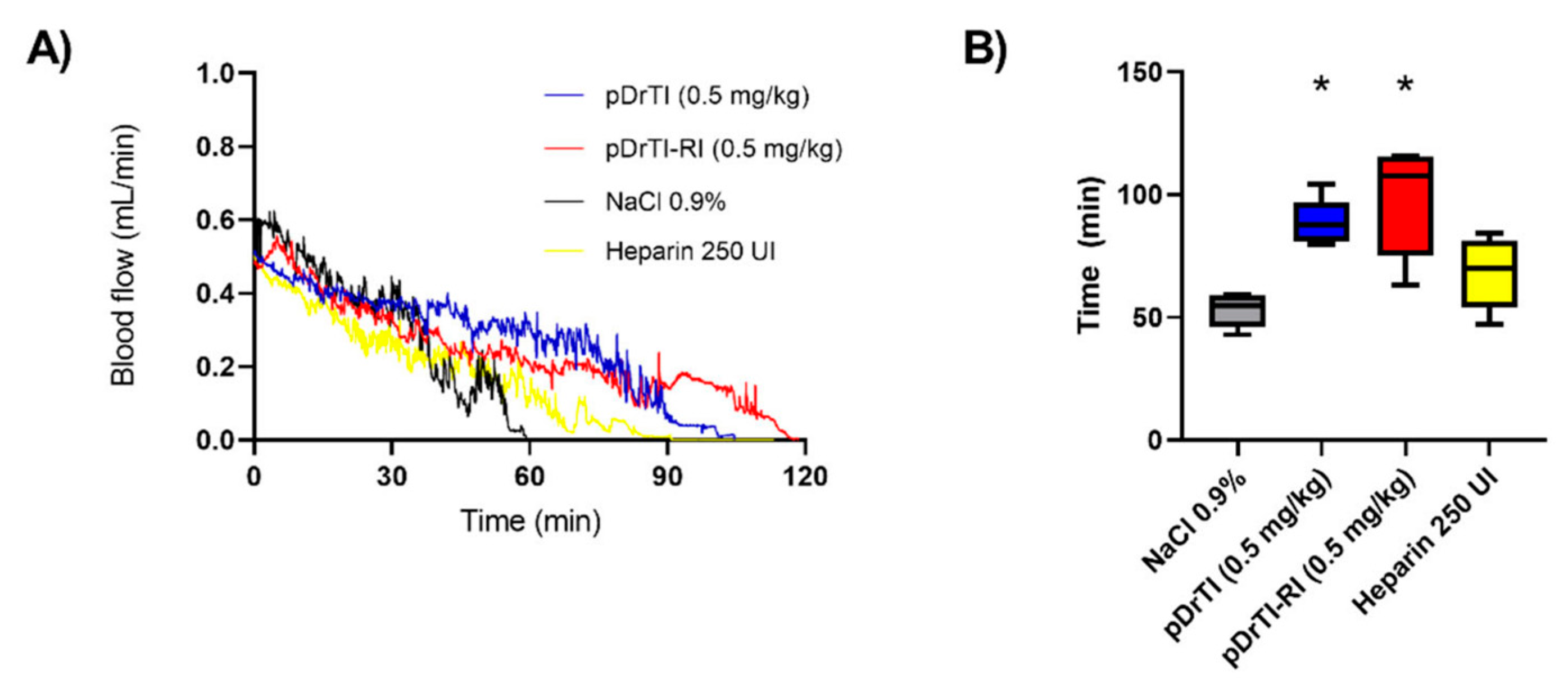
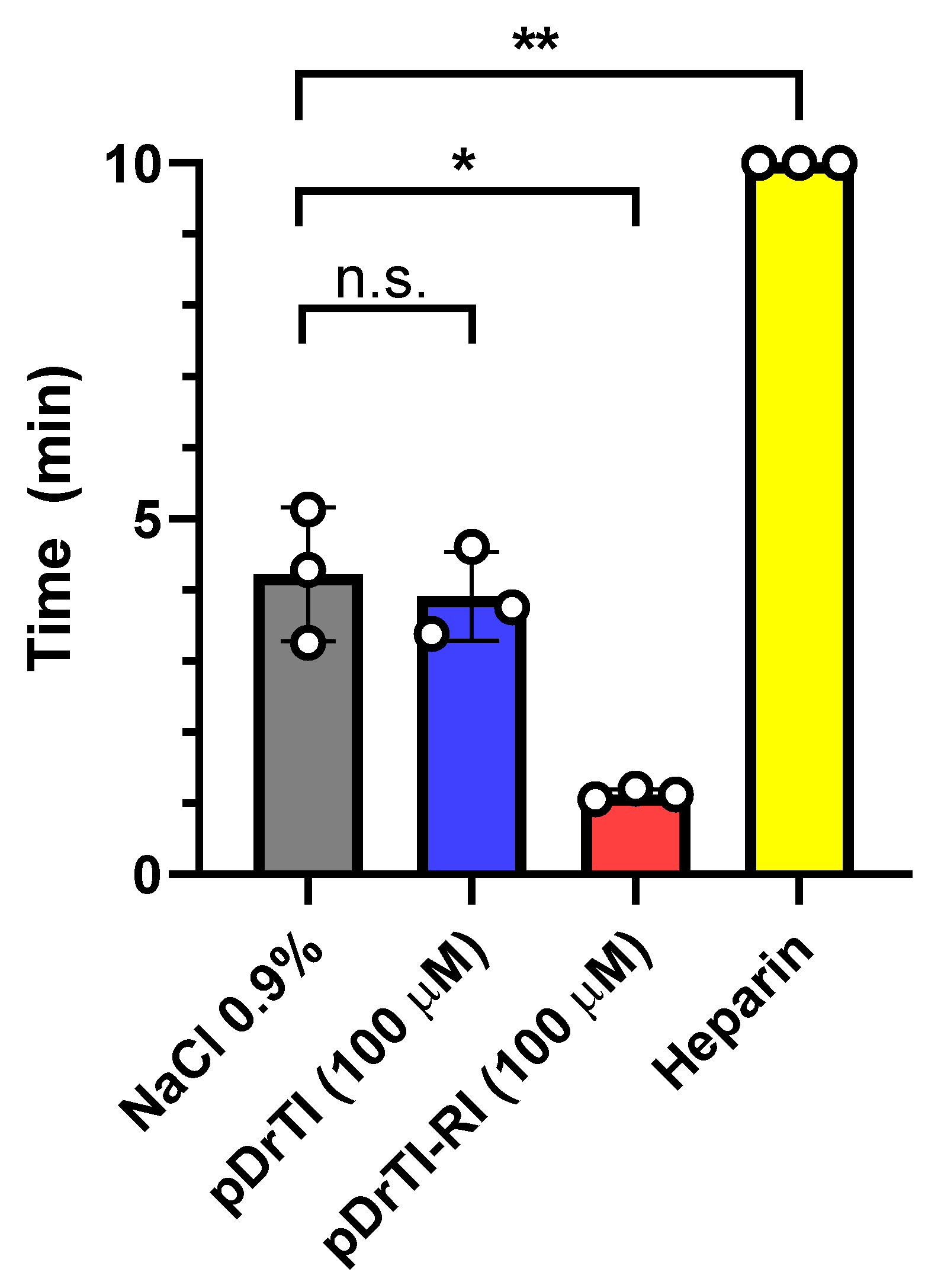
3.6. Bleeding Time
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Noncommunicable Diseases Country Profiles 2018; World Health Organization: Geneva, Switzerland, 2018; ISBN 9789241514620. [Google Scholar]
- Franchini, M.; Mannucci, P.M. The Hemostatic Balance Revisited through the Lessons of Mankind Evolution. Intern. Emerg. Med. 2008, 3, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Eikelboom, J.W.; Chan, N.C. Fifty Years of Research on Antithrombotic Therapy: Achievements and Disappointments. Eur. J. Intern. Med. 2019, 70, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Hulot, J.-S.; Moliterno, D.J.; Harrington, R.A. Antiplatelet and Anticoagulation Therapy for Acute Coronary Syndromes. Circ. Res. 2014, 114, 1929–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anticoagulant Medicines. Available online: https://www.nhs.uk/conditions/anticoagulants/ (accessed on 14 September 2022).
- Macedo, M.L.R.; Ribeiro, S.F.F.; Taveira, G.B.; Gomes, V.M.; de Barros, K.M.C.A.; Maria-Neto, S. Antimicrobial Activity of ILTI, a Kunitz-Type Trypsin Inhibitor from Inga Laurina (SW.) Willd. Curr. Microbiol. 2016, 72, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Bonturi, C.R.; Motaln, H.; Silva, M.C.C.; Salu, B.R.; de Brito, M.V.; de Andrade Luz Cost, L.; Torquato, H.F.V.; Nunes, N.N.D.S.; Paredes-Gamero, E.J.; Turnšek, T.L.; et al. Could a Plant Derived Protein Potentiate the Anticancer Effects of a Stem Cell in Brain Cancer? Oncotarget 2018, 9, 21296–21312. [Google Scholar] [CrossRef] [Green Version]
- Brito, M.V.; de Oliveira, C.; Salu, B.R.; Andrade, S.A.; Malloy, P.M.D.; Sato, A.C.; Vicente, C.P.; Sampaio, M.U.; Maffei, F.H.A.; Oliva, M.L.V. The Kallikrein Inhibitor from Bauhinia Bauhinioides (BbKI) Shows Antithrombotic Properties in Venous and Arterial Thrombosis Models. Thromb. Res. 2014, 133, 945–951. [Google Scholar] [CrossRef]
- Pandey, P.K.; Jamal, F. Bio-Potency of a 21 KDa Kunitz-Type Trypsin Inhibitor from Tamarindus Indica Seeds on the Developmental Physiology of H. Armigera. Pestic. Biochem. Physiol. 2014, 116, 94–102. [Google Scholar] [CrossRef]
- Salu, B.R.; Pando, S.C.; Brito, M.V.D.; Medina, A.F.; Odei-Addo, F.; Frost, C.; Naude, R.; Sampaio, M.U.; Emsley, J.; Maffei, F.H.A.; et al. Improving the Understanding of Plasma Kallikrein Contribution to Arterial Thrombus Formation Using Two Plant Protease Inhibitors. Platelets 2019, 30, 305–313. [Google Scholar] [CrossRef]
- Lobo, Y.A.; Bonazza, C.; Batista, F.P.; Castro, R.A.; Bonturi, C.R.; Salu, B.R.; de Cassia Sinigaglia, R.; Toma, L.; Vicente, C.M.; Pidde, G.; et al. EcTI Impairs Survival and Proliferation Pathways in Triple-Negative Breast Cancer by Modulating Cell-Glycosaminoglycans and Inflammatory Cytokines. Cancer Lett. 2020, 491, 108–120. [Google Scholar] [CrossRef]
- Pando, S.C.; Oliva, M.L.; Sampaio, C.A.; Di Ciero, L.; Novello, J.C.; Marangoni, S. Primary Sequence Determination of a Kunitz Inhibitor Isolated from Delonix Regia Seeds. Phytochemistry 2001, 57, 625–631. [Google Scholar] [CrossRef]
- Krauchenco, S.; Pando, S.C.; Marangoni, S.; Polikarpov, I. Crystal Structure of the Kunitz (STI)-Type Inhibitor from Delonix Regia Seeds. Biochem. Biophys. Res. Commun. 2003, 312, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.L.V.; Grisolia, D.; Sampaio, C.A.M.; Sampaio, M.U. Properties of Highly Purified Human Plasma Kallikrein. Agents Actions 1982, 1, 52–57. [Google Scholar]
- Eitzman, D.T.; Westrick, R.J.; Nabel, E.G.; Ginsburg, D. Plasminogen Activator Inhibitor-1 and Vitronectin Promote Vascular Thrombosis in Mice. Blood 2000, 95, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Perzborn, E.; Strassburger, J.; Wilmen, A.; Pohlmann, J.; Roehrig, S.; Schlemmer, K.-H.; Straub, A. In Vitro and in Vivo Studies of the Novel Antithrombotic Agent BAY 59-7939-an Oral, Direct Factor Xa Inhibitor. J. Thromb. Haemost. 2005, 3, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Kam, C.M.; Copher, J.C.; Powers, J.C. Mechanism-Based Isocoumarin Inhibitors for Trypsin-like Serine Proteases Involved in Blood Coagulation. J. Am. Chem. Soc. 1987, 109, 5044–5045. [Google Scholar] [CrossRef]
- Stalker, T.J.; Traxler, E.A.; Wu, J.; Wannemacher, K.M.; Cermignano, S.L.; Voronov, R.; Diamond, S.L.; Brass, L.F. Hierarchical Organization in the Hemostatic Response and Its Relationship to the Platelet-Signaling Network. Blood 2013, 121, 1875–1885. [Google Scholar] [CrossRef]
- Matsuno, H.; Uematsu, T.; Nagashima, S.; Nakashima, M. Photochemically Induced Thrombosis Model in Rat Femoral Artery and Evaluation of Effects of Heparin and Tissue-Type Plasminogen Activator with Use of This Model. J. Pharmacol. Methods 1991, 25, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Blobaum, A.L.; Marnett, L.J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [Green Version]
- Huff, R.G.; Bayram, E.; Tan, H.; Knutson, S.T.; Knaggs, M.H.; Richon, A.B.; Santago, P.; Fetrow, J.S. Chemical and Structural Diversity in Cyclooxygenase Protein Active Sites. C&B 2005, 2, 1533–1552. [Google Scholar] [CrossRef]
- Xie, Z.; Li, Z.; Shao, Y.; Liao, C. Discovery and Development of Plasma Kallikrein Inhibitors for Multiple Diseases. Eur. J. Med. Chem. 2020, 190, 112137. [Google Scholar] [CrossRef]
- Duga, S.; Salomon, O. Congenital Factor XI Deficiency: An Update. Semin. Thromb. Hemost. 2013, 39, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Hovgaard, L.; Frokjaer, S.; van de Weert, M. (Eds.) Pharmaceutical Formulation Development of Peptides and Proteins, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar] [CrossRef]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.-L.; Ye, C.; Zhou, F.; Wang, J.; Yang, D.; Yin, W.; Wang, M.-W.; Xu, H.E.; Jiang, Y. Molecular Basis for Kinin Selectivity and Activation of the Human Bradykinin Receptors. Nat. Struct. Mol. Biol. 2021, 28, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Xu, B. Inspiration from the Mirror: D-Amino Acid Containing Peptides in Biomedical Approaches. Biomol. Concepts 2016, 7, 179–187. [Google Scholar] [CrossRef]
- Li, C.; Voos, K.M.; Pathak, M.; Hall, G.; McCrae, K.R.; Dreveny, I.; Li, R.; Emsley, J. Plasma Kallikrein Structure Reveals Apple Domain Disc Rotated Conformation Compared to Factor XI. J. Thromb. Haemost. 2019, 17, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Roshal, M.; Reyes Gil, M. Activated Partial Thromboplastin Time. In Transfusion Medicine and Hemostasis, 3rd ed.; Shaz, B.H., Hillyer, C.D., Reyes Gil, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Chapter 129; pp. 779–781. ISBN 9780128137260. [Google Scholar]
- Valentin, L.I.; Sicard, G.A.; Freeman, M.B.; Allen, B.T.; McGoff, M.A.; Anderson, C.B. Combined Arachidonic Acid and ADP Platelet Inhibition Maximizes Patency of Small-Diameter Vascular Grafts. Surgery 1988, 104, 178–184. [Google Scholar]
- Fang, C.; Schmaier, A.; Stavrou, E.; Adams, G.; Nieman, M.T.; LaRusch, G.; Chen, A.; Zhou, Y.; Bilodeau, M.; Mahdi, F.; et al. Bradykinin B2 Receptor KO Mice Are Protected from Thrombosis by A Platelet Spreading Defect. Blood 2010, 116, 3198. [Google Scholar] [CrossRef]
- Murphey, L.J.; Malave, H.A.; Petro, J.; Biaggioni, I.; Byrne, D.W.; Vaughan, D.E.; Luther, J.M.; Pretorius, M.; Brown, N.J. Bradykinin and Its Metabolite Bradykinin 1-5 Inhibit Thrombin-Induced Platelet Aggregation in Humans. J. Pharmacol. Exp. Ther. 2006, 318, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Paredes-Gamero, E.J.; Medeiros, V.P.; Farias, E.H.C.; Justo, G.Z.; Trindade, E.S.; Andrade-Lopes, A.L.; Godinho, R.O.; de Miranda, A.; Ferreira, A.T.; Tersariol, I.L.S.; et al. Heparin induces rat aorta relaxation via integrin-dependent activation of muscarinic M3 receptors. Hypertension 2010, 56, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, W.A.; Luettgen, J.M.; Quan, M.L.; Seiffert, D.A. Inhibition of Factor XIa as a New Approach to Anticoagulation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 388–392. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence |
|---|---|
| pDrTI | Ac-S-P-F-R-V-V-F-V-K-P-NH2 |
| pDrTI-RI | Ac-P *-K *-V *-F *-V *-V *-R *-F *-P *-S *-NH2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Souza, D.A.; Salu, B.R.; Nogueira, R.S.; de Carvalho Neto, J.C.S.; Maffei, F.H.d.A.; Oliva, M.L.V. Peptides Derived from a Plant Protease Inhibitor of the Coagulation Contact System Decrease Arterial Thrombus Formation in a Murine Model, without Impairing Hemostatic Parameters. J. Clin. Med. 2023, 12, 1810. https://doi.org/10.3390/jcm12051810
De Souza DA, Salu BR, Nogueira RS, de Carvalho Neto JCS, Maffei FHdA, Oliva MLV. Peptides Derived from a Plant Protease Inhibitor of the Coagulation Contact System Decrease Arterial Thrombus Formation in a Murine Model, without Impairing Hemostatic Parameters. Journal of Clinical Medicine. 2023; 12(5):1810. https://doi.org/10.3390/jcm12051810
Chicago/Turabian StyleDe Souza, Daniel Alexandre, Bruno Ramos Salu, Ruben Siedlarczyk Nogueira, José Carlos Sá de Carvalho Neto, Francisco Humberto de Abreu Maffei, and Maria Luiza Vilela Oliva. 2023. "Peptides Derived from a Plant Protease Inhibitor of the Coagulation Contact System Decrease Arterial Thrombus Formation in a Murine Model, without Impairing Hemostatic Parameters" Journal of Clinical Medicine 12, no. 5: 1810. https://doi.org/10.3390/jcm12051810
APA StyleDe Souza, D. A., Salu, B. R., Nogueira, R. S., de Carvalho Neto, J. C. S., Maffei, F. H. d. A., & Oliva, M. L. V. (2023). Peptides Derived from a Plant Protease Inhibitor of the Coagulation Contact System Decrease Arterial Thrombus Formation in a Murine Model, without Impairing Hemostatic Parameters. Journal of Clinical Medicine, 12(5), 1810. https://doi.org/10.3390/jcm12051810