
Biochemical Mechanisms beyond Glycosphingolipid Accumulation in Fabry Disease: Might They Provide Additional Therapeutic Treatments?

 , , , and
, , , and {kind=link}
{kind=link}
Abstract
:1. Introduction
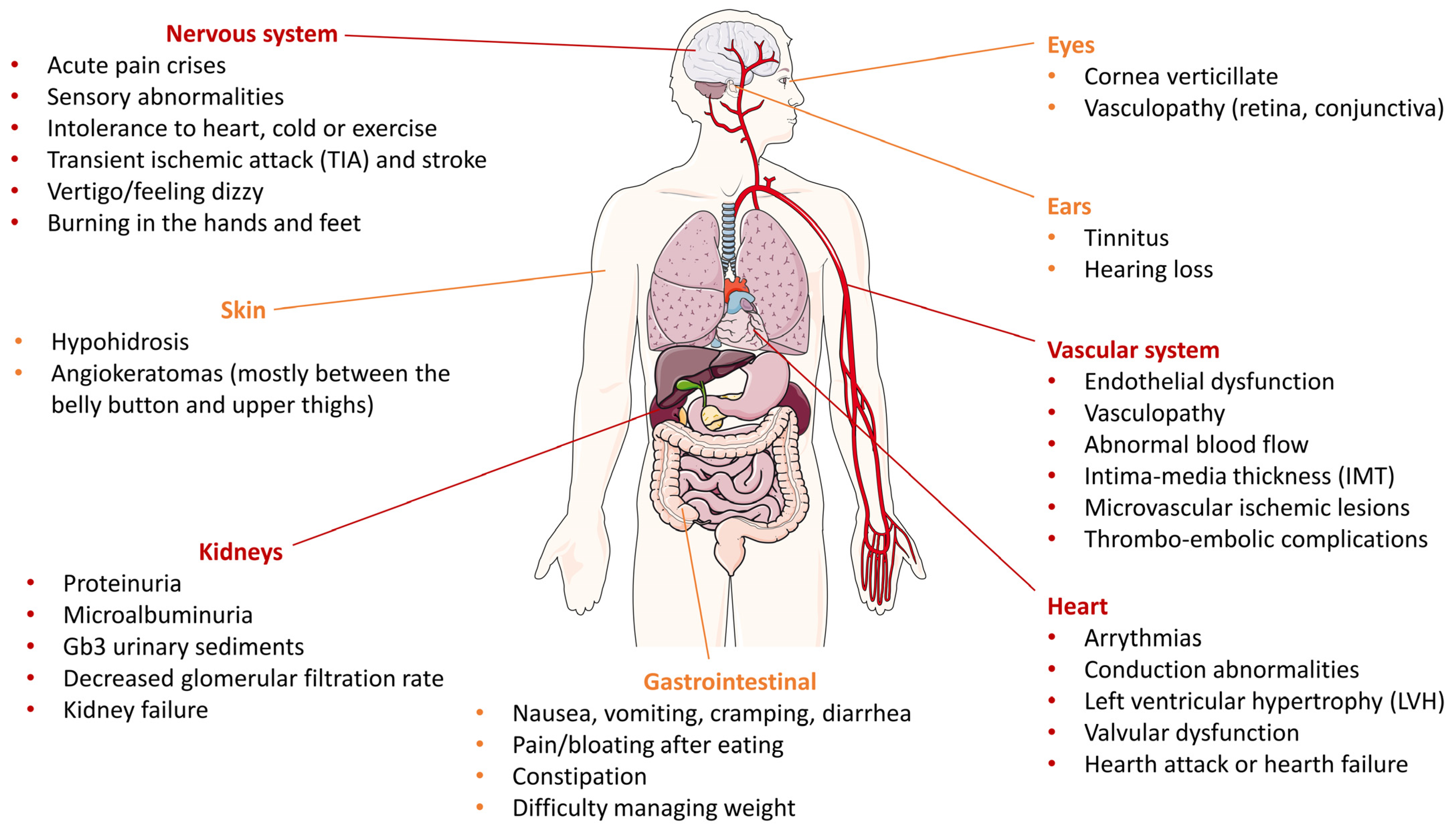
2. Altered Pathways in Fabry Disease
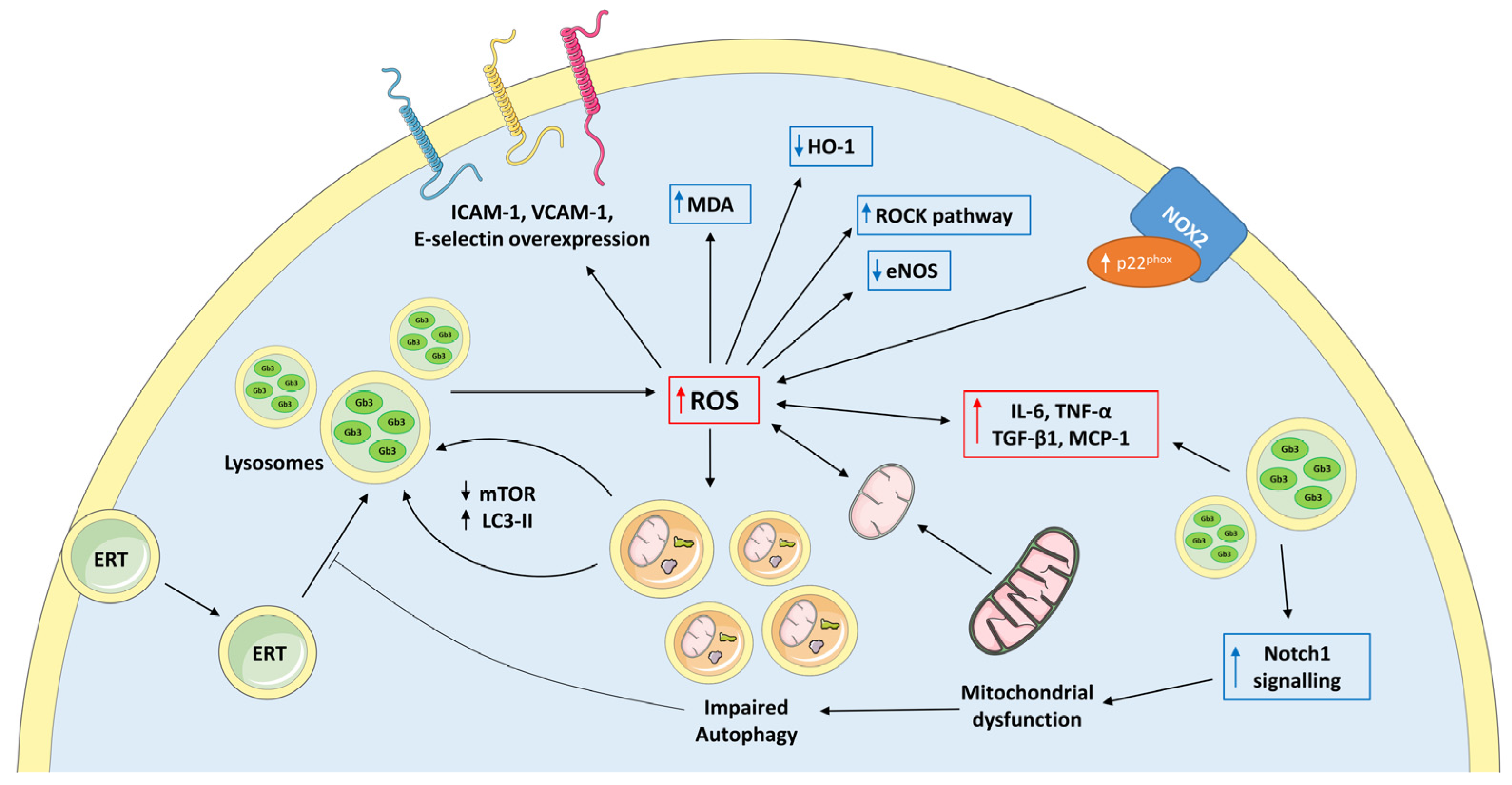
2.1. Oxidative Stress in Fabry Disease
2.2. Mithocondrial Dysfunction in Fabry Disease
2.3. Impaired Autophagy in Fabry Disease
3. Potential Additional Therapeutic Strategies in Fabry Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional Characterisation of Alpha-Galactosidase A Mutations as a Basis for a New Classification System in Fabry Disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry Disease, an under-Recognized Multisystemic Disorder: Expert Recommendations for Diagnosis, Management, and Enzyme Replacement Therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Gibas, A.L.; Klatt, R.; Johnson, J.; Clarke, J.T.R.; Katz, J. Disease Rarity, Carrier Status, and Gender: A Triple Disadvantage for Women with Fabry Disease. J. Genet. Couns. 2008, 17, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn Screening for Fabry Disease in Taiwan Reveals a High Incidence of the Later-Onset GLA Mutation c.936+919G\textgreaterA (IVS4+919G\textgreaterA). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guce, A.I.; Clark, N.E.; Salgado, E.N.; Ivanen, D.R.; Kulminskaya, A.A.; Brumer, H.; Garman, S.C. Catalytic Mechanism of Human α-Galactosidase. J. Biol. Chem. 2010, 285, 3625–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eijk, M.; Ferraz, M.J.; Boot, R.G.; Aerts, J.M.F.G. Lyso-Glycosphingolipids: Presence and Consequences. Essays Biochem. 2020, 64, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Dekker, N.; Bouwman, M.G.; Linthorst, G.E.; Zwinderman, A.H.; Wijburg, F.A.; Kuiper, S.; vd Bergh Weerman, M.A.; Groener, J.E.M.; Poorthuis, B.J.; et al. Plasma Globotriaosylsphingosine: Diagnostic Value and Relation to Clinical Manifestations of Fabry Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; Van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated Globotriaosylsphingosine Is a Hallmark of Fabry Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P. Fabry Disease. Orphanet J. Rare Dis. 2010, 5, 1–49. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry Disease Revisited: Management and Treatment Recommendations for Adult Patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Matern, D.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S. Newborn Screening for Lysosomal Storage Disorders. Semin. Perinatol. 2015, 39, 206–216. [Google Scholar] [CrossRef]
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia De Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry Disease Defined: Baseline Clinical Manifestations of 366 Patients in the Fabry Outcome Survey. Eur. J. Clin. Investig. 2004, 34, 236–242. [Google Scholar] [CrossRef]
- Waldek, S.; Patel, M.R.; Banikazemi, M.; Lemay, R.; Lee, P. Life Expectancy and Cause of Death in Males and Females with Fabry Disease: Findings from the Fabry Registry. Genet. Med. 2009, 11, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Yousef, Z.; Elliott, P.M.; Cecchi, F.; Escoubet, B.; Linhart, A.; Monserrat, L.; Namdar, M.; Weidemann, F. Left Ventricular Hypertrophy in Fabry Disease: A Practical Approach to Diagnosis. Eur. Heart J. 2013, 34, 802–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linhart, A. The Heart in Fabry Disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 1-903539-03-X. [Google Scholar]
- Sunder-Plassmann, G. Renal Manifestations of Fabry Disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 1-903539-03-X. [Google Scholar]
- Sharma, A.; Sartori, M.; Zaragoza, J.J.; Villa, G.; Lu, R.; Faggiana, E.; Brocca, A.; Di Lullo, L.; Feriozzi, S.; Ronco, C. Fabry’s Disease: An Example of Cardiorenal Syndrome Type 5. Heart Fail. Rev. 2015, 20, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Siegenthaler, M.; Huynh-Do, U.; Krayenbuehl, P.; Pollock, E.; Widmer, U.; Debaix, H.; Olinger, E.; Frank, M.; Namdar, M.; Ruschitzka, F.; et al. Impact of Cardio-Renal Syndrome on Adverse Outcomes in Patients with Fabry Disease in a Long-Term Follow-Up. Int. J. Cardiol. 2017, 249, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Üçeyler, N.; Ganendiran, S.; Kramer, D.; Sommer, C. Characterization of Pain in Fabry Disease. Clin. J. Pain 2014, 30, 915–920. [Google Scholar] [CrossRef]
- Burand, A.J.J.; Stucky, C.L. Fabry Disease Pain: Patient and Preclinical Parallels. Pain 2021, 162, 1305. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of Inflammatory Pathways to Fabry Disease Pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef]
- Lee, M.H.; Choi, E.N.; Jeon, Y.J.; Jung, S.-C. Possible Role of Transforming Growth Factor-Β1 and Vascular Endothelial Growth Factor in Fabry Disease Nephropathy. Int. J. Mol. Med. 2012, 30, 1275–1280. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine Actions on Human Glomerular Podocytes: Implications for Fabry Nephropathy. Nephrol. Dial. Transplant. 2011, 26, 1797–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, Y.J.; Jung, N.; Park, J.-W.; Park, H.-Y.; Jung, S.-C. Epithelial–Mesenchymal Transition in Kidney Tubular Epithelial Cells Induced by Globotriaosylsphingosine and Globotriaosylceramide. PLoS ONE 2015, 10, e0136442. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Chien, Y.; Wang, K.-L.; Leu, H.-B.; Hsiao, C.-Y.; Lai, Y.-H.; Wang, C.-Y.; Chang, Y.-L.; Lin, S.-J.; Niu, D.-M.; et al. Evaluation of Proinflammatory Prognostic Biomarkers for Fabry Cardiomyopathy With Enzyme Replacement Therapy. Can. J. Cardiol. 2016, 32, 1221.e1–1221.e9. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Hanawa, H.; Jiao, S.; Hasegawa, G.; Ohno, Y.; Yoshida, K.; Suzuki, T.; Kashimura, T.; Obata, H.; Tanaka, K.; et al. Elevated Endomyocardial Biopsy Macrophage-Related Markers in Intractable Myocardial Diseases. Inflammation 2015, 38, 2288–2299. [Google Scholar] [CrossRef]
- Lidove, O.; Kaminsky, P.; Hachulla, E.; Leguy-Seguin, V.; Lavigne, C.; Marie, I.; Maillot, F.; Serratrice, C.; Masseau, A.; Chérin, P.; et al. Fabry Disease ‘The New Great Imposter’: Results of the French Observatoire in Internal Medicine Departments (FIMeD). Clin. Genet. 2012, 81, 571–577. [Google Scholar] [CrossRef]
- Lenders, M.; Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef]
- Kant, S.; Atta, M.G. Therapeutic Advances in Fabry Disease: The Future Awaits. Biomed. Pharmacother. 2020, 131, 110779. [Google Scholar] [CrossRef]
- Hughes, D.A.; Elliott, P.M.; Shah, J.; Zuckerman, J.; Coghlan, G.; Brookes, J.; Mehta, A.B. Effects of Enzyme Replacement Therapy on the Cardiomyopathy of Anderson–Fabry Disease: A Randomised, Double-Blind, Placebo-Controlled Clinical Trial of Agalsidase Alfa. Heart 2008, 94, 153–158. [Google Scholar] [CrossRef]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A.; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme Replacement Therapy in Fabry Disease: A Randomized Controlled Trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef]
- Rombach, S.M.; Smid, B.E.; Bouwman, M.G.; Linthorst, G.E.; Dijkgraaf, M.G.W.; Hollak, C.E.M. Long Term Enzyme Replacement Therapy for Fabry Disease: Effectiveness on Kidney, Heart and Brain. Orphanet J. Rare Dis. 2013, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, L.J.; Wyatt, K.M.; Henley, W.; Nikolaou, V.; Waldek, S.; Hughes, D.A.; Pastores, G.M.; Logan, S. Long-Term Effectiveness of Enzyme Replacement Therapy in Fabry Disease: Results from the NCS-LSD Cohort Study. J. Inherit. Metab. Dis. 2014, 37, 969–978. [Google Scholar] [CrossRef]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the Pharmacologic Chaperone Migalastat in a Subset of Male Patients with the Classic Phenotype of Fabry Disease and Migalastat-Amenable Variants: Data from the Phase 3 Randomized, Multicenter, Double-Blind Clinical Trial and Extension Study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [CrossRef] [Green Version]
- Ravarotto, V.; Simioni, F.; Carraro, G.; Bertoldi, G.; Pagnin, E.; Calò, L.A. Oxidative Stress and Cardiovascular-Renal Damage in Fabry Disease: Is There Room for a Pathophysiological Involvement? J. Clin. Med. 2018, 7, 409. [Google Scholar] [CrossRef] [Green Version]
- Das, A.M.; Naim, H.Y. Biochemical Basis of Fabry Disease with Emphasis on Mitochondrial Function and Protein Trafficking. Adv. Clin. Chem. 2009, 49, 57–71. [Google Scholar] [CrossRef]
- Ivanova, M. Altered Sphingolipids Metabolism Damaged Mitochondrial Functions: Lessons Learned from Gaucher and Fabry Diseases. J. Clin. Med. 2020, 9, 1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Oxidative Stress; Academic Press: London, UK, 1985; ISBN 978-0-12-642760-8. [Google Scholar]
- Ravarotto, V.; Bertoldi, G.; Innico, G.; Gobbi, L.; Calò, L.A. The Pivotal Role of Oxidative Stress in the Pathophysiology of Cardiovascular-Renal Remodeling in Kidney Disease. Antioxidants 2021, 10, 1041. [Google Scholar] [CrossRef]
- Ravarotto, V.; Bertoldi, G.; Stefanelli, L.F.; Nalesso, F.; Calò, L.A. Pathomechanism of Oxidative Stress in Cardiovascularrenal Remodeling and Therapeutic Strategies. Kidney Res. Clin. Pract. 2022, 41, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.S.; Meng, X.L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide Induces Oxidative Stress and Up-Regulates Cell Adhesion Molecule Expression in Fabry Disease Endothelial Cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Chiou, C.C.; Chang, P.Y.; Chan, E.C.; Wu, T.L.; Tsao, K.C.; Wu, J.T. Urinary 8-Hydroxydeoxyguanosine and Its Analogs as DNA Marker of Oxidative Stress: Development of an ELISA and Measurement in Both Bladder and Prostate Cancers. Clin. Chim. Acta 2003, 334, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Chou, Y.C.; Hsiao, C.Y.; Chien, Y.; Wang, K.L.; Lai, Y.H.; Chang, Y.L.; Niu, D.M.; Yu, W.C. Amelioration of Serum 8-OHdG Level by Enzyme Replacement Therapy in Patients with Fabry Cardiomyopathy. Biochem. Biophys. Res. Commun. 2017, 486, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Scopelliti, F.; Vulpis, E.; Tafani, M.; Villanova, L.; Verardo, R.; De Paulis, R.; Russo, M.A.; Frustaci, A. Increased Oxidative Stress Contributes to Cardiomyocyte Dysfunction and Death in Patients with Fabry Disease Cardiomyopathy. Hum. Pathol. 2015, 46, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Biancini, G.B.; Moura, D.J.; Manini, P.R.; Faverzani, J.L.; Netto, C.B.O.; Deon, M.; Giugliani, R.; Saffi, J.; Vargas, C.R. DNA Damage in Fabry Patients: An Investigation of Oxidative Damage and Repair. Mutat. Research. Genet. Toxicol. Environ. Mutagen. 2015, 784–785, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Vivekanandan-Giri, A.; Pennathur, S.; Smid, B.E.; Aerts, J.M.F.G.; Hollak, C.E.M.; Shayman, J.A. Establishing 3-Nitrotyrosine as a Biomarker for the Vasculopathy of Fabry Disease. Kidney Int. 2014, 86, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M.; Gebhard, C.; Studiger, R.; Shi, Y.; Mocharla, P.; Schmied, C.; Brugada, P.; Lüscher, T.F.; Camici, G.G. Globotriaosylsphingosine Accumulation and Not Alpha-Galactosidase-A Deficiency Causes Endothelial Dysfunction in Fabry Disease. PloS ONE 2012, 7, e36373. [Google Scholar] [CrossRef]
- Sun, J.; Druhan, L.J.; Zweier, J.L. Reactive Oxygen and Nitrogen Species Regulate Inducible Nitric Oxide Synthase Function Shifting the Balance of Nitric Oxide and Superoxide Production. Arch. Biochem. Biophys. 2010, 494, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.F.; Scott, L.T.C.; Gladwin, M.T.; Altarescu, G.; Kaneski, C.; Suzuki, K.; Pease-Fye, M.; Ferri, R.; Brady, R.O.; Herscovitch, P.; et al. Regional Cerebral Hyperperfusion and Nitric Oxide Pathway Dysregulation in Fabry Disease: Reversal by Enzyme Replacement Therapy. Circulation 2001, 104, 1506–1512. [Google Scholar] [CrossRef] [Green Version]
- Bodary, P.F.; Shen, Y.; Vargas, F.B.; Bi, X.; Ostenso, K.A.; Gu, S.; Shayman, J.A.; Eitzman, D.T. α-Galactosidase A Deficiency Accelerates Atherosclerosis in Mice with Apolipoprotein E Deficiency. Circulation 2005, 111, 629–632. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.S.; Arning, E.; West, M.L.; Day, T.S.; Chen, S.; Meng, X.L.; Forni, S.; McNeill, N.; Goker-Alpan, O.; Wang, X.; et al. Tetrahydrobiopterin Deficiency in the Pathogenesis of Fabry Disease. Hum. Mol. Genet. 2017, 26, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Biancini, G.B.; Vanzin, C.S.; Rodrigues, D.B.; Deon, M.; Ribas, G.S.; Barschak, A.G.; Manfredini, V.; Netto, C.B.O.; Jardim, L.B.; Giugliani, R.; et al. Globotriaosylceramide Is Correlated with Oxidative Stress and Inflammation in Fabry Patients Treated with Enzyme Replacement Therapy. Biochim. Biophys. Acta 2012, 1822, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Ravarotto, V.; Carraro, G.; Pagnin, E.; Bertoldi, G.; Simioni, F.; Maiolino, G.; Martinato, M.; Landini, L.; Davis, P.A.; Calò, L.A. Oxidative Stress and the Altered Reaction to It in Fabry Disease: A Possible Target for Cardiovascular-Renal Remodeling? PLoS ONE 2018, 13, e0204618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirásková, A.; Bortolussi, G.; Dostálová, G.; Eremiášová, L.; Golaň, L.; Danzig, V.; Linhart, A.; Vítek, L. Serum Bilirubin Levels and Promoter Variations in HMOX1 and UGT1A1 Genes in Patients with Fabry Disease. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Calò, L.A.; Pessina, A.C. RhoA/Rho-Kinase Pathway: Much More than Just a Modulation of Vascular Tone. Evidence from Studies in Humans. J. Hypertens. 2007, 25, 259. [Google Scholar] [CrossRef]
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular–Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, C.; Torri, S.; Montano, V.; Chico, L.; Gruosso, F.; Tuttolomondo, A.; Pinto, A.; Simonetta, I.; Cianci, V.; Salviati, A.; et al. Oxidative Stress Biomarkers in Fabry Disease: Is There a Room for Them? J. Neurol. 2020, 267, 3741–3752. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Nohl, H.; Gille, L.; Staniek, K. Intracellular Generation of Reactive Oxygen Species by Mitochondria. Biochem. Pharmacol. 2005, 69, 719–723. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol. Med. 2017, 23, 116–134. [Google Scholar] [CrossRef]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef]
- Lücke, T.; Höppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry Disease: Reduced Activities of Respiratory Chain Enzymes with Decreased Levels of Energy-Rich Phosphates in Fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Machann, W.; Breunig, F.; Weidemann, F.; Sandstede, J.; Hahn, D.; Köstler, H.; Neubauer, S.; Wanner, C.; Beer, M. Cardiac Energy Metabolism Is Disturbed in Fabry Disease and Improves with Enzyme Replacement Therapy Using Recombinant Human Galactosidase, A. Eur. J. Heart Fail. 2011, 13, 278–283. [Google Scholar] [CrossRef] [PubMed]
- McDermott-Roe, C.; Ye, J.; Ahmed, R.; Sun, X.-M.; Serafín, A.; Ware, J.; Bottolo, L.; Muckett, P.; Cañas, X.; Zhang, J.; et al. Endonuclease G Is a Novel Determinant of Cardiac Hypertrophy and Mitochondrial Function. Nature 2011, 478, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A Human Stem Cell Model of Fabry Disease Implicates LIMP-2 Accumulation in Cardiomyocyte Pathology. Stem Cell Rep. 2019, 13, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Schumann, A.; Schaller, K.; Belche, V.; Cybulla, M.; Grünert, S.C.; Moers, N.; Sass, J.O.; Kaech, A.; Hannibal, L.; Spiekerkoetter, U. Defective Lysosomal Storage in Fabry Disease Modifies Mitochondrial Structure, Metabolism and Turnover in Renal Epithelial Cells. J. Inherit. Metab. Dis. 2021, 44, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 Activates Notch1 in Human Podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [Green Version]
- Basak, N.P.; Roy, A.; Banerjee, S. Alteration of Mitochondrial Proteome Due to Activation of Notch1 Signaling Pathway. J. Biol. Chem. 2014, 289, 7320–7334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, W.-L.; Chou, S.-J.; Chiang, H.-C.; Wang, M.-L.; Chien, C.-S.; Chen, K.-H.; Leu, H.-B.; Wang, C.-Y.; Chang, Y.-L.; Liu, Y.-Y.; et al. Imbalanced Production of Reactive Oxygen Species and Mitochondrial Antioxidant SOD2 in Fabry Disease-Specific Human Induced Pluripotent Stem Cell-Differentiated Vascular Endothelial Cells. Cell Transpl. 2017, 26, 513–527. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA Mutations in Human Disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Simoncini, C.; Chico, L.; Concolino, D.; Sestito, S.; Fancellu, L.; Boadu, W.; Sechi, G.P.; Feliciani, C.; Gnarra, M.; Zampetti, A.; et al. Mitochondrial DNA Haplogroups May Influence Fabry Disease Phenotype. Neurosci. Lett. 2016, 629, 58–61. [Google Scholar] [CrossRef]
- Song, R.; Hu, X.-Q.; Zhang, L. Mitochondrial MiRNA in Cardiovascular Function and Disease. Cells 2019, 8, 1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, J.; Fiordelisi, A.; Sorriento, D.; Cerasuolo, F.; Buonaiuto, A.; Avvisato, R.; Pisani, A.; Varzideh, F.; Riccio, E.; Santulli, G.; et al. Mitochondrial MicroRNAs Are Dysregulated in Patients with Fabry Disease. J. Pharm. Exp. Ther. 2023, 384, 72–78. [Google Scholar] [CrossRef]
- King, L.; Plun-Favreau, H. Chapter 5—Mitophagy. In Parkinson’s Disease; Verstreken, P., Ed.; Academic Press: San Diego, CA, USA, 2017; pp. 139–177. ISBN 978-0-12-803783-6. [Google Scholar]
- Lavandero, S.; Chiong, M.; Rothermel, B.A.; Hill, J.A. Autophagy in Cardiovascular Biology. J. Clin. Investig. 2015, 125, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; van de Hoek, G.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired Autophagy Bridges Lysosomal Storage Disease and Epithelial Dysfunction in the Kidney. Nat. Commun. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chévrier, M.; Brakch, N.; Céline, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerrière, A.L.; Barbey, F.; et al. Autophagosome Maturation Is Impaired in Fabry Disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Liebau, M.C.; Braun, F.; Höpker, K.; Weitbrecht, C.; Bartels, V.; Müller, R.-U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated Autophagy Contributes to Podocyte Damage in Fabry’s Disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Son, M.; Chae, Y.; Oh, S.; Koh, E.S.; Kim, Y.K.; Shin, S.J.; Park, C.W.; Jung, S.-C.; Kim, H.-S. Fabry Disease Exacerbates Renal Interstitial Fibrosis after Unilateral Ureteral Obstruction via Impaired Autophagy and Enhanced Apoptosis. Kidney Res. Clin. Pr. 2021, 40, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-Lysosome Pathway Associated Neuropathology and Axonal Degeneration in the Brains of Alpha-Galactosidase A-Deficient Mice. Acta Neuropathol. Commun. 2014, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Marenco, M.; Segatto, M.; Sacchetti, M.; Mangiantini, P.; Giovannetti, F.; Plateroti, R. Autophagy-Lysosome Pathway Alteration in Ocular Surface Manifestations in Fabry Disease Patients. Orphanet J. Rare Dis. 2022, 17, 291. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The Cell Biology of Disease: Lysosomal Storage Disorders: The Cellular Impact of Lysosomal Dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Maalouf, K.; Jia, J.; Rizk, S.; Brogden, G.; Keiser, M.; Das, A.; Naim, H.Y. A Modified Lipid Composition in Fabry Disease Leads to an Intracellular Block of the Detergent-Resistant Membrane-Associated Dipeptidyl Peptidase IV. J. Inherit. Metab. Dis. 2010, 33, 445–449. [Google Scholar] [CrossRef]
- Song, H.-Y.; Chien, C.-S.; Yarmishyn, A.A.; Chou, S.-J.; Yang, Y.-P.; Wang, M.-L.; Wang, C.-Y.; Leu, H.-B.; Yu, W.-C.; Chang, Y.-L.; et al. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Labilloy, A.; Youker, R.T.; Bruns, J.R.; Kukic, I.; Kiselyov, K.; Halfter, W.; Finegold, D.; do Monte, S.J.H.; Weisz, O.A. Altered Dynamics of a Lipid Raft Associated Protein in a Kidney Model of Fabry Disease. Mol. Genet. Metab. 2014, 111, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Weissmann, C.; Albanese, A.A.; Contreras, N.E.; Gobetto, M.N.; Castellanos, L.C.S.; Uchitel, O.D. Ion Channels and Pain in Fabry Disease. Mol. Pain 2021, 17, 17448069211033172. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Simonetta, I.; Riolo, R.; Todaro, F.; Di Chiara, T.; Miceli, S.; Pinto, A. Pathogenesis and Molecular Mechanisms of Anderson–Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 10088. [Google Scholar] [CrossRef]
- Prabakaran, T.; Nielsen, R.; Larsen, J.V.; Sørensen, S.S.; Rasmussen, U.F.-; Saleem, M.A.; Petersen, C.M.; Verroust, P.J.; Christensen, E.I. Receptor-Mediated Endocytosis of α-Galactosidase A in Human Podocytes in Fabry Disease. PLoS ONE 2011, 6, e25065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rega, L.R.; De Leo, E.; Nieri, D.; Luciani, A. Defective Cystinosin, Aberrant Autophagy−Endolysosome Pathways, and Storage Disease: Towards Assembling the Puzzle. Cells 2022, 11, 326. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Luciani, A. Chloride Transporters and Receptor-Mediated Endocytosis in the Renal Proximal Tubule. J Physiol 2015, 593, 4151–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimarchi, H.; Ceol, M.; Gianesello, L.; Priante, G.; Iotti, A.; Del Prete, D. Downregulation of Megalin, Cubilin, ClC-5 and Podocin in Fabry Nephropathy: Potential Implications in the Decreased Effectiveness of Enzyme Replacement Therapy. J. Nephrol. 2021, 34, 1307–1314. [Google Scholar] [CrossRef]
- Pieroni, M.; Pieruzzi, F.; Mignani, R.; Graziani, F.; Olivotto, I.; Riccio, E.; Ciabatti, M.; Limongelli, G.; Manna, R.; Bolognese, L.; et al. Potential Resistance to SARS-CoV-2 Infection in Lysosomal Storage Disorders. Clin. Kidney J. 2021, 14, 1488–1490. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of Autophagy and Reformation of Lysosomes Regulated by MTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb Signaling Axis by Arginine Regulates MTORC1 Activity. eLife 2016, 5, e11058. [Google Scholar] [CrossRef]
- Lim, J.-A.; Li, L.; Shirihai, O.S.; Trudeau, K.M.; Puertollano, R.; Raben, N. Modulation of MTOR Signaling as a Strategy for the Treatment of Pompe Disease. EMBO Mol. Med. 2017, 9, 353–370. [Google Scholar] [CrossRef]
- An, J.H.; Hong, S.-E.; Yu, S.-L.; Kang, J.; Park, C.G.; Lee, H.Y.; Lee, S.-K.; Lee, D.C.; Park, H.-W.; Hwang, W.-M.; et al. Ceria-Zirconia Nanoparticles Reduce Intracellular Globotriaosylceramide Accumulation and Attenuate Kidney Injury by Enhancing the Autophagy Flux in Cellular and Animal Models of Fabry Disease. J. Nanobiotechnol. 2022, 20, 125. [Google Scholar] [CrossRef]
- Francini-Pesenti, F.; Ravarotto, V.; Bertoldi, G.; Spinella, P.; Calò, L.A. Could Nutritional Therapy Take Us Further in Our Approaches to Fabry Disease? Nutrition 2020, 72. [Google Scholar] [CrossRef]
- Sakuraba, H.; Igarashi, T.; Shibata, T.; Suzuki, Y. Effect of Vitamin E and Ticlopidine on Platelet Aggregation in Fabry’s Disease. Clin. Genet. 1987, 31, 349–354. [Google Scholar] [CrossRef]
- Moore, D.F.; Ye, F.; Brennan, M.L.; Gupta, S.; Barshop, B.A.; Steiner, R.D.; Rhead, W.J.; Brady, R.O.; Hazen, S.L.; Schiffmann, R. Ascorbate Decreases Fabry Cerebral Hyperperfusion Suggesting a Reactive Oxygen Species Abnormality: An Arterial Spin Tagging Study. J. Magn. Reson. Imaging 2004, 20, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, H.W.; Nam, S.A.; Lee, J.Y.; Cho, H.J.; Kim, T.M.; Kim, Y.K. Human Kidney Organoids Reveal the Role of Glutathione in Fabry Disease. Exp. Mol. Med. 2021, 53, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Biancini, G.B.; Jacques, C.E.; Hammerschmidt, T.; de Souza, H.M.; Donida, B.; Deon, M.; Vairo, F.P.; Lourenço, C.M.; Giugliani, R.; Vargas, C.R. Biomolecules Damage and Redox Status Abnormalities in Fabry Patients before and during Enzyme Replacement Therapy. Clin. Chim. Acta 2016, 461, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Bertoldi, G.; Carraro, G.; Ravarotto, V.; Di Vico, V.; Baldini Anastasio, P.; Vitturi, N.; Francini, F.; Stefanelli, L.F.; Calò, L.A. The Effect of Green Tea as an Adjuvant to Enzyme Replacement Therapy on Oxidative Stress in Fabry Disease: A Pilot Study. Front. Nutr. 2022, 9, 924710. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertoldi, G.; Caputo, I.; Driussi, G.; Stefanelli, L.F.; Di Vico, V.; Carraro, G.; Nalesso, F.; Calò, L.A. Biochemical Mechanisms beyond Glycosphingolipid Accumulation in Fabry Disease: Might They Provide Additional Therapeutic Treatments? J. Clin. Med. 2023, 12, 2063. https://doi.org/10.3390/jcm12052063
Bertoldi G, Caputo I, Driussi G, Stefanelli LF, Di Vico V, Carraro G, Nalesso F, Calò LA. Biochemical Mechanisms beyond Glycosphingolipid Accumulation in Fabry Disease: Might They Provide Additional Therapeutic Treatments? Journal of Clinical Medicine. 2023; 12(5):2063. https://doi.org/10.3390/jcm12052063
Chicago/Turabian StyleBertoldi, Giovanni, Ilaria Caputo, Giulia Driussi, Lucia Federica Stefanelli, Valentina Di Vico, Gianni Carraro, Federico Nalesso, and Lorenzo A. Calò. 2023. "Biochemical Mechanisms beyond Glycosphingolipid Accumulation in Fabry Disease: Might They Provide Additional Therapeutic Treatments?" Journal of Clinical Medicine 12, no. 5: 2063. https://doi.org/10.3390/jcm12052063