
An Exercise Immune Fitness Test to Unravel Disease Mechanisms—A Proof-of-Concept Heart Failure Study


 ,
,  , add
Show full author list
, add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
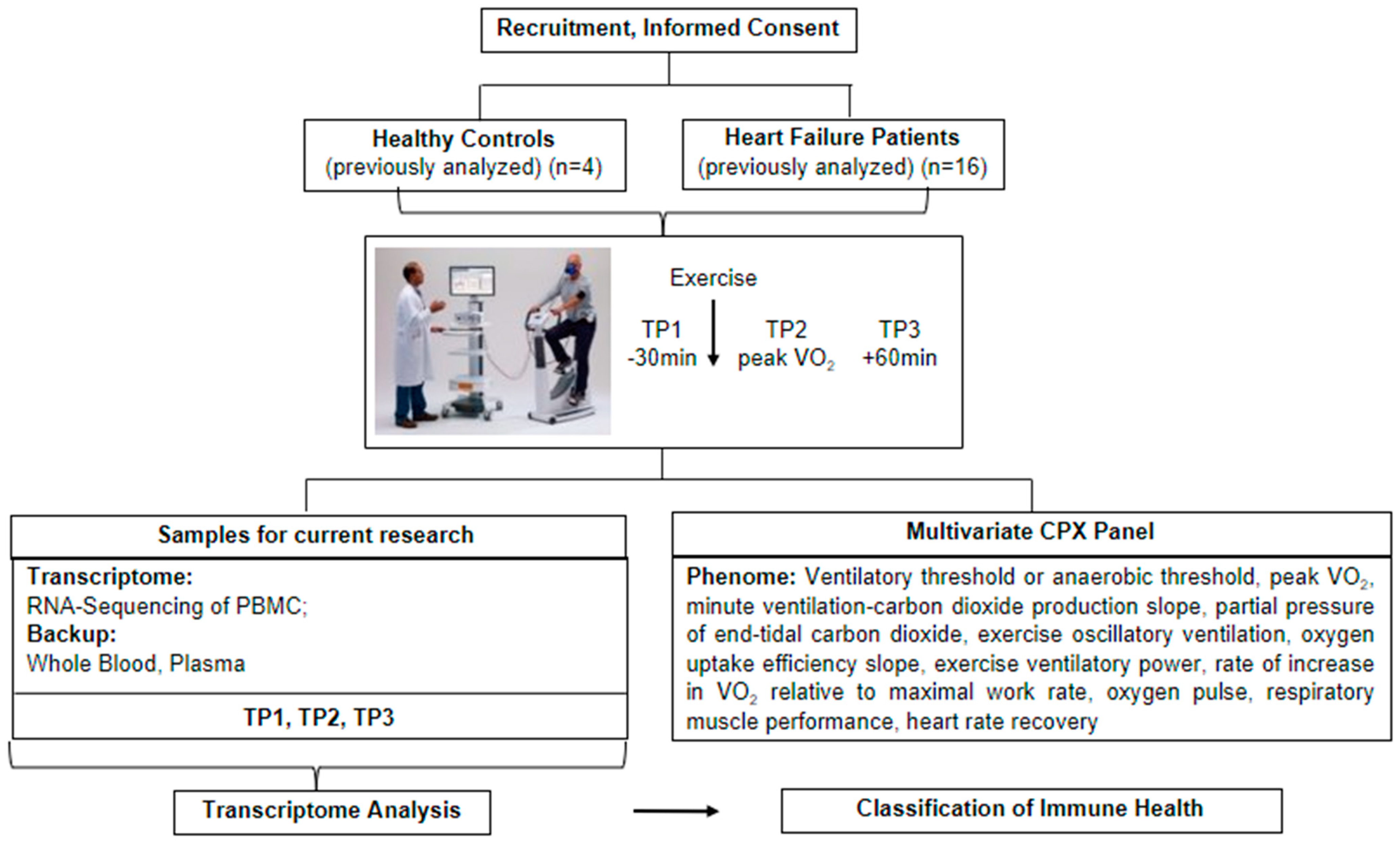
2. Methods and Design
2.1. Subject Population
2.2. Blood Processing
2.3. Statistical Analysis
3. Results
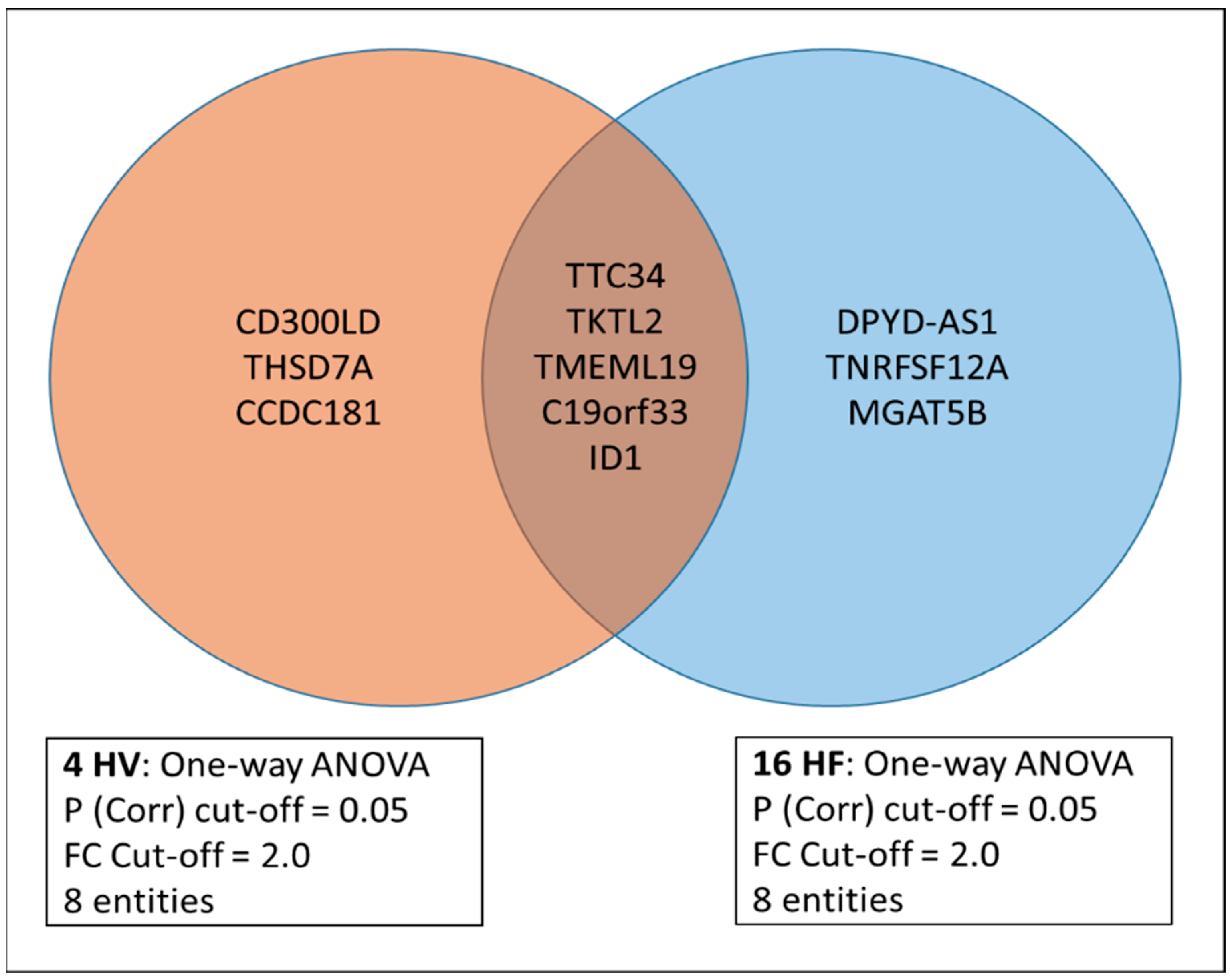
3.1. Differentially Expressed Genes (DEGs) before/at Max/1 h Post Max Exercise in the Combined Cohort
3.2. Differential Behavior of Genes in the HV versus Mild HF1 and Severe HF2 Cohorts (Peak VO2/Percent Predicted VO2)
3.3. Correlations between the DEG/Clinical Profiles (Peak VO2/Percent Predicted VO2) and Survival/Heart Transplant Outcomes
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortega, E. The “bioregulatory effect of exercise” on the innate/inflammatory responses. J. Physiol. Biochem. 2016, 72, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Clausen, J.S.R.; Marott, J.L.; Holtermann, A.; Gyntelberg, F.; Jensen, M.T. Midlife cardiorespiratory fitness and the long-term risk of mortality: 46 years of follow-up. J. Am. Coll. Cardiol. 2018, 72, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.N.; Kohl, H.W.; Paffenbarger, R.S.; Clark, D.G.; Cooper, K.H.; Gibbons, L.W. Physical fitness and all-cause mortality: A prospective study of healthy men and women. JAMA 1989, 262, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Saito, K.; Tanaka, S.; Maki, M.; Yachi, Y.; Asumi, M.; Sugawara, A.; Totsuka, K.; Shimano, H.; Ohashi, Y.; et al. Cardiorespiratory fitness as a quantitative predictor of all-cause mortality and cardiovascular events in healthy men and women: A meta-analysis. JAMA 2009, 301, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Ekelund, L.-G.; Haskell, W.L.; Johnson, J.L.; Whaley, F.S.; Criqui, M.H.; Sheps, D.S.; Lipid Research Clinics Mortality Follow-up, S. Physical fitness as a predictor of cardiovascular mortality in asymptomatic North American men. N. Engl. J. Med. 1988, 319, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.N.; Kohl, H.W.; Barlow, C.E.; Paffenbarger, R.S.; Gibbons, L.W.; Macera, C.A. Changes in physical fitness and all-cause mortality: A prospective study of healthy and unhealthy men. JAMA 1995, 273, 1093–1098. [Google Scholar] [CrossRef]
- Harber, M.P.; Kaminsky, L.A.; Arena, R.; Blair, S.N.; Franklin, B.A.; Myers, J.; Ross, R. Impact of cardiorespiratory fitness on all-cause and disease-specific mortality: Advances since 2009. Prog. Cardiovasc. Dis. 2017, 60, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, T.; Mertens, D.J.; Hamm, L.F.; Beyene, J.; Kennedy, J.; Corey, P.; Shephard, R.J. Prediction of long-term prognosis in 12 169 men referred for cardiac rehabilitation. Circulation 2002, 106, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Arem, H.; Moore, S.C.; Patel, A.; Hartge, P.; De Gonzalez, A.B.; Visvanathan, K.; Campbell, P.T.; Freedman, M.; Weiderpass, E.; Adami, H.O.; et al. Leisure time physical activity and mortality: A detailed pooled analysis of the dose-response relationship. JAMA Intern. Med. 2015, 175, 959–967. [Google Scholar] [CrossRef]
- Myers, J.; Prakash, M.; Froelicher, V.; Do, D.; Partington, S.; Atwood, J.E. Exercise capacity and mortality among men referred for exercise testing. N. Engl. J. Med. 2002, 346, 793–801. [Google Scholar] [CrossRef]
- Ross, R.; Blair, S.N.; Arena, R.; Church, T.S.; Després, J.-P.; Franklin, B.A.; Haskell, W.L.; Kaminsky, L.A.; Levine, B.D.; Lavie, C.J.; et al. Importance of assessing cardiorespiratory fitness in clinical practice: A case for fitness as a clinical vital sign: A scientific statement from the American Heart Association. Circulation 2016, 134, e653–e699. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Shiroma, E.J.; Lobelo, F.; Puska, P.; Blair, S.N.; Katzmarzyk, P.T. Effect of physical inactivity on major non-communicable diseases worldwide: An analysis of burden of disease and life expectancy. Lancet 2012, 380, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.E.; Bittner, V.; Appel, L.; Blair, S.N.; Church, T.; Després, J.-P.; Franklin, B.A.; Miller, T.D.; Pate, R.R.; Taylor-Piliae, R.E.; et al. The National Physical Activity Plan: A call to action from the American Heart Association: A science advisory from the American Heart Association. Circulation 2015, 131, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.P.; Gleeson, M.; Pyne, D.B.; Nieman, D.C.; Dhabhar, F.S.; Shephard, R.J.; Oliver, S.J.; Bermon, S.; Kajeniene, A. Position statement part two: Maintaining immune health. Exerc. Immunol. Rev. 2011, 17, 64–103. [Google Scholar] [PubMed]
- Walsh, N.P.; Gleeson, M.; Shephard, R.J.; Gleeson, M.; Woods, J.A.; Bishop, N.; Fleshner, M.; Green, C.; Pedersen, B.K.; Hoffman-Goete, L.; et al. Position statement part one: Immune function and exercise. Exerc. Immunol. Rev. 2011, 17, 6–63. [Google Scholar] [PubMed]
- World Health Organization. Global Recommendations on Physical Activity for Health; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Gjevestad, G.O.; Holven, K.B.; Ulven, S.M. Effects of exercise on gene expression of inflammatory markers in human peripheral blood cells: A systematic review. Curr. Cardiovasc. Risk Rep. 2015, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Huber, Y.; Gehrke, N.; Biedenbach, J.; Helmig, S.; Simon, P.; Straub, B.K.; Bergheim, I.; Huber, T.; Schuppan, D.; Galle, P.R.; et al. Voluntary distance running prevents TNF-mediated liver injury in mice through alterations of the intrahepatic immune milieu. Cell Death Dis. 2017, 8, e2893. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, G.; McFarlin, B.K.; O’Connor, D.P.; Smith, P.J.W.; Pircher, H.; Simpson, R.J. Aerobic fitness is associated with lower proportions of senescent blood T-cells in man. Brain Behav. Immun. 2011, 25, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.E. Is immunosenescence influenced by our lifetime “dose” of exercise? Biogerontology 2016, 17, 581–602. [Google Scholar] [CrossRef]
- Shockett, P.E.; Khanal, J.; Sitaula, A.; Oglesby, C.; Meachum, W.A.; Castracane, V.D.; Kraemer, R.R. Plasma cell-free mitochondrial DNA declines in response to prolonged moderate aerobic exercise. Physiol. Rep. 2016, 4, e12672. [Google Scholar] [CrossRef]
- Mann, D.L. Inflammatory mediators and the failing heart: Past, present, and the foreseeable future. Circ. Res. 2002, 91, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood J. Am. Soc. Hematol. 2003, 101, 2461–2463. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of inflammation in heart failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Coats, A.J.S. The “muscle hypothesis” of chronic heart failure. J. Mol. Cell. Cardiol. 1996, 28, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Doehner, W.; Frenneaux, M.; Anker, S.D. Metabolic impairment in heart failure: The myocardial and systemic perspective. J. Am. Coll. Cardiol. 2014, 64, 1388–1400. [Google Scholar] [CrossRef] [PubMed]
- Larun, L.; Brurberg, K.G.; Odgaard-Jensen, J.; Price, J.R. Exercise therapy for chronic fatigue syndrome. Cochrane Database Syst. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Loy, B.D.; O’Connor, P.J.; Dishman, R.K. Effect of acute exercise on fatigue in people with ME/CFS/SEID: A meta-analysis. Med. Sci. Sports Exerc. 2016, 48, 2003. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Pugin, J. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 2018, 9, 342896. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Nawroth, P.P. Linking stress to inflammation. Anesthesiol. Clin. N. Am. 2006, 24, 325–340. [Google Scholar] [CrossRef]
- Dhabhar, F.S. Effects of stress on immune function: The good, the bad, and the beautiful. Immunol. Res. 2014, 58, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Mancini, D.M.; Eisen, H.; Kussmaul, W.; Mull, R.; Edmunds, L.H., Jr.; Wilson, J.R. Value of peak exercise oxygen consumption for optimal timing of cardiac transplantation in ambulatory patients with heart failure. Circulation 1991, 83, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.C. An Exercise Immune Fitness Test to Unravel Mechanisms of Post-Acute Sequelae of COVID-19; Taylor & Francis: Abingdon, UK, 2023; Volume 19, pp. 693–697. [Google Scholar]
- Bondar, G.; Togashi, R.; Cadeiras, M.; Schaenman, J.; Cheng, R.K.; Masukawa, L.; Hai, J.; Bao, T.-M.; Chu, D.; Chang, E.; et al. Association between preoperative peripheral blood mononuclear cell gene expression profiles, early postoperative organ function recovery potential and long-term survival in advanced heart failure patients undergoing mechanical circulatory support. PLoS ONE 2017, 12, e0189420. [Google Scholar] [CrossRef] [PubMed]
- Radom-Aizik, S.; Zaldivar, F., Jr.; Leu, S.-Y.; Cooper, D.M. A brief bout of exercise alters gene expression and distinct gene pathways in peripheral blood mononuclear cells of early-and late-pubertal females. J. Appl. Physiol. 2009, 107, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Brock, C.O.; Volden, P.A.; Hernandez, K.; Skor, M.; Kocherginsky, M.; Park, J.E.; Brady, M.J.; Conzen, S.D. Glucocorticoid receptor C h IP-sequencing of subcutaneous fat reveals modulation of inflammatory pathways. Obesity 2015, 23, 2286–2293. [Google Scholar] [CrossRef]
- Solskov, L.; Magnusson, N.E.; Kristiansen, S.B.; Jessen, N.; Nielsen, T.T.; Schmitz, O.; Bøtker, H.E.; Lund, S. Microarray expression analysis in delayed cardioprotection: The effect of exercise, AICAR, or metformin and the possible role of AMP-activated protein kinase (AMPK). Mol. Cell. Biochem. 2012, 360, 353–362. [Google Scholar] [CrossRef]
- Hsu, W.B.; Hsu, W.H.; Hung, J.S.; Shen, W.J.; Hsu, R.W. Transcriptome analysis of osteoblasts in an ovariectomized mouse model in response to physical exercise. Bone Jt. Res. 2018, 7, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Yerra, V.G.; Batchu, S.N.; Kabir, G.; Advani, S.L.; Liu, Y.; Siddiqi, F.S.; Connelly, K.A.; Advani, A. Empagliflozin disrupts a Tnfrsf12a-mediated feed forward loop that promotes left ventricular hypertrophy. Cardiovasc. Drugs Ther. 2022, 36, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.A. Coherent somatic mutation in autoimmune disease. PLoS ONE 2014, 9, e101093. [Google Scholar] [CrossRef]
- Saif, M.W.; Smith, M.; Maloney, A. The first case of severe takotsubo cardiomyopathy associated with 5-fluorouracil in a patient with abnormalities of both dihydropyrimidine dehydrogenase (DPYD) and thymidylate synthase (TYMS) genes. Cureus 2016, 8, e783. [Google Scholar] [CrossRef]
- Schwarz, T.; Prieler, B.; Schmid, J.A.; Grzmil, P.; Neesen, J. Ccdc181 is a microtubule-binding protein that interacts with Hook1 in haploid male germ cells and localizes to the sperm tail and motile cilia. Eur. J. Cell Biol. 2017, 96, 276–288. [Google Scholar] [CrossRef]
- Kanamoto, T.; Mizuhashi, K.; Terada, K.; Minami, T.; Yoshikawa, H.; Furukawa, T. Isolation and characterization of a novel plasma membrane protein, osteoblast induction factor (obif), associated with osteoblast differentiation. BMC Dev. Biol. 2009, 9, 70. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, J.; Zhang, P.; Li, L.; Min, M.; Li, T.; He, W. Development and validation of cancer-associated fibroblasts-related gene landscape in prognosis and immune microenvironment of bladder cancer. Front. Oncol. 2023, 13, 1174252. [Google Scholar] [CrossRef]
- Dinh, P.; Peng, J.-H.; Tran, C.; Tran, T.; Pan, S.-L. Construction of miRNAs and gene expression profiles associated with ischemic cardiomyopathy: Bioinformatics analysis. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Almutairi, M.H.; Alrubie, T.M.; Alamri, A.M.; Almutairi, B.O.; Alrefaei, A.F.; Arafah, M.M.; Alanazi, M.; Semlali, A. Cancer-testis gene biomarkers discovered in colon cancer patients. Genes 2022, 13, 807. [Google Scholar] [CrossRef]
- Li, Y.; Wang, D.W.; Chen, Y.; Chen, C.; Guo, J.; Zhang, S.; Sun, Z.; Ding, H.; Yao, Y.; Zhou, L.; et al. Genome-wide association and functional studies identify SCML4 and THSD7A as novel susceptibility genes for coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Heidecker, B.; Kittleson, M.M.; Kasper, E.K.; Wittstein, I.S.; Champion, H.C.; Russell, S.D.; Hruban, R.H.; Rodriguez, E.R.; Baughman, K.L.; Hare, J.M.; et al. Transcriptomic biomarkers for the accurate diagnosis of myocarditis. Circulation 2011, 123, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Yang, C.; Zhao, L.; Wang, S.; Gao, Z.; Ye, Y. Prognostic Biomarker SPOCD1 and Its Correlation with Immune Infiltrates in Colorectal Cancer. Biomolecules 2023, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Y.; Yan, A.; Yang, Y. SPOCD1 accelerates ovarian cancer progression and inhibits cell apoptosis via the PI3K/AKT pathway. OncoTargets Ther. 2020, 13, 351–359. [Google Scholar] [CrossRef]
- Lin, Z.; Chen, L.; Wu, T.; Zhang, Y.; Huang, X.; Chen, Y.; Chen, J.; Xu, Y. Prognostic Value of SPOCD1 in Esophageal Squamous Cell Carcinoma: A Comprehensive Study Based on Bioinformatics and Validation. Front. Genet. 2022, 13, 872026. [Google Scholar] [CrossRef]
- Li, M.; Feng, Z.; Han, R.; Hu, B.; Zhang, R.; Wang, H. Paclitaxel promotes mTOR signaling-mediated apoptosis in esophageal cancer cells by targeting MUC20. Thorac. Cancer 2023, 14, 3089–3096. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Guo, W.-M.; Jia, J.-D.; Kadeerhan, G.; Liu, H.-P.; Bai, T.; Shao, Y.; Wang, D.-W. MUC20 as a novel prognostic biomarker in ccRCC correlating with tumor immune microenvironment modulation. Am. J. Cancer Res. 2022, 12, 695. [Google Scholar] [PubMed]
- Le-Niculescu, H.; Roseberry, K.; Gill, S.S.; Levey, D.F.; Phalen, P.L.; Mullen, J.; Williams, A.; Bhairo, S.; Voegtline, T.; Davis, H.; et al. Precision medicine for mood disorders: Objective assessment, risk prediction, pharmacogenomics, and repurposed drugs. Mol. Psychiatry 2021, 26, 2776–2804. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Diao, H.; Zhao, F.; Xiao, S.; El Zowalaty, A.E.; Dudley, E.A.; Mattson, M.P.; Ye, X. Olfactomedin 1 deficiency leads to defective olfaction and impaired female fertility. Endocrinology 2015, 156, 3344–3357. [Google Scholar] [CrossRef] [PubMed]
- Lipman, A.R.; Fan, X.; Shen, Y.; Chung, W.K. Clinical and genetic characterization of CACNA1A-related disease. Clin. Genet. 2022, 102, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, M.; Barth, M.; Gueden, S.; de Cepoy, P.D.; Aeby, A.; Vilain, C.; Hirsch, E.; de Saint Martin, A.; Des Portes, V.; Lesca, G.; et al. CACNA1A-associated epilepsy: Electroclinical findings and treatment response on seizures in 18 patients. Eur. J. Paediatr. Neurol. 2021, 33, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yanas, A.; Owens, M.C.; Zhang, C.; Fritsch, C.; Fare, C.M.; Copley, K.E.; Shorter, J.; Goldman, Y.E.; Liu, K.F. Sexually dimorphic RNA helicases DDX3X and DDX3Y differentially regulate RNA metabolism through phase separation. Mol. Cell 2022, 82, 2588–2603. [Google Scholar] [CrossRef]
- Dicke, A.-K.; Pilatz, A.; Wyrwoll, M.J.; Punab, M.; Ruckert, C.; Nagirnaja, L.; Aston, K.I.; Conrad, D.F.; Di Persio, S.; Neuhaus, N.; et al. DDX3Y is likely the key spermatogenic factor in the AZFa region that contributes to human non-obstructive azoospermia. Commun. Biol. 2023, 6, 350. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondar, G.; Mahapatra, A.D.; Bao, T.-M.; Silacheva, I.; Hairapetian, A.; Vu, T.; Su, S.; Katappagari, A.; Galan, L.; Chandran, J.; et al. An Exercise Immune Fitness Test to Unravel Disease Mechanisms—A Proof-of-Concept Heart Failure Study. J. Clin. Med. 2024, 13, 3200. https://doi.org/10.3390/jcm13113200
Bondar G, Mahapatra AD, Bao T-M, Silacheva I, Hairapetian A, Vu T, Su S, Katappagari A, Galan L, Chandran J, et al. An Exercise Immune Fitness Test to Unravel Disease Mechanisms—A Proof-of-Concept Heart Failure Study. Journal of Clinical Medicine. 2024; 13(11):3200. https://doi.org/10.3390/jcm13113200
Chicago/Turabian StyleBondar, Galyna, Abhinandan Das Mahapatra, Tra-Mi Bao, Irina Silacheva, Adrian Hairapetian, Thomas Vu, Stephanie Su, Ananya Katappagari, Liana Galan, Joshua Chandran, and et al. 2024. "An Exercise Immune Fitness Test to Unravel Disease Mechanisms—A Proof-of-Concept Heart Failure Study" Journal of Clinical Medicine 13, no. 11: 3200. https://doi.org/10.3390/jcm13113200
APA StyleBondar, G., Mahapatra, A. D., Bao, T.-M., Silacheva, I., Hairapetian, A., Vu, T., Su, S., Katappagari, A., Galan, L., Chandran, J., Adamov, R., Mancusi, L., Lai, I., Rahman, A., Grogan, T., Hsu, J. J., Cappelletti, M., Ping, P., Elashoff, D., ... Deng, M. C. (2024). An Exercise Immune Fitness Test to Unravel Disease Mechanisms—A Proof-of-Concept Heart Failure Study. Journal of Clinical Medicine, 13(11), 3200. https://doi.org/10.3390/jcm13113200