
CAR-T-Cell-Based Cancer Immunotherapies: Potentials, Limitations, and Future Prospects

Abstract
:1. Background
2. Introduction to Cancer: Understanding the Complex Landscape of a Pervasive Disease
3. Current Cancer Immunotherapies
3.1. Monoclonal Antibody Therapy
3.2. Immune Checkpoint Inhibitors (ICIs)
3.3. Cytokine Therapy
3.4. Adoptive Cell Therapies (ACTs) for Cancers
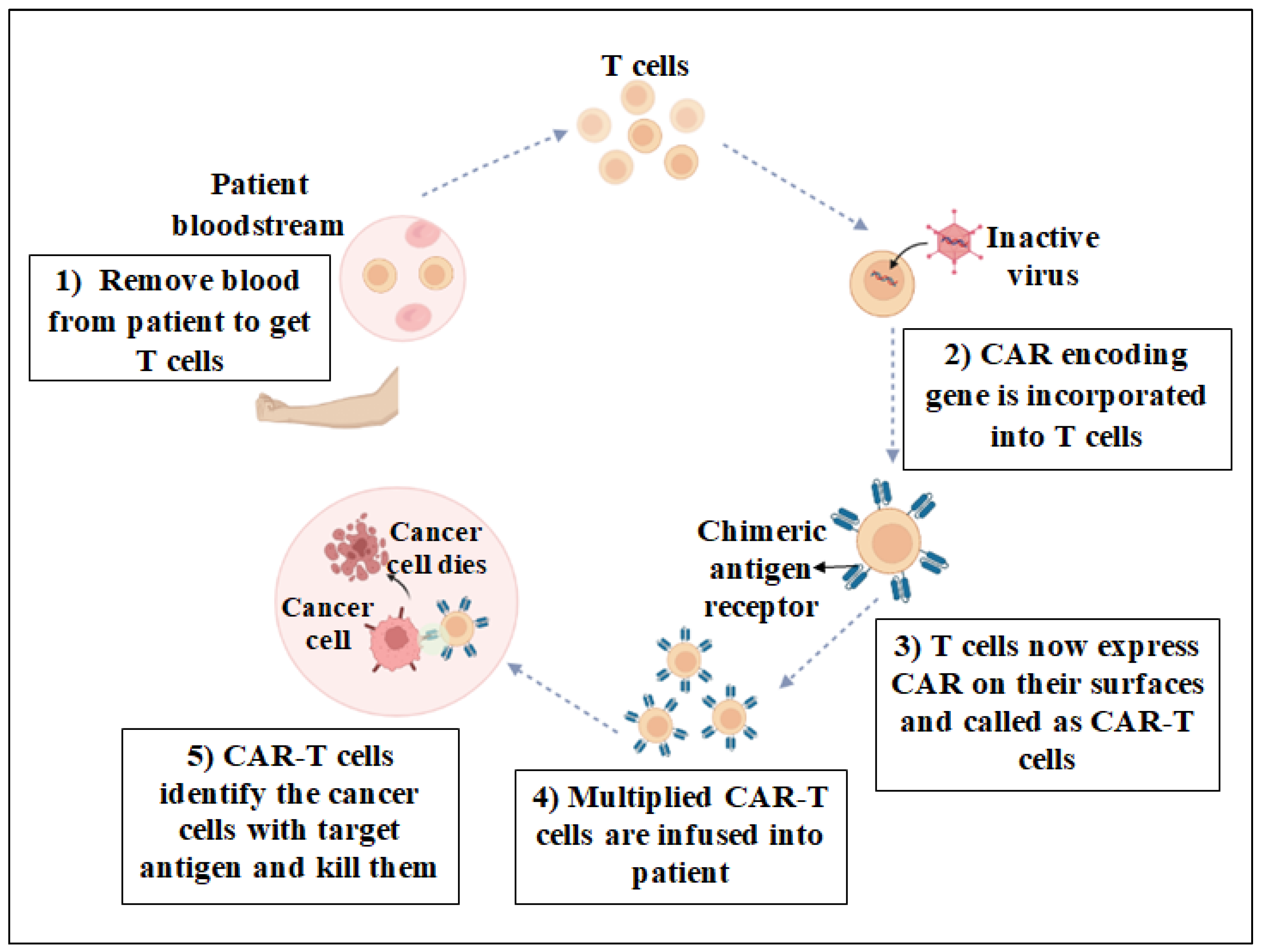
3.5. Chimeric Antigen Receptor T Cell (CAR-T) Therapy
- It shows success in treating specific hematological cancers, including diffuse large B cell lymphoma (DLBCL) and B cell acute lymphoblastic leukemia (B-ALL). It has resulted in high response rates and, in some cases, long-term remissions.
- CAR-T cells are engineered to identify and target specific antigens on the surface of cancer cells. The therapy can be targeted at cancer cells while maintaining normal, healthy cells by tailoring the CARs to a specific antigen.
- CAR-T therapy involves the patient’s own T cells followed by their genetic modification to express CAR specific to cancer cells and their re-infusion into the patient. This tailored approach improves the specificity of the treatment and reduces some side effects.
- Unlike traditional T cell treatments that depend on MHC presentation, CAR-T cells have the ability to detect antigens without the need of the MHC. This fact is particularly beneficial when MHC expression is downregulated by cancer cells.
- In some patients, CAR-T therapy has exhibited durable responses, meaning persistent benefits over a long period of time, potentially providing long-term remission.
- Certain types of CAR-T therapies (such as Kymriah (tisagenlecleucel) and Yescarta (axicabtagene ciloleucel) have received approval from regulatory agencies, including the U.S. Food and Drug Administration (FDA), for the treatment of specific types of leukemia and lymphoma, which allows for insurance reimbursement.
- Ongoing research focuses on improving CAR design, addressing limitations, and expanding the application of CAR-T therapy to other cancer types. Next-generation CARs are being developed to enhance safety, efficacy, and applicability.
3.5.1. CAR-T Structure
3.5.2. Antigen-Binding Domain
3.5.3. Hinge Region
3.5.4. Transmembrane Domain
3.5.5. Intracellular Signaling Domain(s)
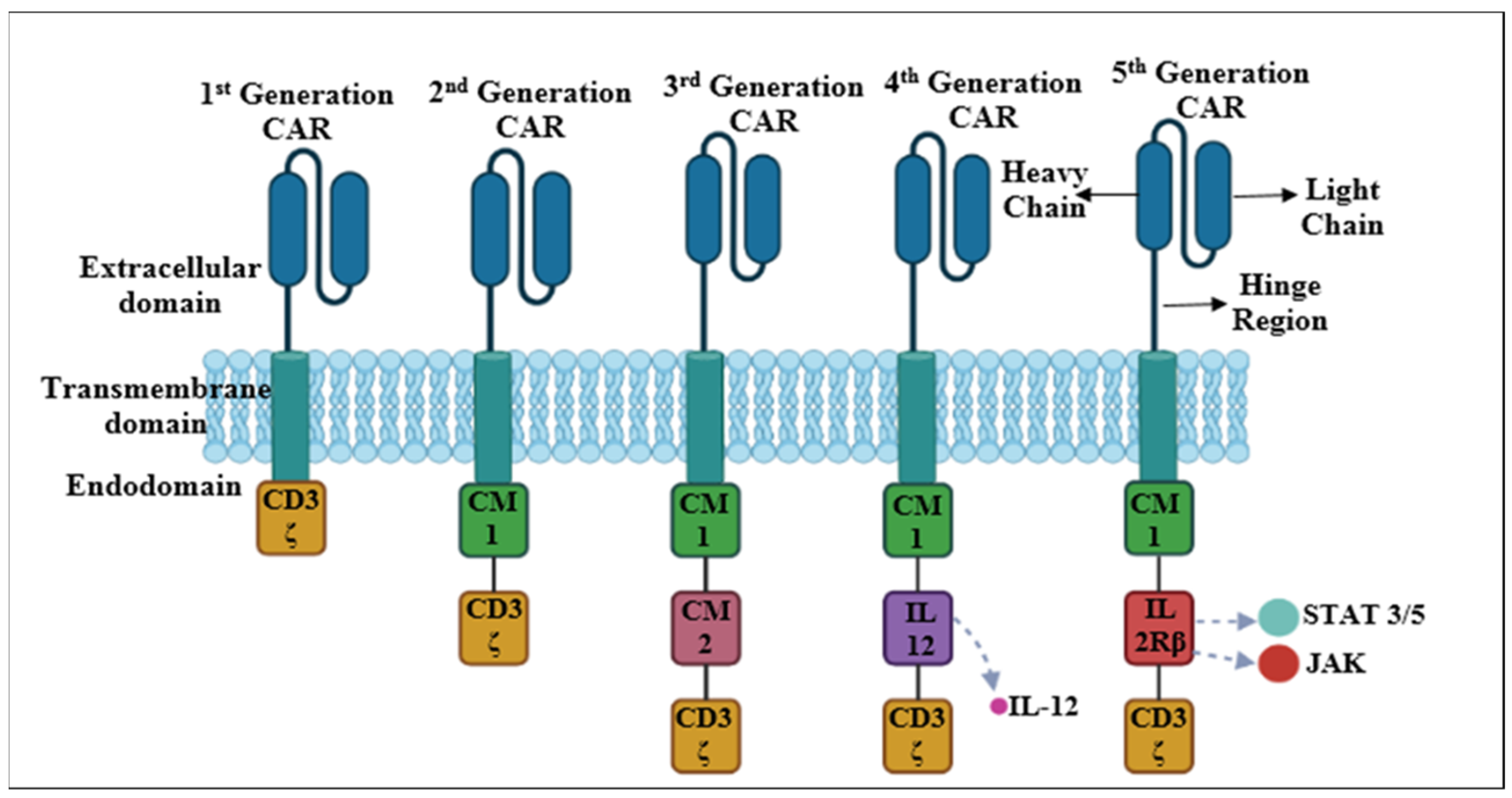
4. Evolution of CAR-T Cells
4.1. First-Generation CARs
- 8.
- Exogenous IL-2 stimulates the survival and persistence of CAR-T cells in the tumor microenvironment by inhibiting premature cell death and improving their antitumor ability;
- 9.
- It enhances CAR-T cell growth, resulting in a bigger pool of effector cells capable of targeting tumor cells;
- 10.
- It stimulates the differentiation of regulatory T cells (Tregs) and effector T cells. It is important to balance these subgroups for efficient antitumor immunity.
- 11.
- It improves the capacity of CAR-T cells to identify and combat cancer cells.
4.2. Second-Generation CARs
4.3. Third-Generation CARs
4.4. Fourth-Generation CARs
4.5. Fifth-Generation CARs
5. Boolean Logic-Gated CARs
6. Manufacturing and Infusion of CAR-T Cells for Cancer Treatment
6.1. Sources of T Cells
6.2. Isolation of T Cells
6.3. T Cell Activation
6.3.1. Cell-Based T Cell Activation
6.3.2. Beads-Based T Cell Activation
6.3.3. Antibody-Coated Magnetic Beads
6.3.4. Antibody-Coated Nanobeads
6.3.5. Expamer Technology
6.3.6. Activation with Anti-CD3 Antibodies
6.4. Genetic Modification of T Cells
6.4.1. Non-Viral Transfer Methods
6.4.2. Viral Transduction
6.4.3. Transposons
6.4.4. CRISPR/Cas9
6.5. Expansion of CAR-T Cells
6.6. Quality Control and Characterization
6.7. Cryopreservation
6.8. Lymphodepletion
6.9. CAR-T Cell Infusion
7. Clinical Applications of CAR-T Cells
7.1. Hematological Malignancies
7.2. Lymphoblastic Leukemia/Lymphoma
7.3. Acute Myeloid Leukemia
7.4. Multiple Myeloma
7.5. Hodgkin’s Lymphoma
8. Solid Tumors
8.1. Prostate Cancer
8.2. Breast Cancer
8.3. Renal and Hepatic Cancers
8.4. Ovarian Cancer
8.5. Colorectal Cancer
8.6. Pancreatic Cancer
8.7. Thoracic Cancer
8.8. Solid Pediatric Tumors
9. Registered Clinical Trials Using CAR-T Cell Therapy
10. Drawbacks of CAR-T Cell Therapy
10.1. Antigen Escape
10.2. On-Target Off-Tumor Effects
10.3. Tumor Infiltration and CAR-T Cell Trafficking
10.4. Immunosuppressive Microenvironment
10.5. Toxicities Associated with CAR-T Cells
10.6. Cost of CAR-T Therapy
11. Methods to Reduce CAR-T-Cell-Therapy-Related Side Effects
11.1. Modifying CAR Structure
11.2. CAR Immunogenicity
11.3. Neurotoxicity and CAR-Transduced T Cell Modification
11.4. CAR Off-Switches
12. Application of Artificial Intelligence in Manufacturing CAR-T Cells
13. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Cooper, G.M.; Hausman, R.E. The development and causes of cancer. Cell A Mol. Approach 2000, 2, 719–728. [Google Scholar]
- Bindea, G.; Mlecnik, B.; Angell, H.K.; Galon, J. The immune landscape of human tumors: Implications for cancer immunotherapy. Oncoimmunology 2014, 3, e27456. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Galon, J.; Dieu-Nosjean, M.-C.; Cremer, I.; Fisson, S.; Damotte, D.; Pagès, F.; Tartour, E.; Sautès-Fridman, C. Immune infiltration in human cancer: Prognostic significance and disease control. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–24. [Google Scholar]
- Varadé, J.; Magadán, S.; González-Fernández, Á. Human immunology and immunotherapy: Main achievements and challenges. Cell. Mol. Immunol. 2021, 18, 805–828. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.L.; Basu, S.; Soni, V.; Jaiswal, R.K. Immunotherapy: An alternative promising therapeutic approach against cancers. Mol. Biol. Rep. 2022, 49, 9903–9913. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R.; Kersten, M.J.; Salles, G.; Abramson, J.S.; Schuster, S.J.; Locke, F.L.; Andreadis, C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am. J. Hematol. 2021, 96, 1295–1312. [Google Scholar] [CrossRef] [PubMed]
- ImmPACT Bio. ImmPACT Bio Announces FDA Clearance of IND for Novel Bispecific CAR to Treat Aggressive B-Cell Lymphoma. 2023. Available online: https://tdg.ucla.edu/immpact-bio-announces-fda-clearance-ind-novel-bispecific-car-treat-aggressive-b-cell-lymphoma#:~:text=(%E2%80%9CImmPACT%20Bio%E2%80%9D)%2C,a%20bispecific%20%E2%80%9COR%2DGate%E2%80%9D (accessed on 2 September 2023).
- Bäckel, N.; Hort, S.; Kis, T.; Nettleton, D.F.; Egan, J.R.; Jacobs, J.J.L.; Grunert, D.; Schmitt, R.H. Elaborating the potential of Artificial Intelligence in automated CAR-T cell manufacturing. Front. Mol. Med. 2023, 3, 1250508. [Google Scholar] [CrossRef]
- Carbone, A. Cancer classification at the crossroads. Cancers 2020, 12, 980. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Bhardwaj, A.; Gupta, S. Cancer treatment therapies: Traditional to modern approaches to combat cancers. Mol. Biol. Rep. 2023, 50, 9663–9676. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Klener, P.; Otahal, P.; Lateckova, L. Immunotherapy approaches in cancer treatment. Curr. Pharm. Biotechnol. 2015, 16, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.; Sun, W. Cancer immunotherapy in clinical practice—The past, present, and future. Chin. J. Cancer 2014, 33, 445. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Puhr, H.C.; Ilhan-Mutlu, A. New emerging targets in cancer immunotherapy: The role of LAG3. ESMO Open 2019, 4, e000482. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Southam, C.M.; Brunschwig, A.; Levin, A.G.; Dizon, Q.S. Effect of leukocytes on transplantability of human cancer. Cancer 1966, 19, 1743–1753. [Google Scholar] [CrossRef]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N. Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. JNCI J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Lora, A.M.G.; Van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, M.C.; Minute, L.; Rodriguez, I.; Garasa, S.; Perez-Ruiz, E.; Inogés, S.; Melero, I.; Berraondo, P. Antibody-dependent cell cytotoxicity: Immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol. 2017, 95, 347–355. [Google Scholar] [CrossRef]
- Moon, D.; Tae, N.; Park, Y.; Lee, S.-W.; Kim, D.H. Development of bispecific antibody for cancer immunotherapy: Focus on T cell engaging antibody. Immune Netw. 2022, 22, e4. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T cell therapy beyond oncology: Autoimmune diseases and viral infections. Biomedicines 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, L.; Cui, H.; Wang, X.; Zhang, G.; Ma, J.; Han, H.; He, W.; Wang, W.; Zhao, Y.; et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci. Rep. 2014, 4, 3571. [Google Scholar] [CrossRef] [PubMed]
- Chailyan, A.; Marcatili, P.; Tramontano, A. The association of heavy and light chain variable domains in antibodies: Implications for antigen specificity. FEBS J. 2011, 278, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [PubMed]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.C.; Riddell, S.R. Designing chimeric antigen receptors to effectively and safely target tumors. Curr. Opin. Immunol. 2015, 33, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Almåsbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; A Munthe, L.; Andersen, J.T.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, M.-R.; Sentman, C.L. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J. Immunol. 2012, 189, 2290–2299. [Google Scholar] [CrossRef]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Heuser, C.; Hombach, A.; Lösch, C.; Manista, K.; Abken, H. T-cell activation by recombinant immunoreceptors: Impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. 2003, 10, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Q.J.; Yang, S.; Kochenderfer, J.N.; Zheng, Z.; Zhong, X.; Sadelain, M.; Eshhar, Z.; Rosenberg, S.A.; Morgan, R.A. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 2009, 183, 5563–5574. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3ζ transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N.C. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [CrossRef] [PubMed]
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef]
- Cai, B.; Guo, M.; Wang, Y.; Zhang, Y.; Yang, J.; Guo, Y.; Dai, H.; Yu, C.; Sun, Q.; Qiao, J.; et al. Co-infusion of haplo-identical CD19-chimeric antigen receptor T cells and stem cells achieved full donor engraftment in refractory acute lymphoblastic leukemia. J. Hematol. Oncol. 2016, 9, 131. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, J.; Wu, Z.; Yu, J.; Cui, Q.; Pu, C.; Liang, B.; Luo, Y.; Shi, J.; Jin, A.; et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J. Hematol. Oncol. 2016, 9, 70. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Marin, V.; Pizzitola, I.; Agostoni, V.; Attianese, G.M.P.G.; Finney, H.; Lawson, A.; Pule, M.; Rousseau, R.; Biondi, A.; Biagi, E. Cytokine-induced killer cells for cell therapy of acute myeloid leukemia: Improvement of their immune activity by expression of CD33-specific chimeric receptors. Haematologica 2010, 95, 2144. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In vivo fate and activity of second-versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Razavi, S.; DeRose, R.; Inoue, T. Synthesizing biomolecule-based Boolean logic gates. ACS Synth. Biol. 2013, 2, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Tousley, A.M.; Rotiroti, M.C.; Labanieh, L.; Rysavy, L.W.; Rietberg, S.P.; de la Serna, E.L.; Sotillo, E.; Weber, E.W.; Rietberg, S.P.; Dalton, G.N.; et al. Coopting T cell proximal signaling molecules enables Boolean logic-gated CAR T cell control. bioRxiv 2022, 615, 507–516. [Google Scholar]
- Savanur, M.A.; Weinstein-Marom, H.; Gross, G. Implementing logic gates for safer immunotherapy of cancer. Front. Immunol. 2021, 12, 780399. [Google Scholar] [CrossRef] [PubMed]
- Dejenie, T.A.; Tiruneh, G.; Medhin, M.; Terefe, G.D.; Admasu, F.T.; Tesega, W.W.; Abebe, E.C. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccines Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1–and CTLA-4–based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef]
- Abou-El-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable manufacturing of CAR T cells for cancer immunotherapy. Blood Cancer Discov. 2021, 2, 408–422. [Google Scholar] [CrossRef]
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther.-Oncolyt. 2016, 3, 16015. [Google Scholar] [CrossRef]
- Wang, X.; Stefanski, J.; Borquez-Ojeda, O.; Qu, J.; Hack, A.; He, Q. (Eds.) Comparison of CTS Dynabeadsc CD3/CD28, Miltenyi TransAct CD3/28 and ExpAct Beads for Large-Scale CAR T Cell Manufacturing; European Society of Gene and Cell Therapy Collaborative Congress: Helsinki, Finland, 2015. [Google Scholar]
- Poltorak, M.P.; Graef, P.; Tschulik, C.; Wagner, M.; Cletiu, V.; Dreher, S.; Borjan, B.; Fraessle, S.P.; Effenberger, M.; Turk, M.; et al. Expamers: A new technology to control T cell activation. Sci. Rep. 2020, 10, 17832. [Google Scholar] [CrossRef]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J. Clin. Oncol. 2016, 34, 1112. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, Z.; Cohen, C.J.; Gattinoni, L.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther. 2006, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.J.; A Seinstra, B.; Nikhamin, Y.; Yeh, R.; Usachenko, Y.; van Leeuwen, D.G.; Purdon, T.; Pegram, H.J.; Brentjens, R.J. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol. Ther. 2015, 23, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Moffett, H.F.; Coon, M.E.; Radtke, S.; Stephan, S.B.; McKnight, L.; Lambert, A.; Stoddard, B.L.; Kiem, H.P.; Stephan, M.T. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 2017, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Piscopo, N.J.; Mueller, K.P.; Das, A.; Hematti, P.; Murphy, W.L.; Palecek, S.P.; Capitini, C.M.; Saha, K. Bioengineering solutions for manufacturing challenges in CAR T cells. Biotechnol. J. 2018, 13, 1700095. [Google Scholar] [CrossRef] [PubMed]
- Tumaini, B.; Lee, D.W.; Lin, T.; Castiello, L.; Stroncek, D.F.; Mackall, C.; Wayne, A.; Sabatino, M. Simplified process for the production of anti–CD19-CAR–engineered T cells. Cytotherapy 2013, 15, 1406–1415. [Google Scholar] [CrossRef]
- Cullen, B.R.; Lomedico, P.T.; Ju, G. Transcriptional interference in avian retroviruses—Implications for the promoter insertion model of leukaemogenesis. Nature 1984, 307, 241–245. [Google Scholar] [CrossRef]
- Jin, Z.; Maiti, S.; Huls, H.; Singh, H.; Olivares, S.; Mátés, L.; Izsvák, Z.; Ivics, Z.; A Lee, D.; E Champlin, R.; et al. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011, 18, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017, 31, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Farooq, A.; Ahmad, F.J.; Akhtar, M.; Choudhery, M.S. Prime editing: A potential treatment option for β-thalassemia. Cell Biol. Int. 2023, 47, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, M.A.; Corn, J.E.; Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods 2017, 121, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhao, Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein Cell 2017, 8, 634–643. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Kudriaeva, A.; Miftakhova, R.; Rybalov, A.; Bulatov, E. Recent Advances in the Development of Bioreactors for Manufacturing of Adoptive Cell Immunotherapies. Bioengineering 2022, 9, 808. [Google Scholar] [CrossRef]
- Campbell, A.; Brieva, T.; Raviv, L.; Rowley, J.; Niss, K.; Brandwein, H.; Oh, S.; Karnieli, O. Concise review: Process development considerations for cell therapy. Stem Cells Transl. Med. 2015, 4, 1155–1163. [Google Scholar] [CrossRef]
- Choudhery, M.S.; Badowski, M.; Muise, A.; Pierce, J.; Harris, D.T. Cryopreservation of whole adipose tissue for future use in regenerative medicine. J. Surg. Res. 2014, 187, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Abraham-Miranda, J.; Menges, M.; Atkins, R.; Mattie, M.; Kanska, J.; Turner, J.; Hidalgo-Vargas, M.J.; Locke, F.L. CAR-T manufactured from frozen PBMC yield efficient function with prolonged in vitro production. Front. Immunol. 2022, 13, 1007042. [Google Scholar] [CrossRef] [PubMed]
- Jandová, M.; Stacey, G.N.; Lánská, M.; Gregor, J.; Rozsívalová, P.; Beková, L.; Ducháová, Z.W.; Belada, D.; Radocha, J.; Měřička, P.; et al. Perspective: The Role of Cryopreservation Techniques in Manufacturing, Transport, and Storage of Car-T Therapy Products. Cryoletters 2023, 44, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Choudhery, M.S.; Badowski, M.; Muise, A.; THarris, D. Utility of cryopreserved umbilical cord tissue for regenerative medicine. Curr. Stem Cell Res. Ther. 2013, 8, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Choudhery, M.S.; Harris, D.T. Cryopreservation can be used as an anti-aging strategy. Cytotherapy 2014, 16, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Bojic, S.; Murray, A.; Bentley, B.L.; Spindler, R.; Pawlik, P.; Cordeiro, J.L.; Bauer, R.; de Magalhães, J.P. Winter is coming: The future of cryopreservation. BMC Biol. 2021, 19, 56. [Google Scholar] [CrossRef] [PubMed]
- Amini, L.; Silbert, S.K.; Maude, S.L.; Nastoupil, L.J.; Ramos, C.A.; Brentjens, R.J.; Sauter, C.S.; Shah, N.N.; Abou-El-Enein, M. Preparing for CAR T cell therapy: Patient selection, bridging therapies and lymphodepletion. Nat. Rev. Clin. Oncol. 2022, 19, 342–355. [Google Scholar] [CrossRef]
- Owen, K.; Ghaly, R.; Shohdy, K.S.; Thistlethwaite, F. Lymphodepleting chemotherapy practices and effect on safety and efficacy outcomes in patients with solid tumours undergoing T cell receptor-engineered T cell (TCR-T) Therapy: A systematic review and meta-analysis. Cancer Immunol. Immunother. 2023, 72, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, X.; Yuan, Z.; Liu, L.; Luo, L.; Li, Y.; Wu, K.; Liu, J.; Yang, C.; Li, Z.; et al. Eradication of T-ALL cells by CD7-targeted universal CAR-T cells and initial test of ruxolitinib-based CRS management. Clin. Cancer Res. 2021, 27, 1242–1246. [Google Scholar] [CrossRef]
- Feng, J.; Xu, H.; Cinquina, A.; Wu, Z.; Chen, Q.; Zhang, P.; Wang, X.; Shan, H.; Xu, L.; Zhang, Q.; et al. Treatment of aggressive T cell lymphoblastic lymphoma/leukemia using anti-CD5 CAR T cells. Stem Cell Rev. Rep. 2021, 17, 652–661. [Google Scholar] [CrossRef]
- Pinz, K.; Liu, H.; Golightly, M.; Jares, A.; Lan, F.; Zieve, G.W.; Hagag, N.; Schuster, M.; E Firor, A.; Jiang, X.; et al. Preclinical targeting of human T-cell malignancies using CD4-specific chimeric antigen receptor (CAR)-engineered T cells. Leukemia 2016, 30, 701–707. [Google Scholar] [CrossRef]
- Salman, H.; Pinz, K.G.; Wada, M.; Shuai, X.; Yan, L.E.; Petrov, J.C.; Ma, Y. Preclinical targeting of human acute myeloid leukemia using CD4-specific chimeric antigen receptor (CAR) T cells and NK cells. J. Cancer 2019, 10, 4408. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Ma, L.; Liu, S.; Chang, L.; Wen, F. Chimeric antigen receptor T cells targeting CD7 in a child with high-risk T-cell acute lymphoblastic leukemia. Int. Immunopharmacol. 2021, 96, 107731. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, M.; Tesio, M.; June, C.H.; Houot, R. CAR T-cells for T-cell malignancies: Challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia 2018, 32, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Maciocia, P.M.; Wawrzyniecka, P.A.; Maciocia, N.C.; Burley, A.; Karpanasamy, T.; Devereaux, S.; Hoekx, M.; O’Connor, D.; Leon, T.E.; Rapoz-D’Silva, T.; et al. Anti-CCR9 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia. Blood J. Am. Soc. Hematol. 2022, 140, 25–37. [Google Scholar] [CrossRef]
- Fanale, M.A.; Horwitz, S.M.; Forero-Torres, A.; Bartlett, N.L.; Advani, R.H.; Pro, B.; Chen, R.W.; Davies, A.; Illidge, T.; Uttarwar, M.; et al. Five-year outcomes for frontline brentuximab vedotin with CHP for CD30-expressing peripheral T-cell lymphomas. Blood J. Am. Soc. Hematol. 2018, 131, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Ball, E.D.; Varki, A. Myeloid precursors and acute myeloid leukemia cells express multiple CD33-related Siglecs. Exp. Hematol. 2006, 34, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.-E.; Loff, S.; Dietrich, J.; Spehr, J.; Jiménez, G.J.; von Bonin, M.; Ehninger, G.; Cartellieri, M.; Ehninger, A. Evaluation of switch-mediated costimulation in trans on universal CAR-T cells (UniCAR) targeting CD123-positive AML. Oncoimmunology 2021, 10, 1945804. [Google Scholar] [CrossRef] [PubMed]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3− ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Xiao, W.; Li, W.; Wang, L.; Yang, S.; Wang, W.; Xu, L.; Liao, S.; Liu, W.; et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Riether, C.; Pabst, T.; Höpner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat. Med. 2020, 26, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T cell therapy in hematological malignancies: Current opportunities and challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef] [PubMed]
- Jetani, H.; Navarro-Bailón, A.; Maucher, M.; Frenz, S.; Verbruggen, C.; Yeguas, A.; Vidriales, M.B.; González, M.; Saborido, J.R.; Kraus, S.; et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2021, 138, 1830–1842. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cao, W.; Que, Y.; Wang, Q.; Xiao, Y.; Gu, C.; Wang, D.; Wang, J.; Jiang, L.; Xu, H.; et al. A phase I study of anti-BCMA CAR T cell therapy in relapsed/refractory multiple myeloma and plasma cell leukemia. Clin. Transl. Med. 2021, 11, e346. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kanapuru, B.; George, B.; Lin, X.; Xu, Z.; Bryan, W.W.; Pazdur, R.; Theoret, M.R. FDA approval summary: Idecabtagene vicleucel for relapsed or refractory multiple myeloma. Clin. Cancer Res. 2022, 28, 1759–1764. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mahendravada, A.; Ballard, B.; Kale, B.; Ramos, C.; West, J.; Maguire, T.; McKay, K.; Lichtman, E.; Tuchman, S.; et al. Safety and efficacy of targeting CD138 with a chimeric antigen receptor for the treatment of multiple myeloma. Oncotarget 2019, 10, 2369. [Google Scholar] [CrossRef]
- Van de Donk, N.W.C.J.; Janmaat, M.L.; Mutis, T.; Lammerts Van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W.H.I. Monoclonal antibodies targeting CD 38 in hematological malignancies and beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.V.; Luetkens, T.; Scherer, S.D.; Davis, P.; Mause, E.R.V.; Olson, M.L.; Yousef, S.; Panse, J.; Abdiche, Y.; Li, K.D.; et al. CD229 CAR T cells eliminate multiple myeloma and tumor propagating cells without fratricide. Nat. Commun. 2020, 11, 798. [Google Scholar] [CrossRef]
- Kriegsmann, K.; Kriegsmann, M.; Cremer, M.; Schmitt, M.; Dreger, P.; Goldschmidt, H.; Müller-Tidow, C.; Hundemer, M. Cell-based immunotherapy approaches for multiple myeloma. Br. J. Cancer 2019, 120, 38–44. [Google Scholar] [CrossRef]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood J. Am. Soc. Hematol. 2017, 130, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-M.; Wu, Z.-Q.; Wang, Y.; Guo, Y.-L.; Dai, H.-R.; Wang, X.-H.; Li, X.; Zhang, Y.-J.; Zhang, W.-Y.; Chen, M.-X.; et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin lymphoma: An open-label phase I trial. Clin. Cancer Res. 2017, 23, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, T.J.; Zhao, B.; Oldan, J.; Hucks, G.; Khandani, A.; Dittus, C.; Smith, J.; Morrison, J.K.; Cheng, C.J.; Ivanova, A.; et al. Pretherapy metabolic tumor volume is associated with response to CD30 CAR T cells in Hodgkin lymphoma. Blood Adv. 2022, 6, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tao, C.; Li, J.; Tang, J.C.-O.; Chan, A.S.-C.; Zhou, Y. Chimeric antigen receptor T cells applied to solid tumors. Front. Immunol. 2022, 13, 984864. [Google Scholar] [CrossRef] [PubMed]
- Hillerdal, V.; Essand, M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs 2015, 29, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.F.; Wang, X.; Hullings, M.; Arauz, G.; Bartido, S.; Lewis, J.S.; Schöder, H.; Zanzonico, P.; Scher, H.I.; Sadelain, M.; et al. Chimeric antigen receptor (CAR+) modified T cells targeting prostate specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). Am. Soc. Clin. Oncol. 2013, 31, 72. [Google Scholar] [CrossRef]
- Weimin, S.; Abula, A.; Qianghong, D.; Wenguang, W. Chimeric cytokine receptor enhancing PSMA-CAR-T cell-mediated prostate cancer regression. Cancer Biol. Ther. 2020, 21, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: Possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Wolf, P.; Alzubi, J.; Gratzke, C.; Cathomen, T. The potential of CAR T cell therapy for prostate cancer. Nat. Rev. Urol. 2021, 18, 556–571. [Google Scholar] [CrossRef]
- Ghoussoub, R.A.; Dillon, D.A.; D’Aquila, T.; Rimm, E.B.; Fearon, E.R.; Rimm, D.L. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1998, 82, 1513–1520. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Liu, J.-W.; Lu, C.; Wei, J.-F. CAR-T cell therapy for breast cancer: From basic research to clinical application. Int. J. Biol. Sci. 2022, 18, 2609. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: Results of phase I trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tang, Q. Recent updates on chimeric antigen receptor T cell therapy for hepatocellular carcinoma. Cancer Gene Therapy 2021, 28, 1075–1087. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Aithal, A.; Rauth, S.; Kshirsagar, P.; Shah, A.; Lakshmanan, I.; Junker, W.M.; Jain, M.; Ponnusamy, M.P.; Batra, S.K. MUC16 as a novel target for cancer therapy. Expert Opin. Ther. Targets 2018, 22, 675–686. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor–modified T-cell therapy for CEA+ liver metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef]
- Katz, S.C.; E Moody, A.; Guha, P.; Hardaway, J.C.; Prince, E.; LaPorte, J.; Stancu, M.; E Slansky, J.; Jordan, K.R.; Schulick, R.D.; et al. HITM-SURE: Hepatic immunotherapy for metastases phase Ib anti-CEA CAR-T study utilizing pressure enabled drug delivery. J. Immunother. Cancer 2020, 8, e001097. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ metastatic colorectal cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; Dekervel, J.; Hendlisz, A.; Anguille, S.; Ahmad, A.; Cerf, E.; Jacques-Hespel, C.; Gauthy, E.; Agaugué, S.; Gilham, D.E.; et al. Updated data from the alloSHRINK phase 1 first-in-Human study evaluating CYAD-101, an innovative non-Gene-Edited allogeneic CAR-T, in metastatic colorectal cancer. J. Clin. Oncol. 2021, 39, 74. [Google Scholar] [CrossRef]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; E Wilentz, R.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 2001, 7, 3862–3868. [Google Scholar] [PubMed]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Nikolaidi, M.; Garces, I.; Lorizio, D.; Castro, N.M.; Caiafa, S.G.; Moore, K.; Brown, N.F.; Kocher, H.M.; Duan, X.; et al. CEACAM7 is an effective target for CAR T-cell therapy of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2021, 27, 1538–1552. [Google Scholar] [CrossRef] [PubMed]
- Patel, U.; Abernathy, J.; Savani, B.N.; Oluwole, O.; Sengsayadeth, S.; Dholaria, B. CAR T cell therapy in solid tumors: A review of current clinical trials. EJHaem 2022, 3, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Zauderer, M.G.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Ngai, D.A.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. Abstract CT036: A phase I clinical trial of malignant pleural disease treated with regionally delivered autologous mesothelin-targeted CAR T cells: Safety and efficacy. Cancer Res. 2019, 79 (Suppl. 13), CT036. [Google Scholar] [CrossRef]
- Burns, I.; Gwynne, W.D.; Suk, Y.; Custers, S.; Chaudhry, I.; Venugopal, C.; Singh, S.K. The Road to CAR T-Cell Therapies for Pediatric CNS Tumors: Obstacles and New Avenues. Front. Oncol. 2022, 12, 815726. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti–PD-1 agent pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Cordoba, S.; Onuoha, S.; Thomas, S.; Pignataro, D.S.; Hough, R.; Ghorashian, S.; Vora, A.; Bonney, D.; Veys, P.; Rao, K.; et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: A phase 1 trial. Nat. Med. 2021, 27, 1797–1805. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.G.; de Vos, S.; et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): A single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Chng, W.J. CAR T-cell therapy in multiple myeloma: More room for improvement. Blood Cancer J. 2021, 11, 84. [Google Scholar] [CrossRef]
- Fowler, N.H.; Dickinson, M.; Dreyling, M.; Martinez-Lopez, J.; Kolstad, A.; Butler, J.; Ghosh, M.; Popplewell, L.; Chavez, J.C.; Bachy, E.; et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: The phase 2 ELARA trial. Nat. Med. 2022, 28, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Jain, P.; Nastoupil, L.; Westin, J.; Lee, H.J.; Navsaria, L.; Steiner, R.E.; Ahmed, S.; Moghrabi, O.; Oriabure, O.; Chen, W.; et al. Outcomes and management of patients with mantle cell lymphoma after progression on brexucabtagene autoleucel therapy. Br. J. Haematol. 2021, 192, e38–e42. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tan, Y.; Wang, G.; Deng, B.; Ling, Z.; Song, W.; Seery, S.; Zhang, Y.; Peng, S.; Xu, J.; et al. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: First-in-human, phase I trial. J. Clin. Oncol. 2021, 39, 3340–3351. [Google Scholar] [CrossRef]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: Review of the literature and challenges ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef]
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; et al. Fully human Bcma targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood 2018, 132 (Suppl. S1), 1011. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef]
- Murad, J.P.; Kozlowska, A.K.; Lee, H.J.; Ramamurthy, M.; Chang, W.-C.; Yazaki, P.; Colcher, D.; Shively, J.; Cristea, M.; Forman, S.J.; et al. Effective targeting of TAG72+ peritoneal ovarian tumors via regional delivery of CAR-engineered T cells. Front. Immunol. 2018, 9, 2268. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.-C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional delivery of chimeric antigen receptor–engineered T cells effectively targets HER2+ breast cancer metastasis to the brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.-C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: Refueling the CAR. Blood J. Am. Soc. Hematol. 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Roex, G.; Timmers, M.; Wouters, K.; Campillo-Davo, D.; Flumens, D.; Schroyens, W.; Chu, Y.; Berneman, Z.N.; Lion, E.; Luo, F.; et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J. Hematol. Oncol. 2020, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Porter, D.L. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology 2016, 2016, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, L.; Sánchez-Escamilla, M.; Perales, M.-A. CAR T cell toxicity: Current management and future directions. Hemasphere 2019, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Moore, T.J.; Zhang, H.; Anderson, G.; Alexander, G.C. Estimated costs of pivotal trials for novel therapeutic agents approved by the US Food and Drug Administration, 2015–2016. JAMA Intern. Med. 2018, 178, 1451–1457. [Google Scholar] [CrossRef]
- Cliff, E.R.S.; Kelkar, A.H.; Russler-Germain, D.A.; Tessema, F.A.; Raymakers, A.J.; Feldman, W.B.; Kesselheim, A.S. high cost of chimeric antigen receptor T-cells: Challenges and solutions. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e397912. [Google Scholar] [CrossRef]
- Milone, M.C.; Bhoj, V.G. The pharmacology of T cell therapies. Mol. Ther.-Methods Clin. Dev. 2018, 8, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Van Der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hill, T.; Shamah, S.M.; I Salter, A.; Chen, Y.; Mohler, K.M.; Riddell, S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 2017, 31, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Lekakis, L.J.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; Timmerman, J.M.; et al. Preliminary results of prophylactic tocilizumab after axicabtageneciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive non-Hodgkin lymphoma (NHL). Blood 2017, 130, 1547. [Google Scholar] [CrossRef]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood J. Am. Soc. Hematol. 2019, 133, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Galimova, R.M.; Buzaev, I.V.; Ramilevich, K.A.; Yuldybaev, L.K.; Shaykhulova, A.F. Artificial intelligence-developments in medicine in the last two years. Chronic Dis. Transl. Med. 2019, 5, 64–68. [Google Scholar] [CrossRef]
- Gottschlich, A.; Thomas, M.; Grünmeier, R.; Lesch, S.; Rohrbacher, L.; Igl, V.; Briukhovetska, D.; Benmebarek, M.-R.; Vick, B.; Dede, S.; et al. Single-cell transcriptomic atlas-guided development of CAR-T cells for the treatment of acute myeloid leukemia. Nat. Biotechnol. 2023, 41, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Bedoya, A.D.; Futoma, J.; Clement, M.E.; Corey, K.; Brajer, N.; Lin, A.; Simons, M.G.; Gao, M.; Nichols, M.; Balu, S.; et al. Machine learning for early detection of sepsis: An internal and temporal validation study. JAMIA Open 2020, 3, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021, 12, 428. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Trial ID | Trial Subjects | Phase | Intervention | Enrollments |
|---|---|---|---|---|
| NCT02659943 | B Cell Lymphoma, NHLs | Phase1 | Biological: Anti-CD19 CAR-T cells, Drug: fudarabine, cyclophosphamide | 27 |
| NCT02926833 | Refractory DLBCL | Phase1, Phase 2 | Biological: KTE-C19, Drug: atezolizumab, cyclophosphamide, fludarabine | 37 |
| NCT02215967 | Myeloma | Phase1 | Biological: anti-BCMA CAR-T cells, Drug: Fudarabine, cyclophosphamide | 30 |
| NCT00924326 | B cell Lymphoma types | Phase1, Phase2 | Biological: Anti-CD19, Drug: fludarabine, cyclophosphamide, aldesleukin, fludarabine, cyclophosphamide | 43 |
| NCT01865617 | Recurrent Adult ALL, CLL, DLBCL, MCL, NHL, and Small Lymphocytic Lymphoma; Refractory ALL, Chronic Lymphocytic Leukemia, DLBCL, MCL, NHL, and Small Lymphocytic Lymphoma | Phase1, Phase2 | Biological: Autologous Anti-CD19CAR-4-1BB-CD3zeta-EGFRt-expressing T lymphocytes | 204 |
| NCT03049449 | Lymphoma, Large-Cell, Anaplastic, Enteropathy-Associated T cell Lymphoma, DLBCL, Extranodal NK-T cell Lymphoma | Phase1 | Biological: anti-tumor necrosis factor receptor superfamily member 8 (CD 30) CAR-T cells, Drug: cyclophosphamide, fludarabine | 26 |
| NCT02348216 | Refractory DLBCL, Relapsed DLBCL, TFL, PMBCL, HGBCL | Phase1, Phase2 | Biological: axicabtagene ciloleucel, Drug: fludarabine, cyclophosphamide | 307 |
| NCT02761915 | Relapsed or Refractory Neuroblastoma | Phase1 | Procedure: leukapheresis, Drug: cyclophosphamide, fludarabine, Genetic: 1RG-CART | 17 |
| NCT01626495 | B Cell Leukemia, B Cell Lymphoma | Phase1, Phase2 | Biological: CART-19 | 73 |
| NCT03338972 | Refractory Plasma Cell Myeloma, Recurrent Plasma Cell Myeloma | Phase1 | Biological: autologous anti-BCMA-CAR-expressing CD4+/CD8+ T-lymphocytes FCARH143, Procedure: leukapheresis Drug: fudarabine, cyclophosphamide | 28 |
| NCT01593696 | Large-Cell Lymphoma, ALL, B Cell Lymphoma, Leukemia, Non-Hodgkin’s Lymphoma | Phase1 | Biological: anti-CD19 CAR-T cells | 53 |
| NCT03289455 | Recurrent Childhood Acute Lymphoblastic Leukemia, B Acute Lymphoblastic Leukemia, B cell Acute Lymphoblastic Leukemia, Refractory Childhood Acute Lymphoblastic Leukemia | Phase1, Phase2 | Biological: AUTO3 (CD19/22) CAR-T cells | 23 |
| NCT03019055 | B Cell Chronic Lymphocytic Leukemia, Non-Hodgkin’s Lymphoma, Small Lymphocytic Lymphoma | Phase1 | Biological: CAR-20/19-T cells, CAR-20/19-T cells, CAR-20/19-T cells. | 26 |
| NCT02030847 | Patients with refractory or relapsed B Cell ALL with no available curative treatment options | Phase2 | Biological: CAR-T19 | 42 |
| NCT02614066 | Refractory/Relapsed B-precursor Acute Lymphoblastic Leukemia | Phase1, Phase2 | Biological: brexucabtagene autoleucel, Drug: cyclophosphamide, fludarabine | 125 |
| NCT03958656 | Myeloma, Multiple Myeloma | Phase1 | Biological: anti-signaling lymphocytic activation molecule f7 (SLAMF7) CAR-T cells Drug: fludarabine, cyclophosphamide, fludarabine, rimiducid, | 13 |
| NCT03483103 | NHL, DLBCL | Phase 2 | Biological: lisocabtagene maraleucel | 74 |
| NCT03761056 | B- ell Lymphoma | Phase2 | Biological: axicabtagene ciloleucel, Drug: fludarabine, cyclophosphamide | 42 |
| NCT04456959 | Precursor Cell Lymphoblastic Leukemia, Lymphoma | Drug: inotuzumab ozogamicin | 28 | |
| NCT04030195 | Non-Hodgkin’s Lymphoma Refractory, Non-Hodgkin’s Lymphoma, Chronic Lymphocytic Leukemia, Lymphoma, Non-Hodgkin’s, Leukemia, B cell Non-Hodgkin’s Lymphoma, B cell Chronic Lymphocytic Leukemia, Small Lymphocytic Lymphoma | Phase1, Phase2 | Genetic: PBCAR20A, Drug: fludarabine, cyclophosphamide | 18 |
| NCT01454596 | Brain Cancer, Malignant Glioma, Glioblastoma, Gliosarcoma | Phase1, Phase2 | Biological: epidermal growth factor receptor (EGFRV)iii CAR-transduced PBL, Drug: fludarabine, aldesleukin, cyclophosphamide | 18 |
| NCT01460901 | Neuroblastoma | Phase1 | Biological: GD2 CAR-modified tri-virus-specific cytotoxic T cells | 5 |
| NCT02601313 | Relapsed/Refractory Mantle Cell Lymphoma | Phase2 | Biological: autoleucel, brexucabtagene Drug: cyclophosphamide, fludarabine, axicabtagene ciloleucel | 105 |
| NCT02650999 | CD19+ DLBCL, Follicular Lymphomas, Mantle Cell Lymphomas | Phase1, Phase2 | Drug: pembrolizumab | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choudhery, M.S.; Arif, T.; Mahmood, R.; Harris, D.T. CAR-T-Cell-Based Cancer Immunotherapies: Potentials, Limitations, and Future Prospects. J. Clin. Med. 2024, 13, 3202. https://doi.org/10.3390/jcm13113202
Choudhery MS, Arif T, Mahmood R, Harris DT. CAR-T-Cell-Based Cancer Immunotherapies: Potentials, Limitations, and Future Prospects. Journal of Clinical Medicine. 2024; 13(11):3202. https://doi.org/10.3390/jcm13113202
Chicago/Turabian StyleChoudhery, Mahmood S., Taqdees Arif, Ruhma Mahmood, and David T. Harris. 2024. "CAR-T-Cell-Based Cancer Immunotherapies: Potentials, Limitations, and Future Prospects" Journal of Clinical Medicine 13, no. 11: 3202. https://doi.org/10.3390/jcm13113202