
Leveraging Cancer Phenotypic Plasticity for Novel Treatment Strategies


 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
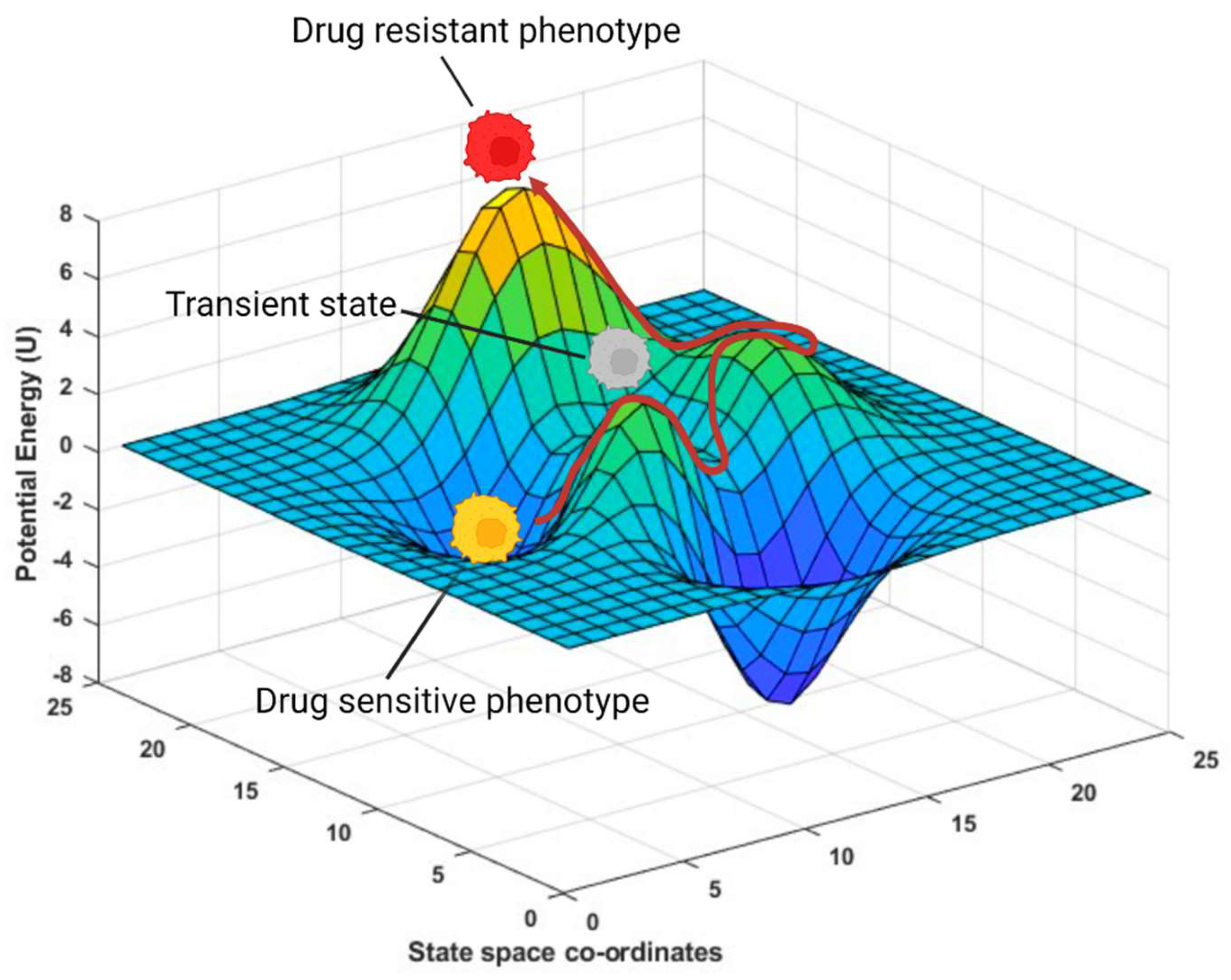
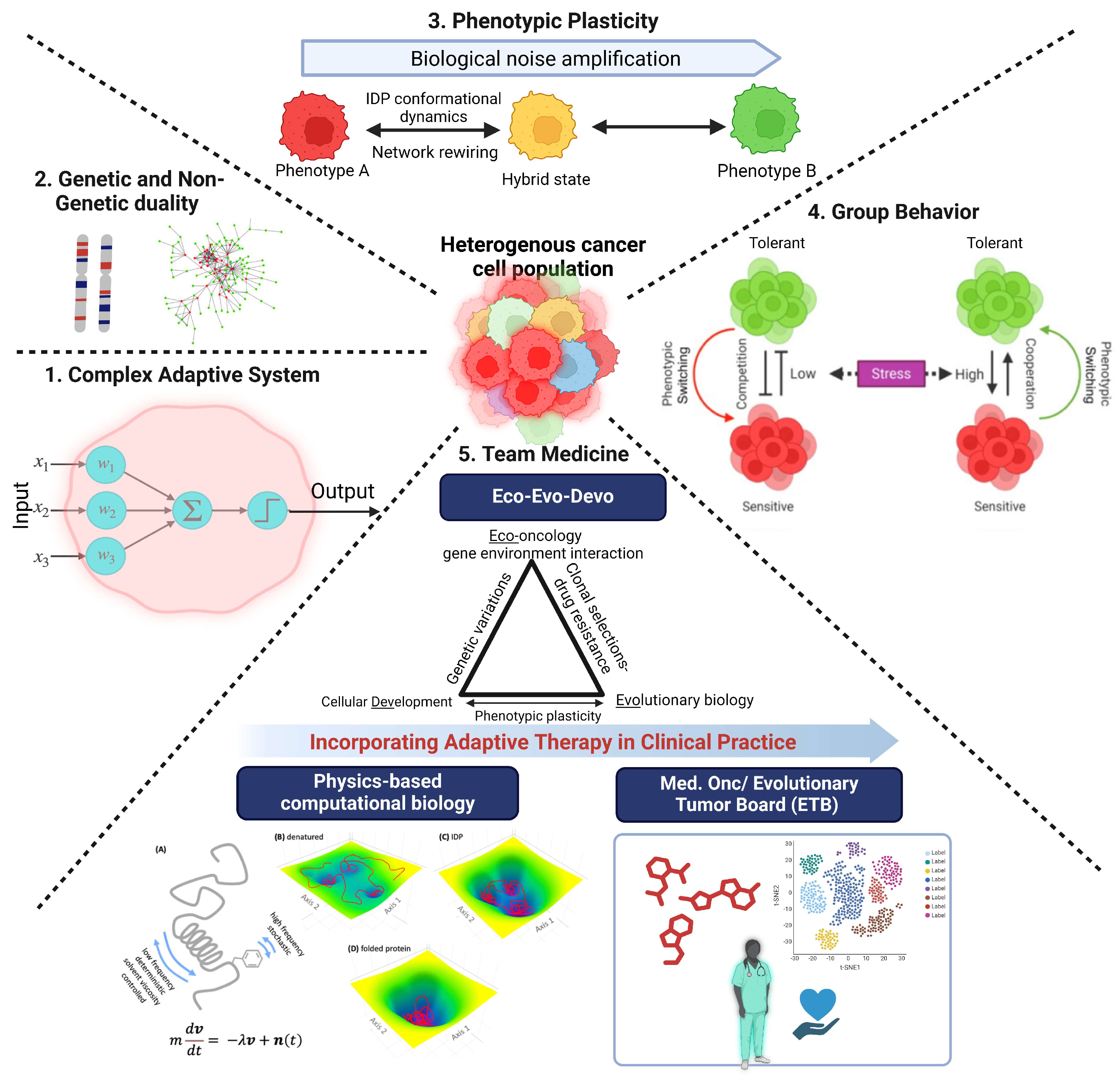
2. Phenotypic Plasticity Is an Emergent Property of Cancer Cells
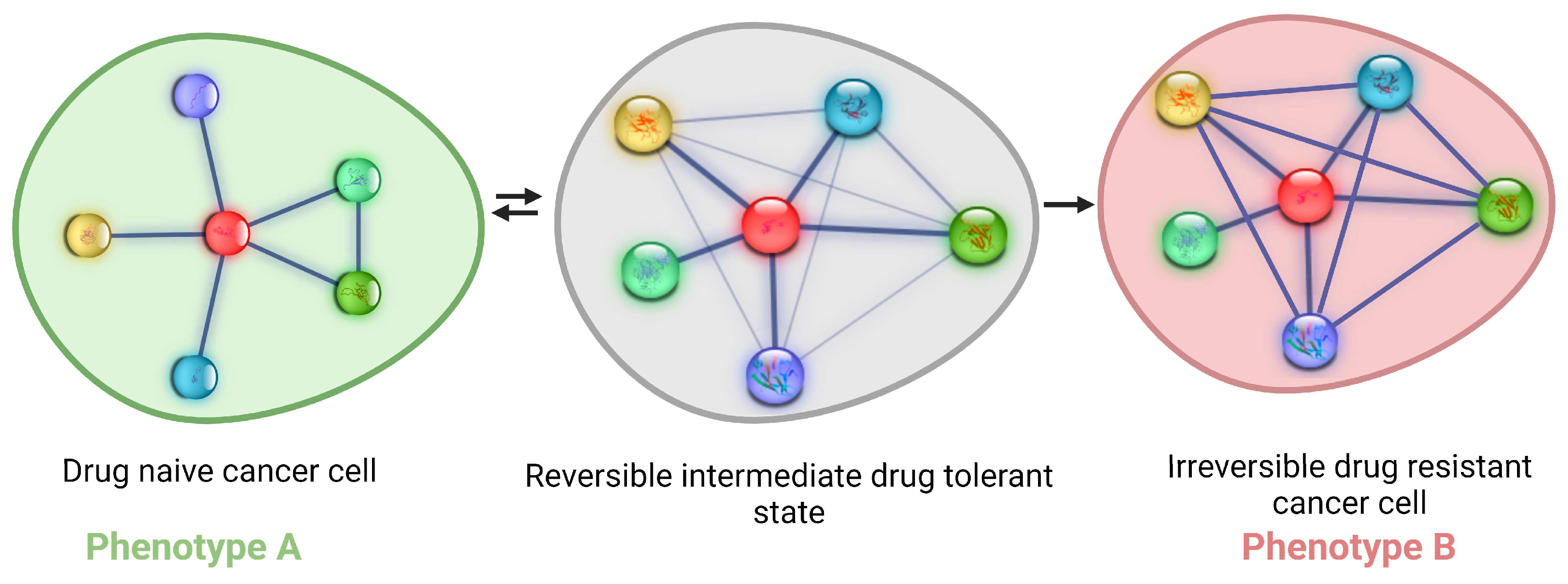
3. Cancer Cells Explore Non-Genetic Mechanisms to Increase Their Fitness
4. Intrinsically Disordered Proteins, Conformational Noise, and Phenotypic Switching
5. Phenotypic Plasticity, Non-Genetic Phenotypic Heterogeneity, and Bet Hedging
6. Leveraging Group Behavior of Cancer Cells to Develop New Therapeutic Strategies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cao, Z.; Prettner, K.; Kuhn, M.; Yang, J.; Jiao, L.; Wang, Z.; Li, W.; Geldsetzer, P.; Bärnighausen, T.; et al. Estimates and Projections of the Global Economic Cost of 29 Cancers in 204 Countries and Territories from 2020 to 2050. JAMA Oncol. 2023, 9, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.J.; Xu, A.; Lythgoe, M.P.; Prasad, V. Cancer Drug Approvals That Displaced Existing Standard-of-Care Therapies, 2016–2021. JAMA Netw. Open 2022, 5, e222265. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Downward, J. Exploiting the therapeutic implications of KRAS inhibition on tumor immunity. Cancer Cell 2024, 42, 338–357. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Kulkarni, P. The Genetic/Non-genetic Duality of Drug ‘Resistance’ in Cancer. Trends Cancer 2018, 4, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Mohanty, A.; Achuthan, S.; Kotnala, S.; Jolly, M.K.; Kulkarni, P.; Salgia, R. Group Behavior and Emergence of Cancer Drug Resistance. Trends Cancer 2021, 7, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Mohanty, A.; Ramisetty, S.; Duvivier, H.; Khan, A.; Shrestha, S.; Tan, T.; Merla, A.; El-Hajjaoui, M.; Malhotra, J.; et al. A Nexus between Genetic and Non-Genetic Mechanisms Guides KRAS Inhibitor Resistance in Lung Cancer. Biomolecules 2023, 13, 1587. [Google Scholar] [CrossRef] [PubMed]
- Hug, S.M.; Gaut, B.S. The phenotypic signature of adaptation to thermal stress in Escherichia coli. BMC Evol. Biol. 2015, 15, 177. [Google Scholar] [CrossRef] [PubMed]
- Tadrowski, A.C.; Evans, M.R.; Waclaw, B. Phenotypic Switching Can Speed up Microbial Evolution. Sci. Rep. 2018, 8, 8941. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Salgia, R. Comprehending phenotypic plasticity in cancer and evolution. iScience 2024, 27, 109308. [Google Scholar] [CrossRef] [PubMed]
- Kavran, A.J.; Stuart, S.A.; Hayashi, K.R.; Basken, J.M.; Brandhuber, B.J.; Ahn, N.G. Intermittent treatment of BRAF(V600E) melanoma cells delays resistance by adaptive resensitization to drug rechallenge. Proc. Natl. Acad. Sci. USA 2022, 119, e2113535119. [Google Scholar] [CrossRef] [PubMed]
- Bray, D. Wetware: A Computer in Every Living Cell; Yale University Press: New Haven, CT, USA, 2009. [Google Scholar]
- Kulkarni, P.; Bhattacharya, S.; Achuthan, S.; Behal, A.; Jolly, M.K.; Kotnala, S.; Mohanty, A.; Rangarajan, G.; Salgia, R.; Uversky, V. Intrinsically Disordered Proteins: Critical Components of the Wetware. Chem. Rev. 2022, 122, 6614–6633. [Google Scholar] [CrossRef] [PubMed]
- Ros-Rocher, N.; Brunet, T. What is it like to be a choanoflagellate? Sensation, processing and behavior in the closest unicellular relatives of animals. Anim. Cogn. 2023, 26, 1767–1782. [Google Scholar] [CrossRef] [PubMed]
- Dinet, C.; Michelot, A.; Herrou, J.; Mignot, T. Linking single-cell decisions to collective behaviours in social bacteria. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2021, 376, 20190755. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kim, S.J.; Hara, M.; Aono, M. Remarkable problem-solving ability of unicellular amoeboid organism and its mechanism. R. Soc. Open Sci. 2018, 5, 180396. [Google Scholar] [CrossRef] [PubMed]
- Tero, A.; Takagi, S.; Saigusa, T.; Ito, K.; Bebber, D.P.; Fricker, M.D.; Yumiki, K.; Kobayashi, R.; Nakagaki, T. Rules for biologically inspired adaptive network design. Science 2010, 327, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, T.; Tero, A.; Nakagaki, T.; Kuramoto, Y. Amoebae anticipate periodic events. Phys. Rev. Lett. 2008, 100, 018101. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.R.; MacDonald, H.; Mann, R.P.; Marshall, J.A.; Latty, T.; Garnier, S. Decision-making without a brain: How an amoeboid organism solves the two-armed bandit. J. R. Soc. Interface 2016, 13, 20160030. [Google Scholar] [CrossRef] [PubMed]
- Nakagaki, T.; Kobayashi, R.; Nishiura, Y.; Ueda, T. Obtaining multiple separate food sources: Behavioural intelligence in the Physarum plasmodium. Proc. Biol. Sci. 2004, 271, 2305–2310. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, A.R.; Ashraf, B.; Jolly, M.K. Biophysical and biochemical attributes of hybrid epithelial/mesenchymal phenotypes. Phys. Biol. 2022, 19, 025001. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, A.R.; Kundnani, D.; Biswas, K.; Ghosh, A.; Hanash, S.M.; Tripathi, S.C.; Jolly, M.K. NFATc Acts as a Non-Canonical Phenotypic Stability Factor for a Hybrid Epithelial/Mesenchymal Phenotype. Front. Oncol. 2020, 10, 553342. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, A.R.; Sahoo, S.; Biswas, K.; Jolly, M.K. A Computational Systems Biology Approach Identifies SLUG as a Mediator of Partial Epithelial-Mesenchymal Transition (EMT). Cells Tissues Organs 2022, 211, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, A.R.; Sahoo, S.; McMullen, I.; Saxena, A.N.; Venugopal, S.K.; Somarelli, J.A.; Jolly, M.K. KLF4 Induces Mesenchymal-Epithelial Transition (MET) by Suppressing Multiple EMT-Inducing Transcription Factors. Cancers 2021, 13, 5135. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, A.R.; Sahoo, S.; Manjunatha, P.; Goyal, S.; Kasiviswanathan, V.A.; Mahesh, Y.; Ramu, S.; McMullen, I.; Somarelli, J.A.; Jolly, M.K. The ELF3 transcription factor is associated with an epithelial phenotype and represses epithelial-mesenchymal transition. J. Biol. Eng. 2023, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P. The failure of protein cancer biomarkers to reach the clinic: Why, and what can be done to address the problem? BMC Med. 2012, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.H.; Fiala, C.A.; Diamandis, E.P.; Kulasingam, V. Pitfalls in Cancer Biomarker Discovery and Validation with Emphasis on Circulating Tumor DNA. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2568–2574. [Google Scholar] [CrossRef]
- Cabús, L.; Lagarde, J.; Curado, J.; Lizano, E.; Pérez-Boza, J. Current challenges and best practices for cell-free long RNA biomarker discovery. Biomark. Res. 2022, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, K.; Dhar, R.; Pethusamy, K.; Srivastava, T.; Shankar, A.; Rath, G.K.; Karmakar, S. The issues and challenges with cancer biomarkers. J. Cancer Res. Ther. 2023, 19, S20–S35. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.scientificamerican.com/article/the-paradox-of-precision-medicine/ (accessed on 1 April 2024).
- Frayling, T.M. Genome-wide association studies: The good, the bad and the ugly. Clin. Med. 2014, 14, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Cyll, K.; Ersvær, E.; Vlatkovic, L.; Pradhan, M.; Kildal, W.; Avranden Kjær, M.; Kleppe, A.; Hveem, T.S.; Carlsen, B.; Gill, S.; et al. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br. J. Cancer 2017, 117, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Gure, A.O.; Chua, R.; Williamson, B.; Gonen, M.; Ferrera, C.A.; Gnjatic, S.; Ritter, G.; Simpson, A.J.; Chen, Y.T.; Old, L.J.; et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8055–8062. [Google Scholar] [CrossRef] [PubMed]
- Bolli, M.; Kocher, T.; Adamina, M.; Guller, U.; Dalquen, P.; Haas, P.; Mirlacher, M.; Gambazzi, F.; Harder, F.; Heberer, M.; et al. Tissue microarray evaluation of Melanoma antigen E (MAGE) tumor-associated antigen expression: Potential indications for specific immunotherapy and prognostic relevance in squamous cell lung carcinoma. Ann. Surg. 2002, 236, 785–793, discussion 793. [Google Scholar] [PubMed]
- Wei, R.; Dean, D.C.; Thanindratarn, P.; Hornicek, F.J.; Guo, W.; Duan, Z. Cancer testis antigens in sarcoma: Expression, function and immunotherapeutic application. Cancer Lett. 2020, 479, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Jäger, E.; Karbach, J.; Gnjatic, S.; Neumann, A.; Bender, A.; Valmori, D.; Ayyoub, M.; Ritter, E.; Ritter, G.; Jäger, D.; et al. Recombinant vaccinia/fowlpox NY-ESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc. Natl. Acad. Sci. USA 2006, 103, 14453–14458. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Robbins, P.F.; Raffeld, M.; Aung, P.P.; Tsokos, M.; Rosenberg, S.A.; Miettinen, M.M.; Lee, C.C. NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: Significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod. Pathol. 2012, 25, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Caballero, O.L.; Chen, Y.T. Cancer/testis (CT) antigens: Potential targets for immunotherapy. Cancer Sci. 2009, 100, 2014–2021. [Google Scholar] [CrossRef]
- Hofmann, O.; Caballero, O.L.; Stevenson, B.J.; Chen, Y.T.; Cohen, T.; Chua, R.; Maher, C.A.; Panji, S.; Schaefer, U.; Kruger, A.; et al. Genome-wide analysis of cancer/testis gene expression. Proc. Natl. Acad. Sci. USA 2008, 105, 20422–20427. [Google Scholar] [CrossRef] [PubMed]
- Sang, W.; Zhou, Y.; Chen, H.; Yu, C.; Dai, L.; Liu, Z.; Chen, L.; Fang, Y.; Ma, P.; Wu, X.; et al. Receptor-interacting Protein Kinase 2 Is an Immunotherapy Target in Pancreatic Cancer. Cancer Discov. 2024, 14, 326–347. [Google Scholar] [CrossRef]
- Bell, C.C.; Gilan, O. Principles and mechanisms of non-genetic resistance in cancer. Br. J. Cancer 2020, 122, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Vander Velde, R.; Shaffer, S.; Marusyk, A. Integrating mutational and nonmutational mechanisms of acquired therapy resistance within the Darwinian paradigm. Trends Cancer 2022, 8, 456–466. [Google Scholar] [CrossRef]
- Mohanty, A.; Nam, A.; Srivastava, S.; Jones, J.; Lomenick, B.; Singhal, S.S.; Guo, L.; Cho, H.; Li, A.; Behal, A.; et al. Acquired resistance to KRAS G12C small-molecule inhibitors via genetic/nongenetic mechanisms in lung cancer. Sci. Adv. 2023, 9, eade3816. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Martelotto, L.; Baslan, T.; Vides, A.; Solomon, M.; Mai, T.T.; Chaudhary, N.; Riely, G.J.; Li, B.T.; Scott, K.; et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat. Med. 2017, 23, 929–937. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudabadi, G.; Rajagopalan, K.; Getzenberg, R.H.; Hannenhalli, S.; Rangarajan, G.; Kulkarni, P. Intrinsically disordered proteins and conformational noise: Implications in cancer. Cell Cycle 2013, 12, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Achuthan, S.; Bhattacharya, S.; Jolly, M.K.; Kotnala, S.; Leite, V.B.P.; Mohanty, A.; Orban, J.; Roy, S.; Rangarajan, G.; et al. Protein conformational dynamics and phenotypic switching. Biophys. Rev. 2021, 13, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Salgia, R.; Rangarajan, G. Intrinsically disordered proteins and conformational noise: The hypothesis a decade later. iScience 2023, 26, 107109. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, A.K.; Jarosz, D.F. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J. Mol. Biol. 2018, 430, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Kutateladze, T.G. Characterization of functional disordered regions within chromatin-associated proteins. iScience 2021, 24, 102070. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Agarwal, P.; Kumar, A. Disordered regions tune order in chromatin organization and function. Biophys. Chem. 2022, 281, 106716. [Google Scholar] [CrossRef] [PubMed]
- Deiana, A.; Forcelloni, S.; Porrello, A.; Giansanti, A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS ONE 2019, 14, e0217889. [Google Scholar] [CrossRef] [PubMed]
- Eva, J.; Lamb, M. Soft inheritance: Challenging the Modern Synthesis. Genet. Mol. Biol. 2008, 31, 389–395. [Google Scholar] [CrossRef]
- Karras, P.; Black, J.R.M.; McGranahan, N.; Marine, J.C. Decoding the interplay between genetic and non-genetic drivers of metastasis. Nature 2024, 629, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Philipp, L.M.; Giaimo, S.; Sebens, S.; Traulsen, A.; Raatz, M. Understanding and leveraging phenotypic plasticity during metastasis formation. NPJ Syst. Biol. Appl. 2023, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Bhat, G.R.; Sethi, I.; Sadida, H.Q.; Rah, B.; Mir, R.; Algehainy, N.; Albalawi, I.A.; Masoodi, T.; Subbaraj, G.K.; Jamal, F.; et al. Cancer cell plasticity: From cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance. Cancer Metastasis Rev. 2024, 43, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, E.B.; De, S.; Leder, K.; Foo, J. Understanding the role of phenotypic switching in cancer drug resistance. J. Theor. Biol. 2020, 490, 110162. [Google Scholar] [CrossRef]
- Cassidy, T.; Nichol, D.; Robertson-Tessi, M.; Craig, M.; Anderson, A.R.A. The role of memory in non-genetic inheritance and its impact on cancer treatment resistance. PLoS Comput. Biol. 2021, 17, e1009348. [Google Scholar] [CrossRef] [PubMed]
- Capp, J.P.; Thomas, F. From developmental to atavistic bet-hedging: How cancer cells pervert the exploitation of random single-cell phenotypic fluctuations. BioEssays 2022, 44, e2200048. [Google Scholar] [CrossRef] [PubMed]
- Brutovský, B. Scales of Cancer Evolution: Selfish Genome or Cooperating Cells? Cancers 2022, 14, 3253. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Gomez, K.; Rabadan, R. A persistent look at how tumours evade therapy. Nature 2021, 596, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Ebi, H. Drug-Tolerant Persister Cells After EGFR Tyrosine Kinase Inhibitor Treatment: Their Origin and the Influences from the Tumor Microenvironment. J. Thorac. Oncol. 2023, 18, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Dhanyamraju, P.K.; Schell, T.D.; Amin, S.; Robertson, G.P. Drug-Tolerant Persister Cells in Cancer Therapy Resistance. Cancer Res. 2022, 82, 2503–2514. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.W.; Liu, B.; Chen, J.C.; Cao, Z.; Chu, F.R.; Lin, X.; Wang, S.Z.; Wu, J.C. Characteristics and molecular mechanism of drug-tolerant cells in cancer: A review. Front. Oncol. 2023, 13, 1177466. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Dedhar, S. Persister cell plasticity in tumour drug resistance. Semin. Cell Dev. Biol. 2024, 156, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Househam, J.; Heide, T.; Cresswell, G.D.; Spiteri, I.; Kimberley, C.; Zapata, L.; Lynn, C.; James, C.; Mossner, M.; Fernandez-Mateos, J.; et al. Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature 2022, 611, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Chahoud, J.; Anderson, A.R.A.; Zhang, J.; Brown, J.; Gatenby, R.A. Evolutionary Dynamics and Intermittent Therapy for Metastatic Cancers. J. Clin. Oncol. 2023, 41, 4469–4471. [Google Scholar] [CrossRef] [PubMed]
- Pearl Mizrahi, S.; Gefen, O.; Simon, I.; Balaban, N.Q. Persistence to anti-cancer treatments in the stationary to proliferating transition. Cell Cycle 2016, 15, 3442–3453. [Google Scholar] [CrossRef]
- Piggot, P. Epigenetic switching: Bacteria hedge bets about staying or moving. Curr. Biol. 2010, 20, R480–R482. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Kessler, D.A.; Ben-Jacob, E.; Levine, H. Growth feedback as a basis for persister bistability. Proc. Natl. Acad. Sci. USA 2014, 111, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Fasani, R.A.; Savageau, M.A. Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. USA 2013, 110, E2528–E2537. [Google Scholar] [CrossRef]
- Rotem, E.; Loinger, A.; Ronin, I.; Levin-Reisman, I.; Gabay, C.; Shoresh, N.; Biham, O.; Balaban, N.Q. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. USA 2010, 107, 12541–12546. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, J.J.; et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019, 17, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ko, M.E.; Cheng, H.; Zhu, R.; Xue, M.; Wang, J.; Lee, J.W.; Frankiw, L.; Xu, A.; Wong, S.; et al. Multi-omic single-cell snapshots reveal multiple independent trajectories to drug tolerance in a melanoma cell line. Nat. Commun. 2020, 11, 2345. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.J.; Sun, L.; Khan, S.; Yuan, S.; Nongyue, H. Versatility of Cancer Associated Fibroblasts: Commendable Targets for Anti-tumor Therapy. Curr. Drug Targets 2018, 19, 1573–1588. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [PubMed]
- Stanková, K.; Brown, J.S.; Dalton, W.S.; Gatenby, R.A. Optimizing Cancer Treatment Using Game Theory: A Review. JAMA Oncol. 2019, 5, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S. Why Darwin would have loved evolutionary game theory. Proc. Biol. Sci. 2016, 283, 20160847. [Google Scholar] [CrossRef] [PubMed]
- Mirnezami, R.; Nicholson, J.; Darzi, A. Preparing for precision medicine. N. Engl. J. Med. 2012, 366, 489–491. [Google Scholar] [CrossRef]
- Cunningham, J.J.; Brown, J.S.; Vincent, T.L.; Gatenby, R.A. Divergent and convergent evolution in metastases suggest treatment strategies based on specific metastatic sites. Evol. Med. Public Health 2015, 2015, 76–87. [Google Scholar] [CrossRef]
- West, J.; Adler, F.; Gallaher, J.; Strobl, M.; Brady-Nicholls, R.; Brown, J.; Roberson-Tessi, M.; Kim, E.; Noble, R.; Viossat, Y.; et al. A survey of open questions in adaptive therapy: Bridging mathematics and clinical translation. eLife 2023, 12, e84263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef] [PubMed]
- McGehee, C.; Mori, Y. A Mathematical Framework for Comparison of Intermittent versus Continuous Adaptive Chemotherapy Dosing in Cancer. bioRxiv 2024. [Google Scholar] [CrossRef]
- Shlyakhtina, Y.; Moran, K.L.; Portal, M.M. Genetic and Non-Genetic Mechanisms Underlying Cancer Evolution. Cancers 2021, 13, 1380. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; de Goeje, P.L.; Peeper, D.S.; van Amerongen, R. Phenotype switching: Tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res. 2014, 74, 5937–5941. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.D.; Pang, K.; Wu, Z.X.; Dong, Y.; Hao, L.; Qin, J.X.; Wang, W.; Chen, Z.S.; Han, C.H. Tumor cell plasticity in targeted therapy-induced resistance: Mechanisms and new strategies. Signal Transduct. Target. Ther. 2023, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M.; Ferraro, D.A.; Christofori, G. Heterogeneity for IGF-II production maintained by public goods dynamics in neuroendocrine pancreatic cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 1833–1838. [Google Scholar] [CrossRef]
- Capp, J.P.; Thomas, F.; Marusyk, A.; MDujon, A.; Tissot, S.; Gatenby, R.; Roche, B.; Ujvari, B.; DeGregori, J.; Brown, J.S.; et al. The paradox of cooperation among selfish cancer cells. Evol. Appl. 2023, 16, 1239–1256. [Google Scholar] [CrossRef] [PubMed]
- Strobl, M.A.R.; Gallaher, J.; Robertson-Tessi, M.; West, J.; Anderson, A.R.A. Treatment of evolving cancers will require dynamic decision support. Ann. Oncol. 2023, 34, 867–884. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.S.; Cisneros, L.H.; Anderson, A.R.A.; Maley, C.C. In Silico Investigations of Multi-Drug Adaptive Therapy Protocols. Cancers 2022, 14, 2699. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Navas, P.M.; Wojtkowiak, J.W.; Gatenby, R.A. Application of Evolutionary Principles to Cancer Therapy. Cancer Res. 2015, 75, 4675–4680. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T. Targeting multiple kinase pathways: A change in paradigm. Clin. Cancer Res. 2010, 16, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Hardin, G. The tragedy of the commons. The population problem has no technical solution; it requires a fundamental extension in morality. Science 1968, 162, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Delbrück, M. A physicist looks at biology. Resonance 1949, 4, 89–102. [Google Scholar]
- Fitzgerald, D.M.; Hastings, P.J.; Rosenberg, S.M. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Koh, B.; Tan, D.J.H.; Ng, C.H.; Fu, C.E.; Lim, W.H.; Zeng, R.W.; Yong, J.N.; Koh, J.H.; Syn, N.; Meng, W.; et al. Patterns in Cancer Incidence Among People Younger Than 50 Years in the US, 2010 to 2019. JAMA Netw. Open 2023, 6, e2328171. [Google Scholar] [CrossRef]
- Ledford, H. Why are so many young people getting cancer? What the data say. Nature 2024, 627, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Jianhui, Z.; Liying, X.; Jing, S.; Mingyang, S.; Lijuan, W.; Shuai, Y.; Yingshuang, Z.; Zhengwei, W.; Susanna, L.; Konstantinos, T.; et al. Global trends in incidence, death, burden and risk factors of early-onset cancer from 1990 to 2019. BMJ Oncol. 2023, 2, e000049. [Google Scholar] [CrossRef]
- Nam, A.; Mohanty, A.; Bhattacharya, S.; Kotnala, S.; Achuthan, S.; Hari, K.; Srivastava, S.; Guo, L.; Nathan, A.; Chatterjee, R.; et al. Dynamic Phenotypic Switching and Group Behavior Help Non-Small Cell Lung Cancer Cells Evade Chemotherapy. Biomolecules 2022, 12, 8. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramisetty, S.; Subbalakshmi, A.R.; Pareek, S.; Mirzapoiazova, T.; Do, D.; Prabhakar, D.; Pisick, E.; Shrestha, S.; Achuthan, S.; Bhattacharya, S.; et al. Leveraging Cancer Phenotypic Plasticity for Novel Treatment Strategies. J. Clin. Med. 2024, 13, 3337. https://doi.org/10.3390/jcm13113337
Ramisetty S, Subbalakshmi AR, Pareek S, Mirzapoiazova T, Do D, Prabhakar D, Pisick E, Shrestha S, Achuthan S, Bhattacharya S, et al. Leveraging Cancer Phenotypic Plasticity for Novel Treatment Strategies. Journal of Clinical Medicine. 2024; 13(11):3337. https://doi.org/10.3390/jcm13113337
Chicago/Turabian StyleRamisetty, Sravani, Ayalur Raghu Subbalakshmi, Siddhika Pareek, Tamara Mirzapoiazova, Dana Do, Dhivya Prabhakar, Evan Pisick, Sagun Shrestha, Srisairam Achuthan, Supriyo Bhattacharya, and et al. 2024. "Leveraging Cancer Phenotypic Plasticity for Novel Treatment Strategies" Journal of Clinical Medicine 13, no. 11: 3337. https://doi.org/10.3390/jcm13113337