Abstract
Background: Leiomyosarcoma (LMS) originating from the adrenal gland is exceedingly rare, constituting a minute fraction of soft tissue sarcomas. Due to its rarity, with less than 50 documented cases in English medical literature, the diagnosis and management of adrenal LMS remain challenging. The aim of this study was to perform a review of the literature, in order to evaluate the prognosis of these rare cancers and report our specific case. Methods: A systematic review of the literature was conducted using PubMed, Web of Science, Google Scholar, and Scopus databases, up to December 2020. The search utilized MeSH terms such as “Adrenal Gland Neoplasms,” “Leiomyosarcoma,” “Adrenalectomy,” and “Smooth Muscle Tumor.” The inclusion criteria focused on studies reporting patients with a histopathological diagnosis of adrenal leiomyosarcoma. The PRISMA guidelines were followed to ensure a comprehensive analysis. Results: Out of 63 identified studies, 43 met the inclusion criteria and were reviewed. These studies highlighted the rarity and aggressive behavior of adrenal leiomyosarcoma. Surgical excision remains the cornerstone of treatment, often complemented by adjuvant therapies. The reviewed case involved a 52-year-old woman who underwent a right laparoscopic adrenalectomy for a 9 × 7 × 6 cm grade 3 leiomyosarcoma. Despite subsequent adjuvant chemotherapy, hepatic metastases were detected, illustrating the aggressive nature of the disease. The literature underscores the importance of histopathological analysis and long-term surveillance for managing disease progression. Conclusions: Optimal management of adrenal leiomyosarcoma requires a multidisciplinary approach and meticulous follow-up. The rarity of the disease poses challenges for standardizing treatment, but surgical excision and tailored adjuvant therapies show promise. Further research is essential to refine treatment strategies and improve prognosis for this rare malignancy.
1. Introduction
Leiomyosarcoma (LMS) is a mesenchymal tumor that originates from smooth muscle cells [1], representing approximately 25% of all soft tissue sarcomas (STS), a heterogeneous group of neoplasms of mesenchymal origin. Among these, adrenal leiomyosarcoma is exceedingly rare, posing significant challenges in diagnosis and management due to its infrequent occurrence and aggressive nature.
Tumors are located more frequently in the retroperitoneum or intra-abdominal region (35% of cases), while those originating in the uterus account for 30% of leiomyosarcomas [2]. A recent review of case reports in the English medical literature revealed fewer than 50 instances documented [3]. Here, we present a case report focusing on a rare instance of adrenal leiomyosarcoma, emphasizing surgical management with laparoscopic adrenalectomy (LA). In fact, this study aims to bridge this knowledge gap by reviewing the existing literature and integrating findings. The case is noteworthy not only for its rarity but also for the diagnostic complexity and the therapeutic approaches it necessitated, including the onset of hepatic metastases post-surgery.
In addition to the primary adrenal lesion, our case report highlights the occurrence of hepatic metastases, indicating the aggressive nature of adrenal leiomyosarcoma and its propensity for systemic spread. This dissemination underscores the necessity for comprehensive treatment approaches, including surgical intervention and adjuvant chemotherapy, to address both the primary tumor and its metastatic deposits. Furthermore, the presence of hepatic metastases further emphasizes the importance of vigilant surveillance and multidisciplinary management to optimize patient outcomes in the face of this challenging disease. Through this comprehensive examination, the manuscript endeavors to enhance the oncological community’s understanding of adrenal LMS, promote early diagnosis, and refine therapeutic strategies to improve prognosis for this rare disease.
2. Case Report
A 52-year-old patient was referred to the General Surgery Division of the University of Campania Luigi Vanvitelli (Naples, Italy) in September 2022 due to the presence of a solid neoformation in the right adrenal gland. The patient presented, in July of the same year, with symptoms including tachycardia, hypertensive crisis, dyspnea, oppressive chest pain, general discomfort, and cramping pain in the abdomen or right flank region. Initial investigations did not reveal significant pathologies. Preoperative laboratory tests assessing adrenal function revealed no significant abnormalities. Plasma and urinary levels of catecholamines (adrenaline, noradrenaline, dopamine) and metanephrines were within normal ranges. Both plasma and urinary cortisol levels were normal, as were aldosterone and renin levels. Adrenal steroids, including dehydroepiandrosterone sulfate (DHEAS), were also likely within normal limits. Abdominal ultrasound showed a solid formation measuring 70 mm adjacent to the inferior vena cava in the right adrenal lodge and retroperitoneal site, with normal kidneys bilaterally.
Subsequent CT imaging with intravenous contrast revealed a 55 × 50 mm solid formation with a necrotic core and irregular peripheral enhancement in the right adrenal lodge. The mass demonstrated adjacency to but no infiltration of the inferior vena cava, with venous drainage observed in the right ovarian vein. The left adrenal gland appeared normal, with no significant lymphadenopathy [Figure 1].
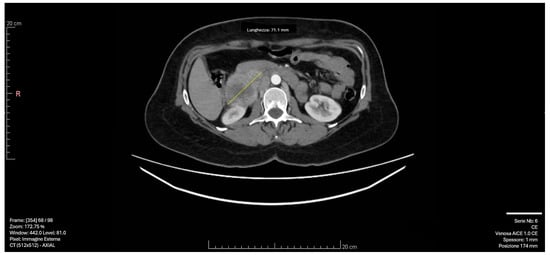
Figure 1.
CT scan image: The right adrenal mass demonstrated adjacency to but no infiltration of the inferior vena cava, with venous drainage observed in the right ovarian vein. The yellow arrow indicates the 55 × 50 mm solid formation with a necrotic core and irregular peripheral enhancement in the right adrenal lodge.
Following multidisciplinary evaluation, a right adrenalectomy was recommended. The laparoscopic procedure was performed under general anesthesia, with pneumoperitoneum initiated using Hasson’s trocar in the paraumbilical area. The highly vascular adrenal mass was densely adhered to the inferior vena cava, exerting external compression on the ipsilateral ureter and kidney, resulting in kidney hypotrophy and complete dislocation [Figure 2].
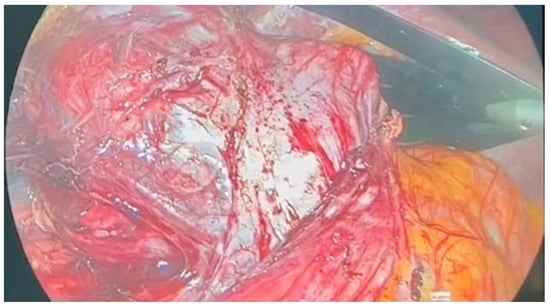
Figure 2.
Intraoperative adrenal mass.
With meticulous dissection and the use of a radiofrequency instrument, the mass was successfully freed from adhesions [Figure 3]. Afferent vessels were ligated and sectioned, and the mass was completely excised using an endobag.
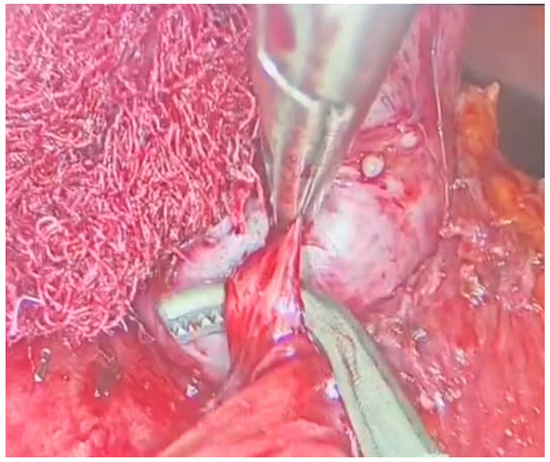
Figure 3.
Laparoscopic adrenalectomy, dissection from the residual adhesions.
Careful hemostasis was achieved, and Hemopatch® and Floseal® were placed. Grossly, the specimen was constituted by a solid nodular mass of 9 × 7 × 6 cm and weighing 148 g, with a grayish cut surface.
Histological examination showed a solid neoplastic proliferation characterized by an expansive growth and constituted by spindle cells arranged in a fascicular pattern (Figure 4A). Some extensive areas of coagulative necrosis (Figure 4B) were present, as well as areas of hyalinization of the stroma (Figure 4C). The neoplastic population was constituted by spindle cells with hyperchromic and irregular nuclei, and frequent mitotic figures (Figure 4D). Immunohistochemistry showed positivity for smooth muscle actin (Figure 4E), desmin (Figure 4F) and calponin, and negativity for cytokeratin, S100, EMA.
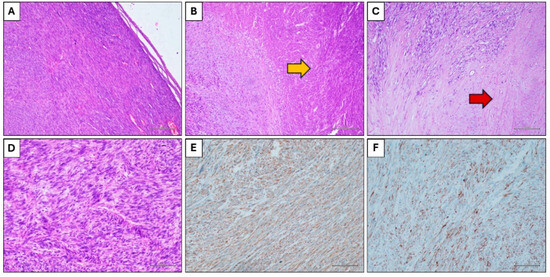
Figure 4.
Histological findings. (A) Spindle cells neoplasms with expansive growth (Hematoxylin and eosin stain, original magnification 100×). (B) Coagulative necrosis (yellow arrow) (Hematoxylin and eosin stain, original magnification 100×). (C) Hyalinization of the stroma (red arrow) (Hematoxylin and eosin stain, original magnification 100×). (D) Neoplastic cells with nuclear atypia and mitotic figures (Hematoxylin and eosin stain, original magnification 200×). (E) Immunohistochemical positivity for smooth muscle actin (Immunohistochemical stain, original magnification 100×). (F) Immunohistochemical positivity for desmin (Immunohistochemical stain, original magnification 100×).
A final diagnosis of grade 3 leiomyosarcoma was rendered.
The patient resumed oral feeding the day after surgery and was discharged after 4 days with a complete resolution of symptoms. After careful multidisciplinary evaluation with various dedicated specialists, a decision was made for close and careful follow-up, pending further imaging tests such as CT or PET-CT at 4 months to continue the therapeutic process.
During the follow-up examination at 4 months post-surgery, a PET-CT scan without contrast conducted at another facility revealed a prominent focal hypermetabolic lesion in the hepatic segment V measuring approximately 51 × 44 mm with a SUV of 14.3, indicative of a secondary lesion. Additionally, osteolytic lesions were observed along the lateral aspect of the third rib on the right hemithorax (SUV max 3.5) and the inferior angle of the left scapula (SUV max 3.2), along with multiple bilateral pulmonary nodules (SUV max 1.2) [Figure 5, Figure 6 and Figure 7]. Hence, the aggressive metastatic dissemination of leiomyosarcoma underscores the imperative for a prompt reassessment of the treatment approach and the contemplation of intensified therapeutic interventions.
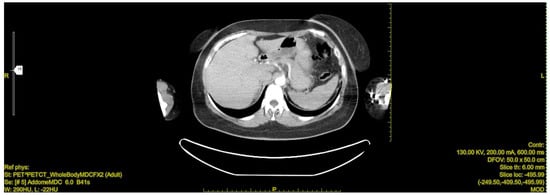
Figure 5.
Postoperative Abdominal CT: Non postoperative complications at the surgical site.
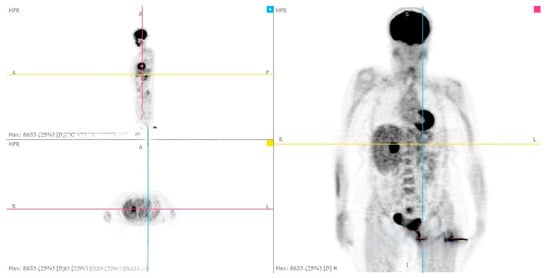
Figure 6.
PET-CT whole body: hepatic metastasis with a SUV of 14.3.
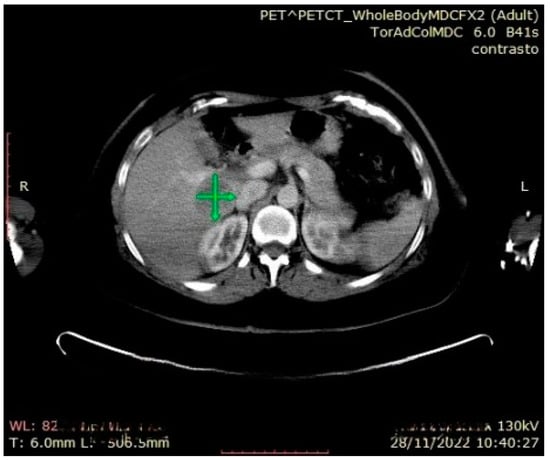
Figure 7.
Abdominal CT: hepatic metastasis identified with green arrows.
In accordance with the described hepatic metastasis presentation, the patient underwent chemotherapy treatment consisting of 9 cycles of Epirubicin + Dacarbazine followed by an additional 3 cycles with Dacarbazine alone.
The follow-up proceeded with the monitoring of symptoms and objective findings for 12 months post-surgical treatment, actively monitoring the secondary manifestations identified in the PET-CT with biochemical and instrumental evaluation, and in constant and diligent multidisciplinary collaboration.
3. Methods
Using the PubMed, Web of Science, Google Scholar and Scopus databases, a systematic review of the current literature was carried out, up to January 2000. The MeSH (Medical Subject Headings) search terms used were: “Adrenal Gland Neoplasms”, “Leyomiosarcoma”, “Adrenalectomy”, “Smooth Muscle Tumor”.
The authors observed that adrenal LMS was an extremely rare neoplasm. The keywords “Adrenal Gland”, “Leiomyosarcomas”, “Mesenchymal Tumors” were used for the research. Several combinations of the keywords and MeSH terms were utilized as showed: “Adrenal Leiomyosarcomas”, “Mesenchymal tumors”. The various terms were substituted during the search. References of the more relevant articles were manually searched. The last research was concluded in December 2020. The search was carried out by two authors, FMM and MP, and the obtained results were discussed with the senior author GC. The final article was realized in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA Statement) guidelines, and was not registered in any systematic review registry [Figure 8] [4]. The following data were extracted from the included studies: first author, year of publication, publishing journal, characteristics of study population, potential combination of surgical and chemoradiotherapeutic treatment, and follow-up (in months).
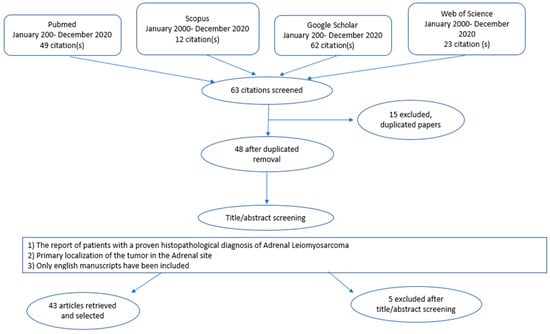
Figure 8.
PRISMA Statement: Flow-Chart. Diagram of the systematic review of the iterate performed in 4 databases from January 2000 up to December 2020. Search terms included: adrenal tumor, Leiomyosarcomas, mesenchymal tumor”. Inclusion criteria are shown in the central box. Major reasons for exclusion were duplicated papers from the different databases (n = 15), the language of the manuscripts included (n = 4). Further reasons for exclusion were primary location of the tumor in the Adrenal site (n = 1). This led to the final selection of 43 studies which fulfilled the inclusion criteria.
The inclusion criteria of the study comprised the reports of patients with a proven histopathological diagnosis of adrenal leiomyosarcoma, with primary localization of the tumor in the adrenal site. All studies that failed to fulfil the established inclusion criteria, and the non English language studies, were excluded. In all the studies, adrenal LMS diagnosis was based on the definitive pathology.
The clinical characteristics included age, sex, localization of the neoplasm, size (in centimeters).
4. Results
Sixty-three suitable studies were identified after the literature review. After the removal of a duplicate study, forty-eight articles were selected for the full-text review. Four studies were excluded because there were in Spanish [5,6,7], and in Korean [8]. Another one was ruled out because the primary tumor was not primary localized in the adrenal gland, but as a secondary metastasis, and also because it was in Japanese [9]. Therefore, forty-three met our inclusion criteria and were enrolled in the current review. The features of the forty-three selected studies were summarized in Table 1.

Table 1.
Literature review of the 44 existing cases (and our case report) of adrenal leiomyosarcoma, including subsequent surgical treatment and follow-up strategies.
4.1. Demographic and Clinicopathological Features
The data revealed that adrenal neoplasms showed no strong preference for any specific age group or sex, with cases reported in individuals ranging from young adolescents to the elderly, and an equal distribution between males and females. This diversity emphasized the importance of including adrenal gland tumors in differential diagnoses for all age groups and both genders.
From the selected studies, forty-five patients with a histological diagnosis of adrenal leiomyosarcoma were identified (24 females and 21 males). The mean age was 51.18 ± 15.26 years, with a median age of 60 ± 15.23 years, ranging from 14 to 78 years. The mean age in female cases was approximately 56.88 with a standard deviation of 14.60, and the median was 60.5 with a standard deviation of 14.60, ranging from 14 to 78 years. In male cases, the mean age was approximately 51.62 with a standard deviation of 14.76, while the median was 50 with a standard deviation of 14.76, ranging from 29 to 75 years. In 21 cases, the tumor was located on the left with one case of multiple metastases, two cases of adjacent organ invasion, one case of renal vein extension, one of inferior vena cava extension, and another with extension of the inferior vena cava and both iliac veins. For three left adrenal leiomyosarcomas, the extent was not specified, and in 12 cases, there was no extension to other organs. Twenty-two adrenal leiomyosarcomas were located on the right, with four cases of inferior vena cava invasion, two cases of inferior vena cava and right atrium invasion, three cases of adjacent organ invasion, one case of pulmonary metastasis and aorta invasion, one case of multiple metastases, and in 3 studies, extension was not specified. In seven cases, there was no extension to other organs. In two cases, the tumor was bilateral without extension to other organs. The mean size of the specimen was 9.35 ± 5.45 cm, but 4 studies did not report the size.
Regarding treatment options, one case underwent adrenalectomy associated with partial nephrectomy without adjuvant chemo-radiotherapy and survived 12 months without recurrence or metastasis [10]. In six cases, adrenalectomy associated with nephrectomy was performed [10,11,17,23,25,26]. Specifically, three cases had no adjuvant therapy [17,25,26] and a mean survival of 10.33 ± 1.79 months without recurrence or metastasis; one case received adjuvant chemoradiotherapy [11], and another one received only adjuvant radiotherapy [23], both alive with metastasis at 9 months follow-up. Also in our study, laparoscopic adrenalectomy and nephrectomy were performed, followed by adjuvant chemotherapy, and the patient was alive with liver metastasis at 12 months follow-up.
In 12 cases, adrenalectomy was performed with thrombectomy [15,19,20,24,48], cavotomy IVC [34,42,48], radiofrequency ablation [28], partial diaphragmatic resection [31], liver partial resection and lymphadenectomy [42,43], distal pancreatectomy with splenectomy [51], hepatic lobectomy and cholecystectomy [16] due to extension to other organs. In 9 of these cases, there was no adjuvant therapy, with 6 patients dying from metastasis at 1 and 12 months follow-up. Two patients were alive without recurrence at 10 months, one patient had a recurrence at 3 months, and in one study, there was no information about follow-up. In 4 cases, extended surgery was followed by adjuvant therapy, with one patient dying shortly after, one patient dying with metastasis at 16 months, one patient alive with metastasis at 6 months, and one patient without follow-up data.
In 20 cases, only adrenalectomy was performed, with one case being bilateral [18]. Our case report and the other four from the literature were laparoscopic [1,40,41,44], within one case converted to open surgery [44]. In 15 cases, adrenalectomy was not followed by adjuvant therapy; furthermore, in three of these cases follow-up data were not available, while the other twelve patients were alive without recurrence or metastasis at a mean follow-up time of 16.91 ± 7.75 months. In one study, exploratory laparotomy was performed without adjuvant therapy, resulting in death after 3 weeks [14]. In one case, palliative chemotherapy and radiotherapy were administered without follow-up information [47]. In one cases, patients underwent chemotherapy, resulting in death at 3 months and survival with metastasis at 9 months, respectively [29,36]. In one case, the patient was treated with radiotherapy and died 11 days later with metastasis [27].
4.2. Definitive Pathology Examination and Immunohistochemistry
The pathology examination and immunohistochemistry findings of various studies on spindle cell tumors exhibited several commonalities and differences [Table 2].
Table 2.
Definitive pathology examination and immunohistochemistry.
Macroscopically, many of these tumors demonstrated central areas of cystic degeneration and necrosis. This central necrosis was a common feature observed in some studies [10,11,15,24], among others. Hemorrhage was also frequently associated with these necrotic areas, as seen in the studies by Lack et al. and Lujan et al. [11,16], indicating vascular involvement and rapid growth of the tumors. However, there were differences in the descriptions of the masses, with some studies reporting multinodular appearances, fibrous substrates, and varying weights, such as the 2400 g mass reported by Candanedo-Gonzalez et al. [21] and the 180 g mass described by Mohanty et al. [23]. In 14 studies, macroscopic characteristics were not described [5,12,13,17,20,22,27,28,29,30,41,43,45,47].
Morphologically, the most consistent feature across these studies was the presence of spindle-shaped cells arranged in fascicles, as consistently reported by Choi et al., Lack et al., Matsui et al., Wang et al., Aoki et al. and Lee et al. [10,11,15,22,24,45], suggesting a mesenchymal origin or differentiation of these tumors. These cells arranged in interlacing fascicles, a pattern observed in studies by Matsui et al., Shao et al., and Zhou et al. [15,30,40]. Atypia and pleomorphism were also common features, indicating the malignant potential and variability in tumor cell morphology. For instance, Lack et al. and Mohanty et al. [11,23] described spindle cells with blunt-ended nuclei and eosinophilic cytoplasm; otherwise, Kato K et al. [19] reported pleomorphic neoplastic cells.
Furthermore, eight reports, including those by Candanedo-Gonzalez et al., Mohanty et al., and Deshmukh et al., described pleomorphic neoplastic cells [16,21,23,32,34,42,46]. High cellularity and nuclear atypia were notable features in studies by Lee et al., Manzano et al., and Mulani et al. [19,47,52]. Candanedo-Gonzalez et al. and Wang et al. described osteoclast-like giant cells [21,24]. Eight studies did not mention the microscopic cellular characteristics [1,12,13,17,20,27,43]. Eight studies did not complete the microscopic cellular information at all, not mentioning mitotic activity either [18,22,28,29,30,39,45,48].
Mitotic activity was another area with significant findings. High mitotic rates were regularly reported, reflecting the aggressive nature of these tumors. Specific figures vary, with Lack et al. reporting 15 mitoses per 10 high power fields (HPFs) and Mohanty et al. noting 12–14 mitoses per HPF, while Lee et al. observed up to 25 mitotic figures per 10 HPFs [11,22,23]. Other studies, such as Deshmukh et al. and Li et al., noted mitotic rates ranging from 10–12/10 HPFs to 18/10 HPFs [32,43]. The Ki-67 proliferation index, which indicates the percentage of tumor cells undergoing mitosis, also varies widely. Lujan et al. reported a Ki-67 index of about 80%, while Gulpinar et al. noted a lower index of 4%, indicating the variability in the proliferative capacities of these tumors [16,33].
Immunohistochemical analysis revealed a consistent profile for adrenal spindle cell neoplasms, which allows for the molecular diagnosis of adrenal leiomyosarcoma. Smooth muscle actin (SMA) was universally positive across studies, including those by Lack et al., Goto et al., and Bhalla et al. [11,25,36]. This positivity was evident in studies by Lack et al., Etten et al., Linos et al., and Mohanty et al., indicating the smooth muscle differentiation of the spindle cell tumors [11,14,16,18]. Desmin positivity was observed in our case and also in 24 reports [1,19,21,22,23,24,26,29,30,32,34,35,36,37,38,39,40,41,43,46,49,50,51,53], while it was negative in three cases [18,31,44]. Vimentin, a marker for mesenchymal origin, was frequently positive, as seen in the studies by Lack et al., Kato et al., Sakellariou et al., Tzaida et al. and Deshmukh et al. [11,19,32,46,50]. On the other hand, markers such as S-100, CD34, and cytokeratins were often negative, helping to exclude other tumor types like neural, endothelial, and epithelial neoplasms. This negativity was consistently reported in studies by Kato et al., Mohanty et al., and Wei et al. [19,23,37]. Seven studies did not explicitly mention immunohistochemistry results [5,10,13,16,17,20,27].
5. Discussion
Adrenal leiomyosarcoma is a rare malignant tumor originating from smooth muscle cells within the adrenal gland [54]. This malignancy is characterized by its complex molecular underpinnings and the signaling pathways implicated in its development. Despite its rarity, the diagnosis and management of this aggressive tumor present a quite challenging [17,20]. The comprehensive analysis of leiomyosarcoma incidence and treatment in adrenal glands necessitate a meticulous examination of individual data points in relation to the aggregate findings, ensuring robust conclusions. Our study’s bibliographic data, meticulously compiled from various sources, serves as a crucial foundation for understanding the landscape of this rare malignancy. The literature on adrenal gland neoplasms from the analyzed cases presents a complex picture of the challenges involved in treating this rare but significant cancer type [1,5,10,11,12,13,14,15,16,18,19,20,21,22,23,24,25,26,27,28,29,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,55]. The variability in tumor characteristics, the need for sometimes extensive surgical intervention, and the potential for adjunct treatments such as chemotherapy and radiation therapy all contribute to the diverse outcomes observed in these patients. This review highlights the heterogeneity in the presentation and management of adrenal gland neoplasms. The variability in tumor characteristics and the consequent choice of treatment significantly impact patient outcomes.
5.1. Demographic Features
Analyzing the intersection of variables such as age, sex, tumor size, side, and extension, it is clear that each factor plays a crucial role in determining the approach to surgical and adjuvant chemoradiation therapies, as well as influencing follow-up outcomes in patients treated for adrenal-related conditions.
Younger patients might be candidates for more aggressive treatments, including extensive surgeries and adjuvant therapies, given their longer expected lifespan and potentially better ability to recover from intense treatments. Conversely, in older patients, clinicians might opt for more conservative treatments, balancing the benefits of aggressive therapy against potential risks and the overall health status of the patient. The age of the patient also impacts follow-up strategies, with more frequent monitoring possibly required for those who undergo aggressive treatments at a younger age to detect recurrences early. Sex may not directly influence the choice of treatment modalities but plays an indirect role through physiological and metabolic differences between males and females that can affect how treatments are tolerated and their effectiveness. These differences might also necessitate tailored follow-up regimens to address sex-specific risks or complications post-treatment. Larger tumors often require more radical surgical approaches, possibly combined with chemotherapy or radiotherapy, to manage the greater risk of local spread or metastasis. In contrast, smaller tumors might be managed with less invasive procedures, relying on the slow-growing nature of some adrenal tumors. The initial size of the tumor heavily influences follow-up outcomes; larger tumors may have a higher risk of recurrence, requiring longer and more intensive monitoring to manage potential complications effectively.
Tumor side (left, right, or bilateral) can affect surgical planning, especially considering the anatomical challenges and potential complications associated with operating on either or both sides of the body. Bilateral tumors may necessitate a more complex and cautious approach, impacting both the immediate and long-term management plans.
The extension of the tumor beyond the adrenal gland into surrounding structures or distant sites significantly impacts the choice of treatment. Tumors with extensive spread are likely treated with combination therapies—surgery to remove as much of the tumor mass as possible, followed by radiation or chemotherapy to address residual disease. The extent of disease at diagnosis is a strong predictor of follow-up needs, as patients with a more extensive disease at the outset may require more rigorous and prolonged surveillance to detect recurrence or manage ongoing issues.
In summary, the interaction of these factors—age, sex, tumor size, side, and extension—shapes the therapeutic approach and follow-up care in managing adrenal-related conditions. Understanding these relationships helps clinicians tailor treatments to individual patient profiles, aiming for the best possible outcomes while minimizing risks and side effects. Aggressive management, including extensive surgery and adjunctive therapies, appears crucial for managing advanced cases.
Case reports provide valuable insights into its clinical presentation, diagnostic challenges, and therapeutic outcomes. Zhou et al. and Wei et al. contributed to the literature with a comprehensive literature review, emphasizing the importance of integrating clinical, radiological, and histopathological findings for accurate diagnosis and optimal treatment planning [5,25,29,31,37,40,43,46,47,56]. Zetler et al. reported a case in an AIDS patient, suggesting a potential link between immunocompromised states and leiomyosarcoma [12]. The rarity of adrenal leiomyosarcoma, particularly in pediatric populations, is exemplified by cases such as the laparoscopic excision of a bilateral primary adrenal leiomyosarcoma in a 14-year-old girl with AIDS, reported by Linos et al. [18]. In fact, ALMS cases have been reported in association with immunocompromised states such as AIDS, suggesting a possible link to Epstein–Barr virus infection, as indicated by Zetler and Boman et al. [12,13]. This underscores the importance of considering the underlying conditions in disease management and follow-up planning. Furthermore, Nagaraj et al. expanded demographic understanding, with a rare case in an Arab male, while Nerli et al. highlighted challenges in young adults [39,49].
However, the optimal treatment strategy should be tailored to individual patient characteristics, considering the invasiveness of the tumor and the patient’s overall health condition.
5.2. Histological Findings
Regarding the histological findings and immunohistochemistry, spindle cell tumors commonly show a spindle-shaped cell morphology, high mitotic activity, and positive immunoreactivity for smooth muscle markers. There are variations in the specific details of cell morphology, mitotic rates, and the expression of additional immunohistochemical markers, reflecting the heterogeneity within this group of tumors. These findings highlight both the shared characteristics and the unique aspects of each case, providing a comprehensive understanding of spindle cell tumors’ pathological and immunohistochemical profiles.
5.3. Molecular Bases and Target Therapy
Its molecular basis involves intricate genetic and epigenetic changes, frequently including alterations in the RB1, TP53, and PTEN genes, which are fundamental to the regulation of the cell cycle, apoptosis, and cellular proliferation [57]. Epigenetic modifications also play a central role in PAL’s pathogenesis, which can lead to the activation of oncogenes and the suppression of tumor suppressor genes, thereby promoting tumor growth and progression [58]. The involvement of microRNAs (miRNAs) in PAL is another significant aspect. The dysregulation of specific miRNAs has been linked to various malignancies, including leiomyosarcomas: In PAL, particular miRNAs may act either as oncogenes or tumor suppressors, though the exact miRNAs involved are still under investigation [59].
Additionally, the tumor microenvironment, which includes interactions with surrounding stromal cells and the extracellular matrix, plays a critical role in influencing the behavior of PAL. These interactions can facilitate tumor growth, angiogenesis, and metastasis through various signaling pathways, such as those mediated by TGF-beta, VEGF, and PDGF [60].
To develop targeted therapies effectively, a deep understanding of these molecular bases is essential. Although challenging, advancements in molecular biology and genetics offer hope for more effective treatment approaches. Ongoing research into the molecular mechanisms governing PAL is crucial for the development of personalized medical strategies and improving patient outcomes with this rare malignancy [61]. Targeted therapies are designed to exploit specific genetic and molecular characteristics of the tumor cells, offering a more precise approach than conventional treatments.
Tyrosine kinase inhibitors (TKIs) have shown promise in treating soft tissue sarcomas due to their ability to interfere with key signaling pathways that promote tumor growth and survival. Drugs such as pazopanib, a multi-targeted TKI, have been approved for advanced soft tissue sarcoma after positive outcomes in clinical trials [62]. These inhibitors target vascular endothelial growth factor receptors (VEGFRs), which are often implicated in the angiogenesis associated with tumor growth.
The mammalian target of the rapamycin (mTOR) pathway is another critical pathway in the progression of various cancers, including LMS. mTOR inhibitors, such as sirolimus and everolimus, have been studied for their effectiveness in slowing down tumor growth by inhibiting cell proliferation and inducing apoptosis [63]. Although results have been mixed, these agents offer a potential treatment avenue, particularly in tumors resistant to conventional chemotherapy.
Monoclonal antibodies targeting growth factor receptors and other tumor-associated antigens are under investigation for LMS. Trabectedin, a drug initially derived from a marine organism, has shown efficacy in LMS by binding to the minor groove of DNA and disrupting the transcription of oncogenes [64].
Recent advances have also focused on targeting the specific genetic mutations and epigenetic alterations found in LMS cells. Agents that modify epigenetic marks, such as HDAC inhibitors, are being explored to re-activate tumor suppressor genes and inhibit oncogenes. These therapies are still largely in the experimental stages, but represent a significant step toward personalized medicine [65,66].
Although in its infancy for LMS, immunotherapy approaches, including checkpoint inhibitors, are being evaluated in clinical trials. These treatments aim to boost the body’s immune response against tumor cells. Early studies suggest a variable response, likely dependent on the immunogenicity of the tumor [67,68].
While the development of targeted therapies for leiomyosarcoma, including PAL, is challenging due to the tumor’s rarity and molecular complexity, ongoing research into these therapies holds significant promise. These targeted treatments aim to improve survival rates and quality of life by tailoring interventions to the specific molecular profiles of leiomyosarcoma tumors.
5.4. Surgical Management
The surgical management of primary adrenal leiomyosarcoma through adrenalectomy is a cornerstone in the treatment of this rare malignancy. It involves various approaches, including laparoscopic adrenalectomy, which has been successfully employed in selected cases. Quildrian et al. reported a case of primary adrenal leiomyosarcoma treated with laparoscopic adrenalectomy, highlighting the feasibility and efficacy of minimally invasive techniques in managing this rare malignancy [41]. Various surgical approaches have been described in the literature, including radical excision, adrenalectomy, and even vena cava resection in cases of tumor invasion, as illustrated by Ozturk et al. [34]. Wang et al. proposed a novel approach to the surgical resection of leiomyosarcoma involving the adrenal vein, emphasizing the importance of meticulous surgical planning and technique [24]. The surgical management of adrenal leiomyosarcoma poses significant challenges due to the tumor’s rare occurrence and aggressive behavior. Among the pivotal decisions in surgical oncology is the choice between open and laparoscopic adrenalectomy; each approach has distinct advantages and considerations that influence treatment outcomes, especially in complex cases like adrenal leiomyosarcoma. The open approach is particularly advantageous in cases where tumors are suspected to invade adjacent organs or the inferior vena cava, as it allows for a more radical resection and the ability to manage intraoperative complications directly [53,69]. Conversely, laparoscopic techniques also provide an enhanced visualization of the surgical field, which can be beneficial in the meticulous dissection required to preserve adrenal function in non-invasive tumors [52,70]. In the last few decades, minimally invasive transperitoneal laparoscopic adrenalectomy has become the standard for the surgical resection of adrenal gland tumors [71].
However, minimally invasive retroperitoneal adrenalectomy has gained popularity as an alternative technique, offering shorter hospital stays, reduced postoperative pain, fewer complications, and improved cosmetic outcomes [22,72,73]. However, the laparoscopic approach requires significant expertise and is limited by the size and invasiveness of the tumor. It is generally preferred for tumors smaller than 6 cm that do not show signs of local invasion. The precision of laparoscopic instruments facilitates careful manipulation and dissection around the adrenal glands, reducing the risk of damaging major vessels [55].
An accurate preoperative examination is essential for selecting eligible patients for laparoscopic adrenalectomy, but also an operative team composed of experienced and skilled surgeons is necessary [30,72,74]. Choi and Liu described the angiographic features of adrenal leiomyosarcoma, providing valuable insights into the vascular characteristics of these tumors that can aid in preoperative planning and intraoperative decision-making [10]. The low risk associated with adrenalectomy, particularly via a laparoscopic approach, provides a definitive diagnosis and treatment without significant risk or cost [75,76].
Studies comparing these approaches show varied results; however, the trend suggests that laparoscopic adrenalectomy can be safely performed with comparable outcomes to open surgery in terms of oncological efficacy when conducted by experienced surgeons. A systematic review highlights that laparoscopic resection for small, localized adrenal leiomyosarcomas achieves outcomes similar to the open approach, with the added benefits of a minimally invasive procedure [77]. Ongoing advances in surgical technology, such as the integration of robotic systems, may further refine the laparoscopic approach, expanding its applicability to more complex adrenal surgeries. Further research and accumulation of case-specific outcomes are necessary to develop clearer guidelines that assist in choosing the most appropriate surgical approach for adrenal leiomyosarcoma [78]. Advanced energy devices, when used responsibly, can enhance surgical outcomes, ensuring cost savings and patient satisfaction [79,80]. The choice of hemostatic device is based on surgeon preference; in our experience, we used a hemostatic flap [74,81]. In conclusion, while laparoscopic adrenalectomy offers significant advantages in selected cases, the choice of surgical approach must be individualized based on a thorough assessment of the tumor’s characteristics and the patient’s overall condition. This tailored approach ensures optimal surgical outcomes and adherence to oncological principles.
5.5. Adjuvant Therapy and Follow-Up
Furthermore, the role of adjuvant therapies, such as chemotherapy and radiotherapy, remain controversial due to the lack of standardized treatment protocols and limited evidence from clinical trials [26,36]. However, in the cases of unresectable or metastatic diseases, systemic chemotherapy may be considered to palliate symptoms and prolong survival, as suggested by Bhalla et al. [36]. Aoki et al. reported a case of primary adrenal leiomyosarcoma in an elderly woman, where the tumor was managed surgically without adjuvant therapy; this was similarly described by Lokanatha et al., where surgery alone was employed for a primary adrenal leiomyosarcoma [45,51].
Lack et al. conducted an immunohistochemical and ultrastructural study of a primary leiomyosarcoma of the adrenal gland, elucidating the histopathological features and immunophenotypic profile of these tumors, which are crucial for definitive diagnosis and prognostication [11,16]. Various histological subtypes of adrenal leiomyosarcoma have been described, including pleomorphic leiomyosarcoma and leiomyosarcoma with osteoclast-like giant cells, underscoring the histopathological heterogeneity of this malignancy [21,23,44]. Histopathological insights from Deshmukh and Gulpinar et al. aid accurate diagnosis, guiding treatment decisions [32,33]. Lee et al. showcased histological diversity, underlining the need for a nuanced pathological evaluation [35].
Regular surveillance is imperative for monitoring disease recurrence, metastasis, and treatment response following surgical resection [28]. Imaging modalities such as 18F-FDG-PET, as highlighted by Van Laarhoven et al., play a crucial role in detecting the early signs of disease progression and guiding subsequent management decisions [27].
A long-term follow-up is essential in the management of adrenal leiomyosarcoma to monitor for recurrence and metastasis [38]. In cases of adrenal leiomyosarcoma with extension into adjacent structures, such as the right atrium and inferior vena cava, highlighting the importance of vigilant surveillance and timely intervention in cases of advanced disease [14,15,19,48]. Such instances highlight the need for vigilance in surveillance imaging and clinical assessment to detect metastatic spread promptly. Moreover, the heterogeneity of ALMS behavior is exemplified by Nerli et al., where despite aggressive treatment, metastasis and adverse outcomes can occur [49]. Conversely, other cases, such as those reported by Nagaraj et al., demonstrate favorable long-term outcomes with appropriate management [39].
In cases of metastatic disease or lymph node involvement, the management becomes even more complex. Onishi et al. reported a case of primary adrenal leiomyosarcoma with lymph node metastasis, highlighting the importance of thorough staging and consideration of systemic therapies [42,82].
The overmentioned chemotherapeutic regimen with Epirubicin + Dacarbazine was selected to address the aggressive metastatic behavior of the leiomyosarcoma, targeting both the hepatic and extrahepatic sites of the involvement identified by imaging. The decision to administer sequential chemotherapy cycles reflects the ongoing effort to optimize disease control and improve the patient’s overall prognosis in the face of advanced disease progression [83].
As evidenced by the case reports gleaned from the scientific literature regarding adrenal leiomyosarcoma, there is an overall average of 10 months of follow-up among all cases reported in Table 1. Specifically, out of 44 cases, survival without metastasis was observed in 17 cases [5,10,12,15,17,24,25,26,30,32,33,35,37,40,41,44,46], survival with metastasis or recurrence in 10 cases [11,21,22,23,34,36,38,43,50] (including our case report), and death occurred in 10 cases, with one case in the perioperative period [16] and the remaining cases during postoperative follow-up [14,15,19,20,27,28,29,45,48] (ranging from 3 weeks to 16 months). Nine patients were lost to follow-up [1,13,18,31,39,47,49,51].
Adrenal leiomyosarcoma exhibits considerable mortality and invasiveness, posing significant challenges in clinical management and prognosis. Many studies have highlighted its aggressive nature and poor outcomes despite therapeutic interventions [84].
5.6. Strengths and Limits
This study utilizes a review of existing literature combined with a detailed case report, offering a unique dual perspective on the rare condition of adrenal leiomyosarcoma. The methodology employed rigorous data collection from established databases, ensuring a robust analysis of available data. We employed multidisciplinary evaluations in our case report, illustrating the complex nature of diagnosis and management in adrenal leiomyosarcoma.
However, the rarity of the disease limits the ability to perform a large-scale, randomized controlled trial and therefore may affect the generalizability of the results. The retrospective nature of the literature review may introduce selection bias, as cases documented in the literature might not be representative of all real-world scenarios.
6. Conclusions
These cases underscore the importance of multidisciplinary collaboration, meticulous surgical technique, and accurate diagnosis in optimizing outcomes for patients with primary adrenal leiomyosarcoma. A correct and precise preoperative diagnosis is very difficult and challenging in most cases. The extensive variability in tumor characteristics and patient outcomes revealed through our review indicates that the successful management of ALMS requires a nuanced approach that integrates advanced surgical techniques and, where appropriate, targeted adjuvant therapies. Despite the challenges posed by its rarity and aggressive nature, ongoing advancements in molecular understanding and surgical methodologies provide a beacon of hope for improving treatment strategies and patient survival rates.
Surgical resection remains the cornerstone of treatment, with the choice between laparoscopic and open adrenalectomy being tailored to the tumor’s specifics and patient condition. The potential of targeted therapies, such as tyrosine kinase inhibitors and mTOR pathway inhibitors, is promising, reflecting a shift towards more personalized treatment plans based on the genetic and molecular landscape of the tumor. Furthermore, regular and vigilant follow-up is crucial for monitoring disease recurrence and progression, which is pivotal in adjusting management strategies promptly.
In conclusion, while the challenges in managing adrenal leiomyosarcoma are formidable, a multidisciplinary approach that leverages the latest research and clinical innovations holds the key to optimizing outcomes. By continuing to refine surgical techniques and explore new therapeutic avenues, there is potential to significantly enhance both the quality of life and the survival rates for patients afflicted with this formidable cancer.
Author Contributions
All authors contributed significantly to the present research and reviewed the entire manuscript. G.C., M.P. and F.M.M. participated substantially in the conception, design and execution of the study and in the analysis and interpretation of the data, and also participated substantially in the drafting and editing of the manuscript. A.C. (Alessandra Conzo), A.C. (Antonio Catauro), L.F. and L.D. participated substantially in the statistical analysis. A.R. (Alessandro Romano), F.M.M., R.P. and R.E. prepared the tables. F.M.M., M.P., G.N., A.R. (Andrea Ronchi) and G.C. participated substantially in the analysis and interpretation of the data. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable; data retrospectively obtained from clinical records.
Informed Consent Statement
All patients gave written informed consent to publish. Informed consent was obtained from all individual participants included in the study.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Waack, A.; Jaggernauth, S.; Vattipally, V. Primary adrenal leiomyosarcoma. Radiol. Case Rep. 2022, 18, 741–744. [Google Scholar] [CrossRef]
- Kannan, S.; Chong, H.H.; Chew, B.; Ferguson, J.D.; Galloway, E.; McCulloch, T.; Rankin, K.S.; Alshford, R.U. Leiomyosarcoma in the extremities and trunk wall: Systematic review and meta-analysis of the oncological outcomes. World J. Surg. Oncol. 2022, 20, 124. [Google Scholar] [CrossRef]
- Sakellariou, M.; Dellaportas, D.; Peppa, M.; Schizas, D.; Pikoulis, E.; Nastos, K. Review of the literature on leiomyoma and leiomyosarcoma of the adrenal gland: A systematic analysis of case reports. In Vivo 2020, 34, 2233–2248. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef]
- Pereira-Beceiro, J.; Rodríguez-Alonso, A.; Janeiro-Pais, J.M.; Alvarez-Fernández, J.C.; Durana Tonder, C. Adrenal pleomorphic leiomyosarcoma, an uncommon malignant suprarenal tumour. Cir. Esp. 2015, 93, 256–258. [Google Scholar] [CrossRef]
- Rodríguez Gómez, I.; Rodríguez-Rivera García, J.; Alvarez Costelo, L.; Gómez Veiga, F.; Lancina Martín, A.; Chantada Abal, V.; González Martín, M. Leiomiosarcoma de cava inferior. Hallazgo incidental [Leiomyosarcoma of the inferior vena cava. Incidental finding]. Arch. Esp. Urol. 2007, 60, 1127–1131. [Google Scholar] [CrossRef]
- Dekou, A.; N’dah, J.K.; Kouame, B.; Kohou, L.; Abroulaye, F.; Gowe, E.; Kassanyou, S.; Manzan, K. Léiomyosarcome primitive de la surrénale chez une noire africaine: Aspects diagnostiques et thérapeutiques [Primary leiomyosarcoma of adrenal gland, in black African woman: Diagnosis and therapeutically aspects]. Prog Urol. 2013, 23, 421–424. [Google Scholar] [CrossRef]
- Yoon, H.R.; Park, D.H. 부신의 원발성 평활근육종의 영상 소견: 증례 보고 [Imaging Findings of Primary Adrenal Leiomyosarcoma: A Case Report]. Taehan Yongsang Uihakhoe Chi. 2020, 81, 459–464. [Google Scholar]
- Seki, M.; Imao, T.; Amano, T.; Takemae, K. [Adrenalectomy for metastatic adrenal tumor from uterine leiomyosarcoma: A case report]. Hinyokika Kiyo 2011, 57, 435–438. [Google Scholar] [PubMed]
- Choi, S.H.; Liu, K. Leiomyosarcoma of the adrenal gland and its angiographic features: A case report. J. Surg. Oncol. 1981, 16, 145–148. [Google Scholar] [CrossRef]
- Lack, E.E.; Graham, C.W.; Azumi, N.; Bitterman, P.; Rusnock, E.J.; O’Brien, W.; Lynch, J.H. Primary leiomyosarcoma of adrenal gland. Case report with immunohistochemical and ultrastructural study. Am. J. Surg. Pathol. 1991, 15, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Zetler, P.J.; Filipenko, J.D.; Bilbey, J.H.; Schmidt, N. Primary adrenal leiomyosarcoma in a man with acquired immunodeficiency syndrome (AIDS). Further evidence for an increase in smooth muscle tumors related to Epstein-Barr infection in AIDS. Arch. Pathol. Lab. Med. 1995, 119, 1164–1167. [Google Scholar]
- Boman, F.; Gultekin, H.; Dickman, P.S. Latent Epstein-Barr virus infection demonstrated in low-grade leiomyosarcomas of adults with acquired immunodeficiency syndrome, but not in adjacent Kaposi’s lesion or smooth muscle tumors in immunocompetent patients. Arch. Pathol. Lab. Med. 1997, 121, 834–838. [Google Scholar] [PubMed]
- Etten, B.; van Ijken, M.G.; Mooi, W.J.; Oudkerk, M.; van Geel, A.N. Primary leiomyosarcoma of the adrenal gland. Sarcoma. 2001, 5, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Fujikawa, K.; Oka, H.; Fukuzawa, S.; Takeuchi, H. Adrenal leiomyosarcoma extending into the right atrium. Int. J. Urol. 2002, 9, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Lujan, M.G.; Hoang, M.P. Pleomorphic leiomyosarcoma of the adrenal gland. Arch. Pathol. Lab. Med. 2003, 127, e32–e35. [Google Scholar] [CrossRef] [PubMed]
- Thamboo, T.P.; Liew, L.C.; Raju, G.C. Adrenal leiomyosarcoma: A case report and literature review. Pathology 2003, 35, 47–49. [Google Scholar] [PubMed]
- Linos, D.; Kiriakopoulos, A.C.; Tsakayannis, D.E.; Theodoridou, M.; Chrousos, G. Laparoscopic excision of bilateral primary adrenal leiomyosarcoma in a 14-year old girl with acquired immunodeficiency syndrome (AIDS). Surgery 2004, 136, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kato, T.; Sakamoto, S.; Kobayashi, T.; Ikeda, R.; Nakamura, T.; Akakura, K.; Hikage, T.; Inoue, T. Primary adrenal leiomyosarcoma with inferior vena cava thrombosis. Int. J. Clin. Oncol. 2004, 9, 189–192. [Google Scholar] [CrossRef]
- Wong, C.; Von Oppell, U.O.; Scott-Coombes, D. Cold feet from adrenal leiomyosarcoma. J. R. Soc. Med. 2005, 98, 418–420. [Google Scholar] [CrossRef]
- Candanedo-Gonzalez, F.A.; Vela Chavez, T.; Cerbulo-Vazquez, A. Pleomorphic leiomyosarcoma of the adrenal gland with osteoclast-like giant cells. Endocr. Pathol. 2005, 16, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Tsang, Y.M.; Liu, K.L. Primary adrenal leiomyosarcoma. Abdom. Imaging 2006, 31, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.K.; Balani, J.P.; Parwani, A.V. Pleomorphic leiomyosarcoma of the adrenal gland: Case report and review of the literature. Urology 2007, 70, 591.e5–591.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.S.; Ocal, I.T.; Salem, R.R.; Elefteriades, J.; Sosa, J.A. Leiomyosarcoma of the adrenal vein: A novel approach to surgical resection. World J. Surg. Oncol. 2007, 5, 109. [Google Scholar] [CrossRef] [PubMed]
- Goto, J.; Otsuka, F.; Kodera, R.; Miyoshi, T.; Kinomura, M.; Otani, H.; Mimura, Y.; Ogura, T.; Yanai, H.; Nasu, Y.; et al. A rare tumor in the adrenal region: Neuron-specific enolase (NSE)-producing leiomyosarcoma in an elderly hypertensive patient. Endocr. J. 2008, 55, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Mencoboni, M.; Bergaglio, M.; Truini, M.; Varaldo, M. Primary adrenal leiomyosarcoma: A case report and literature review. Clin. Med. Oncol. 2008, 2, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, H.W.; Vinken, M.; Mus, R.; Flucke, U.; Oyen, W.J.; Van der Graaf, W.T. The diagnostic hurdle of an elderly male with bone pain: How 18F-FDG-PET led to diagnosis of a leiomyosarcoma of the adrenal gland. Anticancer. Res. 2009, 29, 469–472. [Google Scholar] [PubMed]
- Hamada, S.; Ito, K.; Tobe, M.; Otsuki, H.; Hama, Y.; Kato, Y.; Sugiura, Y.; Kaji, T.; Asano, T.; Hayakawa, M. Bilateral adrenal leiomyosarcoma treated with multiple local therapies. Int. J. Clin. Oncol. 2009, 14, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Karaosmanoglu, A.; Gee, M.S. Sonographic findings of an adrenal leiomyosarcoma. J. Ultrasound Med. 2010, 29, 1369–1373. [Google Scholar] [CrossRef]
- Shao, I.H.; Lee, W.C.; Chen, T.D.; Chiang, Y.J. Leiomyosarcoma of the adrenal vein. Chang. Gung Med. J. 2012, 35, 428–431. [Google Scholar]
- Kanthan, R.; Senger, J.L.; Kanthan, S. Three uncommon adrenal incidentalomas: A 13-year surgical pathology review. World J. Surg. Oncol. 2012, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.D.; Babanagare, S.V.; Anand, M.; Pande, D.P.; Yavalkar, P. Primary adrenal leiomyosarcoma: A case report with immunohistochemical study and review of literature. J. Cancer Res. Ther. 2013, 9, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Gulpinar, M.T.; Yildirim, A.; Gucluer, B.; Atis, R.G.; Atis, R.G.; Canakci, C.; Gurbuz, C.; Caskurlu, T. Primary leiomyosarcoma of the adrenal gland: A case report with immunohistochemical study and literature review. Case Rep. Urol. 2014, 2014, 489630. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, H. Vena Cava invasion by Adrenal Leiomyosarcoma. Rare Tumors 2014, 6, 5275. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Tanawit, G.D.; Lopez, R.A.; Zamuco, J.T.; Cheng, B.G.; Siozon, M.V. Primary leiomyosarcoma of adrenal gland with tissue eosinophilic infiltration. Korean J. Pathol. 2014, 48, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, A.; Sandhu, F.; Sieber, S. Primary adrenal leiomyosarcoma: A case report and review of the literature. Conn. Med. 2014, 78, 403–407. [Google Scholar] [PubMed]
- Wei, J.; Sun, A.; Tao, J.; Wang, C.; Liu, F. Primary adrenal leiomyosarcoma: Case report and review of the literature. Int. J. Surg. Pathol. 2014, 22, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Manzano, A.J.; Nose, V.; Fernandez-Castro, G.L.; Ayala, A. Recurrent primary adrenal leiomyosarcoma: A complete literature review and presentation of a rare adrenal tumor. AACE Clin. Case Rep. 2015, 1, e8–e11. [Google Scholar] [CrossRef]
- Nagaraj, V.; Mustafa, M.; Amin, E.; Ali, W.; Naji Sarsam, S.; Darwish, A. Primary adrenal leiomyosarcoma in an arab male: A rare case report with immunohistochemistry study. Case Rep. Surg. 2015, 2015, 702541. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tang, Y.; Tang, J.; Deng, F.; Gong, G.; Dai, Y. Primary adrenal leiomyosarcoma: A case report and review of literature. Int. J. Clin. Exp. Pathol. 2015, 8, 4258–4263. [Google Scholar]
- Quildrian, S.; Califano, I.; Carrizo, F.; Daffinoti, A.; Calónico, N. Primary adrenal leiomyosarcoma treated by laparoscopic adrenalectomy. Endocrinol. Nutr. 2015, 62, 472–473. [Google Scholar] [CrossRef]
- Onishi, T.; Yanagihara, Y.; Kikugawa, T.; Miura, N.; Noda, T.; Kakuda, T.; Kitazawa, R.; Tanji, N. Primary adrenal leiomyosarcoma with lymph node metastasis: A case report. World J. Surg. Oncol. 2016, 14, 176. [Google Scholar] [CrossRef]
- Li, C.-C.; Liu, W.-Y.; Chang, C.-P.; Kuo, C.-Y.; Kuo, J.-H. Rare care report: Primary leiomyosarcoma of the adrenal gland and literature review. Urol. Sci. 2016, 27, s58. [Google Scholar] [CrossRef]
- Rudin, A.; Torres-Mora, J.; Natt, N.; Richards, M.L. Primary adrenal dedifferentiated leiomyosarcoma: A low grade leiomyosarcoma with a high-grade undifferentiated component: Case report. Clin. Surg. 2016, 1, 1193. [Google Scholar]
- Aoki, C.; Tanaka, S.; Kunihiro, S.; Sagara, M.; Aso, Y. Primary adrenal leiomyosarcoma in an aged japanese woman: A rare case report. J. Clin. Case Rep. 2017, 7, 935. [Google Scholar] [CrossRef]
- Tzaida, O.; Papazian, M.; Kokkinos, C.; Provatas, I.; Markouizou, A.; Novkovic, N.; Nomikos, I. Adrenal gland: An anatomical site of surprises: On occasion of a primary leiomyosarcoma. Ann. Clin. Exp. Metabol. 2017, 2, 1014. [Google Scholar]
- Mulani, S.R.; Stoner, P.; Schlachterman, A.; Ghayee, H.K.; Lu, L.; Gupte, A. First reported case of endoscopic ultrasound-guided core biopsy yielding diagnosis of primary adrenal leiomyosarcoma. Case Rep. Gastrointest. Med. 2018, 2018, 8196051. [Google Scholar] [CrossRef] [PubMed]
- Doppalapudi, S.K.; Shah, T.; Fitzhugh, V.A.; Bargman, V. Primary adrenal leiomyosarcoma with inferior vena cava extension in a 70-year-old man. BMJ Case Rep. 2019, 12, e227670. [Google Scholar] [CrossRef]
- Nerli, R.B.; Ghagane, S.; Dixit, N.S.; Hiremath, M.B.; Deole, S. Adrenal leiomyosarcoma in a young adult male. Int. Cancer Conf. J. 2019, 9, 14–17. [Google Scholar] [CrossRef]
- Sakellariou, M.; Dellaportas, D.; Grapsa, E.; Tzikanoulas, M.; Dellis, A.; Theodosopoulos, T.; Nastos, C. Primary adrenal leiomyosarcoma: A case report. Mol. Clin. Oncol. 2020, 12, 317–320. [Google Scholar] [CrossRef]
- Lokanatha, D.; Jacob, L.; Babu, M.; Lokesh, K.N.; Krishna Sai, R.; Rudresha, A.H.; Rajeev, L.K.; Saldanha, S.; Suma, M.N.; Usha, A. Primary adrenal leiomyosarcoma: An extremely rare mesenchymal tumor. Indian J. Med. Paediatr. Oncol. 2019, 40, 559–562. [Google Scholar] [CrossRef]
- Brunt, L.M. The positive impact of laparoscopic adrenalectomy on complications of adrenal surgery. Surg. Endosc. 2002, 16, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Usta, M.A.; Ulusahin, M.; Alhan, E.; Cinel, A.; Nuhoglu, I. Open adrenalectomy: A 20-year review of our experience in a developing country. Ann. Afr. Med. 2020, 19, 26–30. [Google Scholar] [PubMed]
- Tomasich, F.D.; Luz Mde, A.; Kato, M.; Targa, G.Z.; Dias, L.A.; Zucoloto, F.J.; Ogata, D.C. Leiomiossarcoma primário de adrenal [Primary adrenal leiomyosarcoma]. Arq. Bras. Endocrinol. Metabol. 2008, 52, 1510–1514. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hayes, G. Update on Adrenalectomy. Vet. Clin. North. Am. Small Anim. Pract. 2022, 52, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Samsel, R.; Cichocki, A.; Roszkowska-Purska, K.; Falkowski, S. Mięśniakomięsak gładkokomórkowy nadnercza—opis dwóch przypadków [Leiomyosarcoma of the adrenal gland—two cases report]. Pol. Merkur. Lekarski. 2018, 45, 189–191. [Google Scholar] [PubMed]
- Anoshkin, K.I.; Karandasheva, K.O.; Goryacheva, K.M.; Pyankov, D.V.; Koshkin, P.A.; Pavlova, T.V.; Bobin, A.N.; Shpot, E.V.; Chernov, Y.N.; Vinarov, A.Z.; et al. Multiple Chromoanasynthesis in a Rare Case of Sporadic Renal Leiomyosarcoma: A Case Report. Front. Oncol. 2020, 10, 1653. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef]
- Coley, H.M. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 2008, 34, 378–390. [Google Scholar] [CrossRef]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef] [PubMed]
- van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. EORTC Soft Tissue and Bone Sarcoma Group; PALETTE study group. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Pautier, P.; Italiano, A.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Firmin, N.; Boudou-Rouquette, P.; Bertucci, F.; Balleyguier, C.; Lebrun-Ly, V.; et al. Doxorubicin alone versus doxorubicin with trabectedin followed by trabectedin alone as first-line therapy for metastatic or unresectable leiomyosarcoma (LMS-04): A andomized, multicentre, open-label phase 3 trial. Lancet Oncol. 2022, 23, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Roulleaux Dugage, M.; Nassif, E.F.; Italiano, A.; Bahleda, R. Improving Immunotherapy Efficacy in Soft-Tissue Sarcomas: A Biomarker Driven and Histotype Tailored Review. Front. Immunol. 2021, 12, 775761. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hwedieh, D.O.; Sedlacek, A.L.; Hernandez, L.M.; Yamoah, A.A.; Iyer, S.G.; Weiss, K.R.; Binder, R.J. Immunosurveillance shapes the emergence of neo-epitope landscapes of sarcomas, revealing prime targets for immunotherapy. JCI Insight 2023, 8, e170324. [Google Scholar] [CrossRef] [PubMed]
- Taffurelli, G.; Ricci, C.; Casadei, R.; Selva, S.; Minni, F. Open adrenalectomy in the era of laparoscopic surgery: A review. Updates Surg. 2017, 69, 135–143. [Google Scholar] [CrossRef]
- Assalia, A.; Gagner, M. Laparoscopic adrenalectomy. Br. J. Surg. 2004, 91, 1259–1274. [Google Scholar] [CrossRef]
- Gaujoux, S.; Weinandt, M.; Bonnet, S.; Reslinger, V.; Bertherat, J.; Dousset, B. Surgical treatment of adrenal carcinoma. J. Visc. Surg. 2017, 154, 335–343. [Google Scholar] [CrossRef]
- Gan, L.; Meng, C.; Li, K.; Lei, P.; Li, J.; Wu, J.; Li, Y. Safety and effectiveness of minimally invasive adrenalectomy versus open adrenalectomy in patients with large adrenal tumors (≥5 cm): A meta-analysis and systematic review. Int. J. Surg. 2022, 104, 106779. [Google Scholar] [CrossRef] [PubMed]
- Yip, L.; Duh, Q.Y.; Wachtel, H.; Jimenez, C.; Sturgeon, C.; Lee, C.; Velázquez-Fernández, D.; Berber, E.; Hammer, G.D.; Bancos, I.; et al. American Association of Endocrine Surgeons Guidelines for Adrenalectomy: Executive Summary. JAMA Surg. 2022, 157, 870–877. [Google Scholar] [CrossRef]
- Agrusa, A.; Romano, G.; Navarra, G.; Conzo, G.; Pantuso, G.; Buono, G.D.; Citarrella, R.; Galia, M.; Monte, A.L.; Cucinella, G.; et al. Innovation in endocrine surgery: Robotic versus laparoscopic adrenalectomy. Meta-analysis and systematic literature review. Oncotarget 2017, 8, 102392–102400. [Google Scholar] [CrossRef]
- Danwang, C.; Agbor, V.N.; Bigna, J.J. Obesity and postoperative outcomes of the patients with laparoscopic adrenalectomy: A systematic review and meta-analysis. BMC Surg. 2020, 20, 194. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef]
- Heger, P.; Probst, P.; Hüttner, F.J.; Gooßen, K.; Proctor, T.; Müller-Stich, B.P.; Strobel, O.; Büchler, M.W.; Diener, M.K. Evaluation of Open and Minimally Invasive Adrenalectomy: A Systematic Review and Network Meta-analysis. World J. Surg. 2017, 41, 2746–2757. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Gao, Y.; Zhao, Z.; Zhao, G.; Liu, R.; Lau, W.Y. Robotic resection of benign nonadrenal retroperitoneal tumors: A consecutive case series. Int. J. Surg. 2018, 55, 188–192. [Google Scholar] [CrossRef]
- Patrone, R.; Gambardella, C.; Romano, R.M.; Gugliemo, C.; Offi, C.; Andretta, C.; Vitiello, A.; Tartaglia, E.; Flagiello, L.; Conzo, A. The impact of the ultrasonic, bipolar and integrated energy devices in the adrenal gland surgery: Literature review and our experience. BMC Surg. 2019, 18 (Suppl. S1), 123. [Google Scholar] [CrossRef]
- Conzo, G.; Patrone, R.; Flagiello, L.; Catauro, A.; Conzo, A.; Cacciatore, C.; Mongardini, F.M.; Cozzolino, G.; Esposito, R.; Pasquali, D.; et al. Impact of Current Technology in Laparoscopic Adrenalectomy: 20 Years of Experience in the Treatment of 254 Consecutive Clinical Cases. J. Clin. Med. 2023, 12, 4384. [Google Scholar] [CrossRef]
- Coppola Bottazzi, E.; Gambardella, C.; Mongardini, F.M.; Vanella, S.; Noviello, A.; Palma, T.; Murano, R.; De Chiara, G.; Conzo, G.; Docimo, L.; et al. Prognosis of Adrenal Oncocytic Neoplasms (AONs): Literature Review of 287 Cases and Presentation of the Oldest Patient. J. Clin. Med. 2023, 12, 6925. [Google Scholar] [CrossRef] [PubMed]
- McLeod, M.K. Complications following adrenal surgery. J. Natl. Med. Assoc. 1991, 83, 161–164. [Google Scholar] [PubMed]
- Grimer, R.; Judson, I.; Peake, D.; Seddon, B. Guidelines for the management of soft tissue sarcomas. Sarcoma 2010, 2010, 506182. [Google Scholar] [CrossRef] [PubMed]
- Duregon, E.; Volante, M.; Giorcelli, J.; Terzolo, M.; Lalli, E.; Papotti, M. Adrenal leiomyosarcoma: Clues for a correct diagnosis. Endocr. Pathol. 2018, 29, 253–257. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).