
Application of Ophthalmic Electrophysiology in Inflammatory Disorders of Retina and Optic Nerve


Abstract
:1. Introduction
2. Clinical Applications of Electrophysiology
2.1. Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
2.2. Acute Zonal Occult Outer Retinopathy (AZOOR)
2.3. Adamantiades–Behçet Disease
2.4. Autoimmune Retinopathy and Neuro-Retinopathy
2.5. Birdshot Chorioretinopathy
2.6. Multiple Evanescent White Dot Syndrome
2.7. Vogt–Koyanagi–Harada Disease
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Liu, H.; Milla-Navarro, S.; Villa, P.; Germain, F. Origin of Retinal Oscillatory Potentials in the Mouse, a Tool to Specifically Locate Retinal Damage. Int. J. Mol. Sci. 2023, 24, 3126. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Palmowski-Wolfe, A.M.; International Society for Clinical Electrophysiology of Vision. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 2012, 124, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.B.; Bach, M.; Kondo, M.; Li, S.; Walker, S.; Holopigian, K.; Viswanathan, S.; Robson, A.G. ISCEV standard for clinical multifocal electroretinography (mfERG) (2021 update). Doc. Ophthalmol. 2021, 142, 5–16. [Google Scholar] [CrossRef]
- Parisi, V.; Ziccardi, L.; Stifano, G.; Montrone, L.; Gallinaro, G.; Falsini, B. Impact of regional retinal responses on cortical visually evoked responses: Multifocal ERGs and VEPs in the retinitis pigmentosa model. Clin. Neurophysiol. 2010, 121, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Odom, J.V.; Bach, M.; Brigell, M.; Holder, G.E.; McCulloch, D.L.; Mizota, A.; Tormene, A.P.; International Society for Clinical Electrophysiology of Vision. ISCEV standard for clinical visual evoked potentials: (2016 update). Doc. Ophthalmol. 2016, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Constable, P.A.; Bach, M.; Frishman, L.J.; Jeffrey, B.G.; Robson, A.G.; International Society for Clinical Electrophysiology of Vision. ISCEV Standard for clinical electro-oculography (2017 update). Doc. Ophthalmol. 2017, 134, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.D. Acute posterior multifocal placoid pigment epitheliopathy. Arch. Ophthalmol. 1968, 80, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.V.; Mitchell, P. Acute posterior multifocal placoid pigment epitheliopathy: A long-term study. Aust. N. Z. J. Ophthalmol. 1997, 25, 277–281. [Google Scholar] [CrossRef]
- Vianna, R.; van Egmond, J.; Priem, H.; Kestelyn, P. Natural history and visual outcome in patients with APMPPE. Bull. Soc. Belge. Ophtalmol. 1993, 248, 73–76. [Google Scholar]
- de Laey, J.J. Placoid epitheliopathy and serpiginous choroidopathy. Bull. Soc. Belge. Ophtalmol. 1989, 230, 105–122. [Google Scholar] [PubMed]
- Xerri, O.; Salah, S.; Monnet, D.; Brezin, A.P. Untreated Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): A case series. BMC Ophthalmol. 2018, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Brydak-Godowska, J.; Turczynska, M.; Przybys, M.; Brydak, L.B.; Kecik, D. Ocular Complications in Influenza Virus Infection. Ocul. Immunol. Inflamm. 2018, 27, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Mangeon, M.; Zett, C.; Amaral, C.; Novais, E.; Muccioli, C.; Andrade, G.; Nascimento, H.; Belfort, R., Jr. Multimodal Evaluation of Patients with Acute Posterior Multifocal Placoid Pigment Epitheliopathy and Serpiginous Choroiditis. Ocul. Immunol. Inflamm. 2018, 26, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Goen, T.M.; Terry, J.E. Acute posterior multifocal placoid pigment epitheliopathy. J. Am. Optom. Assoc. 1987, 58, 112–117. [Google Scholar] [PubMed]
- Cozubas, R.; Ungureanu, E.; Instrate, S.L.; Alexandrescu, C.; Nanu, R.V.; Carstocea, L.; Voinea, L.M.; Ciuluvica, R. Similarities and differences between three different types of white dot syndrome and the therapeutic possibilities. Rom. J. Ophthalmol. 2018, 62, 183–187. [Google Scholar] [CrossRef]
- Thomson, S.P.; Roxburgh, S.T. Acute posterior multifocal placoid pigment epitheliopathy associated with adenovirus infection. Eye 2003, 17, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Azar, P., Jr.; Gohd, R.S.; Waltman, D.; Gitter, K.A. Acute posterior multifocal placoid pigment epitheliopathy associated with an adenovirus type 5 infection. Am. J. Ophthalmol. 1975, 80, 1003–1005. [Google Scholar] [CrossRef]
- Daniele, S.; Daniele, C.; Orcidi, F.; Tavano, A. Progression of choroidal atrophy in acute posterior multifocal placoid pigment epitheliopathy. Ophthalmologica 1998, 212, 66–72. [Google Scholar] [CrossRef]
- Yamamoto, S.; Hayashi, M.; Tsuruoka, M.; Yamamoto, T.; Tsukahara, I.; Takeuchi, S. S-cone electroretinograms in multiple evanescent white dot syndrome. Doc. Ophthalmol. 2003, 106, 117–120. [Google Scholar] [CrossRef]
- Aoyagi, R.; Hayashi, T.; Gekka, T.; Kozaki, K.; Tsuneoka, H. Multifocal electroretinographic evaluation of macular function in acute posterior multifocal placoid pigment epitheliopathy. Doc. Ophthalmol. 2013, 126, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Dutta Majumder, P. Current understanding of acute zonal occult outer retinopathy (AZOOR). Indian J. Ophthalmol. 2024, 72, 935–937. [Google Scholar] [CrossRef]
- Gass, J.D. Acute zonal occult outer retinopathy. Donders Lecture: The Netherlands Ophthalmological Society, Maastricht, Holland, June 19, 1992. J. Clin. Neuroophthalmol. 1993, 13, 79–97. [Google Scholar]
- Jampol, L.M.; Wiredu, A. MEWDS, MFC, PIC, AMN, AIBSE, and AZOOR: One disease or many? Retina 1995, 15, 373–378. [Google Scholar]
- Gass, J.D. The acute zonal outer retinopathies. Am. J. Ophthalmol. 2000, 130, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.D. Are acute zonal occult outer retinopathy and the white spot syndromes (AZOOR complex) specific autoimmune diseases? Am. J. Ophthalmol. 2003, 135, 380–381. [Google Scholar] [CrossRef]
- Francis, P.J.; Marinescu, A.; Fitzke, F.W.; Bird, A.C.; Holder, G.E. Acute zonal occult outer retinopathy: Towards a set of diagnostic criteria. Br. J. Ophthalmol. 2005, 89, 70–73. [Google Scholar] [CrossRef]
- Heckenlively, J.R.; Ferreyra, H.A. Autoimmune retinopathy: A review and summary. Semin. Immunopathol. 2008, 30, 127–134. [Google Scholar] [CrossRef] [PubMed]
- He, S.X.; Othman, M.; Hurley, M.; Heckenlively, J.R. Auto Antibodies against Retinal Pigment Epithelium in Patients with Atypical Retinopathies. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3771. [Google Scholar]
- Tagami, M.; Matsumiya, W.; Imai, H.; Kusuhara, S.; Honda, S.; Azumi, A. Autologous antibodies to outer retina in acute zonal occult outer retinopathy. Jpn. J. Ophthalmol. 2014, 58, 462–472. [Google Scholar] [CrossRef]
- Qian, C.X.; Wang, A.; DeMill, D.L.; Jayasundera, T.; Branham, K.; Abalem, M.F.; Khan, N.; Heckenlively, J.R. Prevalence of Antiretinal Antibodies in Acute Zonal Occult Outer Retinopathy: A Comprehensive Review of 25 Cases. Am. J. Ophthalmol. 2017, 176, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, K.A.; Schuerch, K.; Zhao, J.; Lee, W.; Cabral, T.; Yannuzzi, L.A.; Tsang, S.H.; Sparrow, J.R. Quantitative Autofluorescence Intensities in Acute Zonal Occult Outer Retinopathy vs Healthy Eyes. JAMA Ophthalmol. 2017, 135, 1330–1338. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, L.; Yan, W.; Wei, W.; Lai, T.Y.Y. Fundus Autofluorescence Imaging in the Assessment of Acute Zonal Occult Outer Retinopathy. Ophthalmologica 2017, 237, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Mrejen, S.; Khan, S.; Gallego-Pinazo, R.; Jampol, L.M.; Yannuzzi, L.A. Acute zonal occult outer retinopathy: A classification based on multimodal imaging. JAMA Ophthalmol. 2014, 132, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Imamura, Y.; Giovinazzo, V.J.; Spaide, R.F. Fundus autofluorescence and optical coherence tomographic findings in acute zonal occult outer retinopathy. Retina 2010, 30, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Ishiko, S.; Kagokawa, H.; Fukui, K.; Takahashi, A.; Yoshida, A. Morphological study of acute zonal occult outer retinopathy (AZOOR) by multiplanar optical coherence tomography. Acta Ophthalmol. 2009, 87, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.C.; Chen, N.; Tsai, R.K. Acute Zonal Occult Outer Retinopathy (AZOOR): A case report of vision improvement after intravitreal injection of Ozurdex. BMC Ophthalmol. 2017, 17, 236. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Horiguchi, M.; Tanikawa, A. Unilaterally extinguished electroretinograms in an eye with normal visual acuity and visual field. Doc. Ophthalmol. 2021, 142, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Cruz, R.D.; Adachi-Usami, E.; Kakisu, Y. Flash electroretinograms and pattern visually evoked cortical potentials in Behcet’s disease. Jpn. J. Ophthalmol. 1990, 34, 142–148. [Google Scholar]
- Stubiger, N.; Besch, D.; Deuter, C.M.; Zierhut, M.; Kotter, I. Multifocal ERG changes in patients with ocular Behcet’s disease during therapy with interferon alpha 2a. Adv. Exp. Med. Biol. 2003, 528, 529–532. [Google Scholar] [CrossRef]
- Magrys, A.; Anekonda, T.; Ren, G.; Adamus, G. The role of anti-alpha-enolase autoantibodies in pathogenicity of autoimmune-mediated retinopathy. J. Clin. Immunol. 2007, 27, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Weleber, R.G.; Watzke, R.C.; Shults, W.T.; Trzupek, K.M.; Heckenlively, J.R.; Egan, R.A.; Adamus, G. Clinical and electrophysiologic characterization of paraneoplastic and autoimmune retinopathies associated with antienolase antibodies. Am. J. Ophthalmol. 2005, 139, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Adamus, G. Cellular targets of anti-alpha-enolase autoantibodies of patients with autoimmune retinopathy. J. Autoimmun. 2004, 23, 161–167. [Google Scholar] [CrossRef]
- Adamus, G. Autoantibody-induced apoptosis as a possible mechanism of autoimmune retinopathy. Autoimmun. Rev. 2003, 2, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Adamus, G. Antirecoverin antibodies and autoimmune retinopathy. Arch. Ophthalmol. 2000, 118, 1577–1578. [Google Scholar] [CrossRef] [PubMed]
- Mizener, J.B.; Kimura, A.E.; Adamus, G.; Thirkill, C.E.; Goeken, J.A.; Kardon, R.H. Autoimmune retinopathy in the absence of cancer. Am. J. Ophthalmol. 1997, 123, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Heckenlively, J.R.; Fawzi, A.A.; Oversier, J.; Jordan, B.L.; Aptsiauri, N. Autoimmune retinopathy: Patients with antirecoverin immunoreactivity and panretinal degeneration. Arch. Ophthalmol. 2000, 118, 1525–1533. [Google Scholar] [CrossRef]
- Adamus, G.; Karren, L. Autoimmunity against carbonic anhydrase II affects retinal cell functions in autoimmune retinopathy. J. Autoimmun. 2009, 32, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Adamus, G.; Yang, S.; Weleber, R.G. Unique epitopes for carbonic anhydrase II autoantibodies related to autoimmune retinopathy and cancer-associated retinopathy. Exp. Eye Res. 2016, 147, 161–168. [Google Scholar] [CrossRef]
- Adamus, G.; Ren, G.; Weleber, R.G. Autoantibodies against retinal proteins in paraneoplastic and autoimmune retinopathy. BMC Ophthalmol. 2004, 4, 5. [Google Scholar] [CrossRef]
- Adamus, G.; Brown, L.; Schiffman, J.; Iannaccone, A. Diversity in autoimmunity against retinal, neuronal, and axonal antigens in acquired neuro-retinopathy. J. Ophthalmic Inflamm. Infect. 2011, 1, 111–121. [Google Scholar] [CrossRef]
- Radhakrishnan, S.S.; Forma, G.; Carboni, G.; Mutolo, M.G.; Adamus, G.; Iannaccone, A. Patterns of Visual Function Loss in Autoimmune Neuro-Retinopathy (AINR): Psychophysical and Electrophysiological Findings [ARVO abstract]. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3547. [Google Scholar]
- Iyer, S.S.; Forma, G.; Carboni, G.; Brown, L.; Birg, A.; Adamus, G.; Iannaccone, A. Presentation Of Autoimmune Neuro-Retinopathy (AINR) Patients With Anti-Carbonic Anhydrase II Auto-antibodies [ARVO Abstract]. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2924. [Google Scholar]
- Epstein, R.S.; Sollenberger, E.; Adamus, G.; Iannaccone, A. Clinical, Functional, and Imaging Characteristics of Cancer-Associated Retinopathy and Optic Neuropathy. In Proceedings of the American Academy of Ophthalmology Meeting, Chicago, IL, USA, 18–22 October 2014. [Google Scholar]
- Grange, L.; Dalal, M.; Nussenblatt, R.B.; Sen, H.N. Autoimmune retinopathy. Am. J. Ophthalmol. 2014, 157, 266–272.e1. [Google Scholar] [CrossRef]
- Comlekoglu, D.U.; Thompson, I.A.; Sen, H.N. Autoimmune retinopathy. Curr. Opin. Ophthalmol. 2013, 24, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, H.A.; Jayasundera, T.; Khan, N.W.; He, S.; Lu, Y.; Heckenlively, J.R. Management of autoimmune retinopathies with immunosuppression. Arch. Ophthalmol. 2009, 127, 390–397. [Google Scholar] [CrossRef]
- Iannaccone, A.; Birnbaum, F.A.; Champaigne, R.; Adamus, G. Basal Cell Carcinoma-Associated Retinopathy and Optic Neuropathy (BARN): A Novel Paraneoplastic Entity. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2780. [Google Scholar]
- Turaka, K.; Kietz, D.; Krishnamurti, L.; Mitchell, E.; Scanga, H.; Fu, V.L.; Sylvester, C. Carcinoma-associated retinopathy in a young teenager with immature teratoma of the ovary. J. AAPOS 2014, 18, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Suhler, E.B.; Chan, C.C.; Caruso, R.C.; Schrump, D.S.; Thirkill, C.; Smith, J.A.; Nussenblatt, R.B.; Buggage, R.R. Presumed teratoma-associated paraneoplastic retinopathy. Arch. Ophthalmol. 2003, 121, 133–137. [Google Scholar] [CrossRef]
- Ko, A.C.; Hernandez, J.; Brinton, J.P.; Faidley, E.A.; Mugge, S.A.; Mets, M.B.; Kardon, R.H.; Folk, J.C.; Mullins, R.F.; Stone, E.M. Anti-gamma-enolase autoimmune retinopathy manifesting in early childhood. Arch. Ophthalmol. 2010, 128, 1590–1595. [Google Scholar] [CrossRef]
- Polans, A.S.; Witkowska, D.; Haley, T.L.; Amundson, D.; Baizer, L.; Adamus, G. Recoverin, a photoreceptor-specific calcium-binding protein, is expressed by the tumor of a patient with cancer-associated retinopathy. Proc. Natl. Acad. Sci. USA 1995, 92, 9176–9180. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, F.; Moscato, S.; Sabbatini, A.; Chimenti, D.; Bombardieri, S.; Migliorini, P. Autoantibodies specific for alpha-enolase in systemic autoimmune disorders. J. Rheumatol. 2000, 27, 109–115. [Google Scholar]
- Andreasson, S.; Ponjavic, V.; Ehinger, B. Full-field electroretinogram in a patient with cutaneous melanoma-associated retinopathy. Acta Opthalmologica 1993, 71, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Milam, A.H.; Saari, J.C.; Jacobson, S.G.; Lubinski, W.P.; Feun, L.G.; Alexander, K.R. Autoantibodies against retinal bipolar cells in cutaneous melanoma-associated retinopathy. Investig. Ophthalmol. Vis. Sci. 1993, 34, 91–100. [Google Scholar]
- Lei, B.; Bush, R.A.; Milam, A.H.; Sieving, P.A. Human melanoma-associated retinopathy (MAR) antibodies alter the retinal ON-response of the monkey ERG in vivo. Investig. Ophthalmol. Vis. Sci. 2000, 41, 262–266. [Google Scholar]
- Eksandh, L.; Adamus, G.; Mosgrove, L.; Andreasson, S. Autoantibodies against bestrophin in a patient with vitelliform paraneoplastic retinopathy and a metastatic choroidal malignant melanoma. Arch. Ophthalmol. 2008, 126, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Carboni, G.; Forma, G.; Bond, A.D.; Adamus, G.; Iannaccone, A. Bilateral paraneoplastic optic neuropathy and unilateral retinal compromise in association with prostate cancer: A differential diagnostic challenge in a patient with unexplained visual loss. Doc. Ophthalmol. 2012, 125, 63–70. [Google Scholar] [CrossRef]
- Rothova, A.; Berendschot, T.T.; Probst, K.; van Kooij, B.; Baarsma, G.S. Birdshot chorioretinopathy: Long-term manifestations and visual prognosis. Ophthalmology 2004, 111, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Holak, H.M.; Szymaniec, S.; Holak, S.A. The pathogenesis of birdshot chorioretinopathy. Surv. Ophthalmol. 2006, 51, 446–447. [Google Scholar] [CrossRef]
- Gasch, A.T.; Smith, J.A.; Whitcup, S.M. Birdshot retinochoroidopathy. Br. J. Ophthalmol. 1999, 83, 241–249. [Google Scholar] [CrossRef]
- Leung, E.; Palestine, A.; O’Keefe, G.D.; Tripathy, K.; Karth, P.A.; Read, R.W.; Shah, V.A. Birdshot Retinochoroidopathy. In EyeWiki; Karth, P.A., O’Keefe, G.D., Eds.; American Academy of Ophthalmology: San Francisco, CA, USA, 2019. [Google Scholar]
- Holder, G.E.; Robson, A.G.; Pavesio, C.; Graham, E.M. Electrophysiological characterisation and monitoring in the management of birdshot chorioretinopathy. Br. J. Ophthalmol. 2005, 89, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Kiss, S.; Ahmed, M.; Letko, E.; Foster, C.S. Long-term follow-up of patients with birdshot retinochoroidopathy treated with corticosteroid-sparing systemic immunomodulatory therapy. Ophthalmology 2005, 112, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Thorne, J.E.; Jabs, D.A.; Kedhar, S.R.; Peters, G.B.; Dunn, J.P. Loss of visual field among patients with birdshot chorioretinopathy. Am. J. Ophthalmol. 2008, 145, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Jampol, L.M.; Sieving, P.A.; Pugh, D.; Fishman, G.A.; Gilbert, H. Multiple evanescent white dot syndrome. I. Clinical findings. Arch. Ophthalmol. 1984, 102, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Raven, M.L.; Ringeisen, A.L.; Yonekawa, Y.; Stem, M.S.; Faia, L.J.; Gottlieb, J.L. Multi-modal imaging and anatomic classification of the white dot syndromes. Int. J. Retin. Vitr. 2017, 3, 12. [Google Scholar] [CrossRef]
- dell’Omo, R.; Pavesio, C.E. Multiple evanescent white dot syndrome (MEWDS). Int. Ophthalmol. Clin. 2012, 52, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.T. Multiple evanescent white dot syndrome: A review and case report. Clin. Exp. Optom. 2010, 93, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Haen, S.P.; Spaide, R.F. The white dot syndromes. Compr. Ophthalmol. Update 2007, 8, 179–200, discussion 203–174. [Google Scholar] [PubMed]
- Quillen, D.A.; Davis, J.B.; Gottlieb, J.L.; Blodi, B.A.; Callanan, D.G.; Chang, T.S.; Equi, R.A. The white dot syndromes. Am. J. Ophthalmol. 2004, 137, 538–550. [Google Scholar] [CrossRef]
- Cicinelli, M.V.; Ramtohul, P.; Marchese, A.; Bandello, F.; Bailey Freund, K.; Miserocchi, E.; Jampol, L.M. Latest advances in white spot syndromes: New findings and interpretations. Prog. Retin. Eye Res. 2023, 97, 101207. [Google Scholar] [CrossRef]
- Li, D.; Kishi, S. Restored photoreceptor outer segment damage in multiple evanescent white dot syndrome. Ophthalmology 2009, 116, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Feigl, B.; Haas, A.; El-Shabrawi, Y. Multifocal ERG in multiple evanescent white dot syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 2002, 240, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Martidis, A.; Baumal, C.R. Transient multifocal electroretinogram dysfunction in multiple evanescent white dot syndrome. Ophthalmic Surg. Lasers 2002, 33, 246–249. [Google Scholar] [CrossRef] [PubMed]
- da Silva, F.T.; Hirata, C.E.; Olivalves, E.; Oyamada, M.K.; Yamamoto, J.H. Fundus-based and electroretinographic strategies for stratification of late-stage Vogt-Koyanagi-Harada disease patients. Am. J. Ophthalmol. 2009, 148, 939–945.e933. [Google Scholar] [CrossRef]
- Sakata, V.M.; Lavezzo, M.M.; da Silva, F.T.; Rodriguez, E.E.C.; Morita, C.; Abdallah, S.F.; Oyamada, M.K.; Hirata, C.E.; Yamamoto, J.H. Full-field electroretinogram behavior in Vogt-Koyanagi-Harada disease: A 24-month longitudinal study in patients from acute onset evaluated with multimodal analysis. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Zhou, C.; Cao, Q.; Du, Z.; Hu, R.; Wang, Y.; Kijlstra, A.; Yang, P. Longitudinal Study of Visual Function in Vogt-Koyanagi-Harada Disease Using Full-Field Electroretinography. Am. J. Ophthalmol. 2018, 191, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Chee, S.P.; Luu, C.D.; Cheng, C.L.; Lim, W.K.; Jap, A. Visual function in Vogt-Koyanagi-Harada patients. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 785–790. [Google Scholar] [CrossRef]
- Yang, P.; Fang, W.; Wang, L.; Wen, F.; Wu, W.; Kijlstra, A. Study of macular function by multifocal electroretinography in patients with Vogt-Koyanagi-Harada syndrome. Am. J. Ophthalmol. 2008, 146, 767–771. [Google Scholar] [CrossRef]

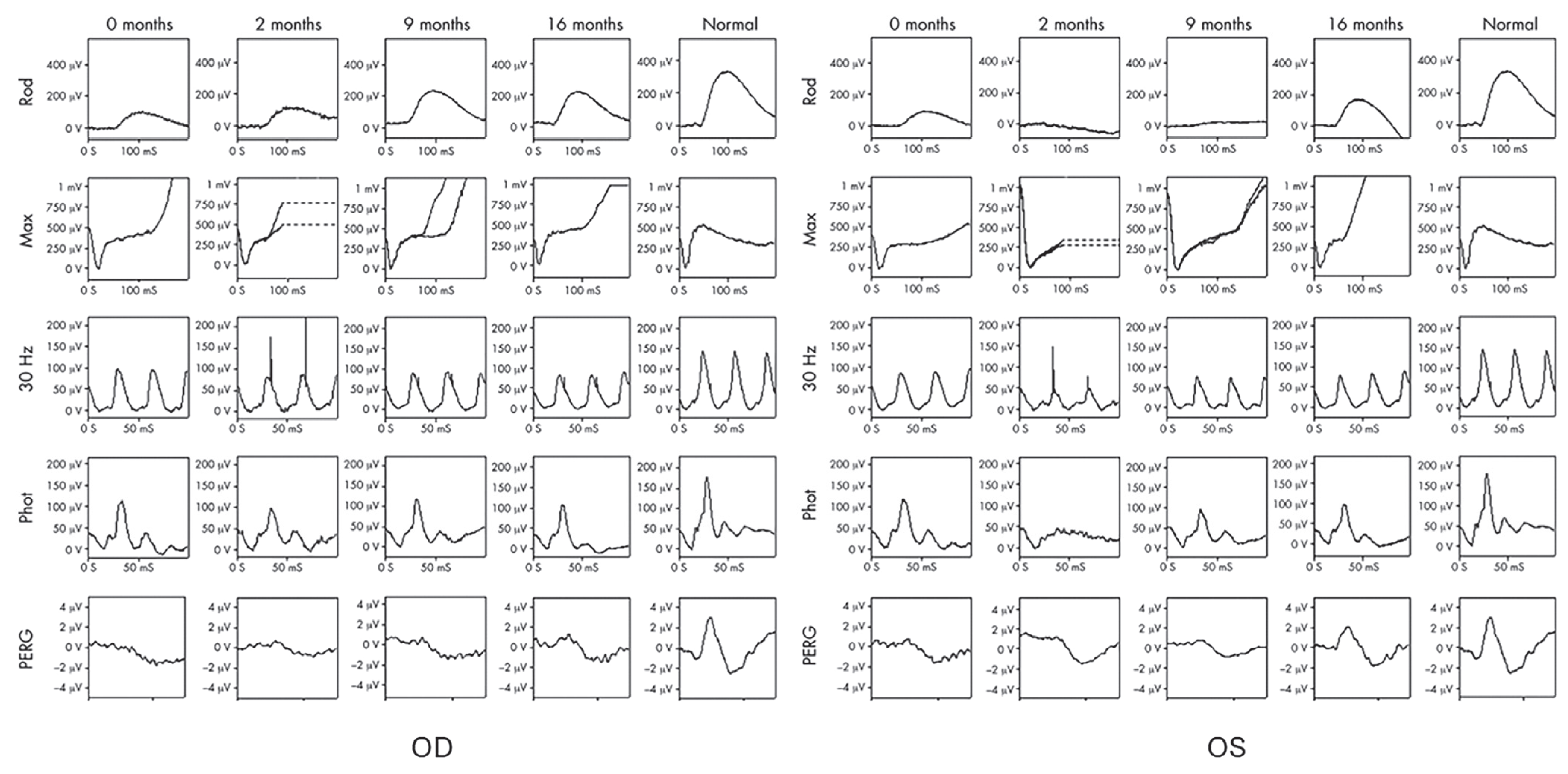
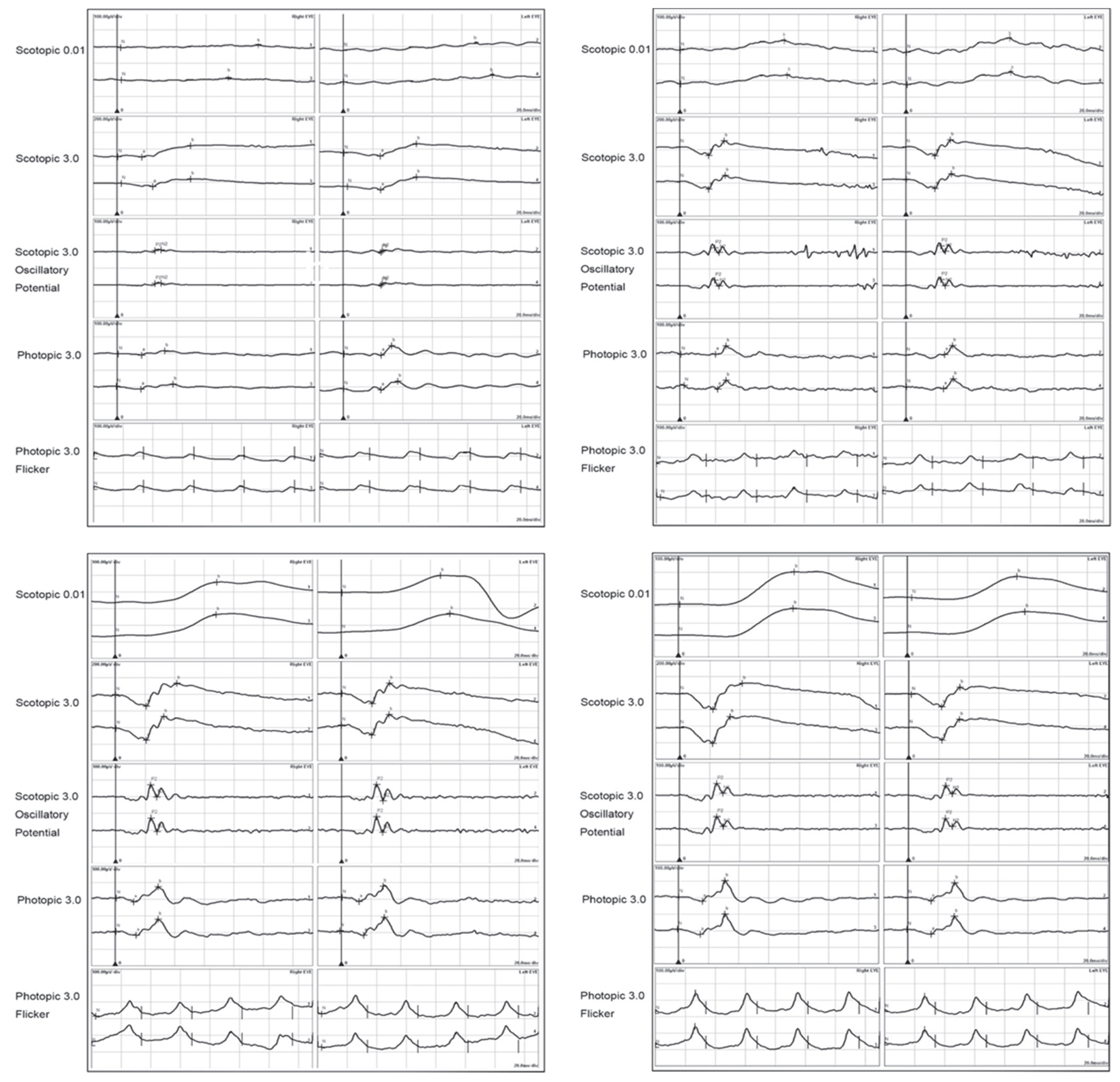
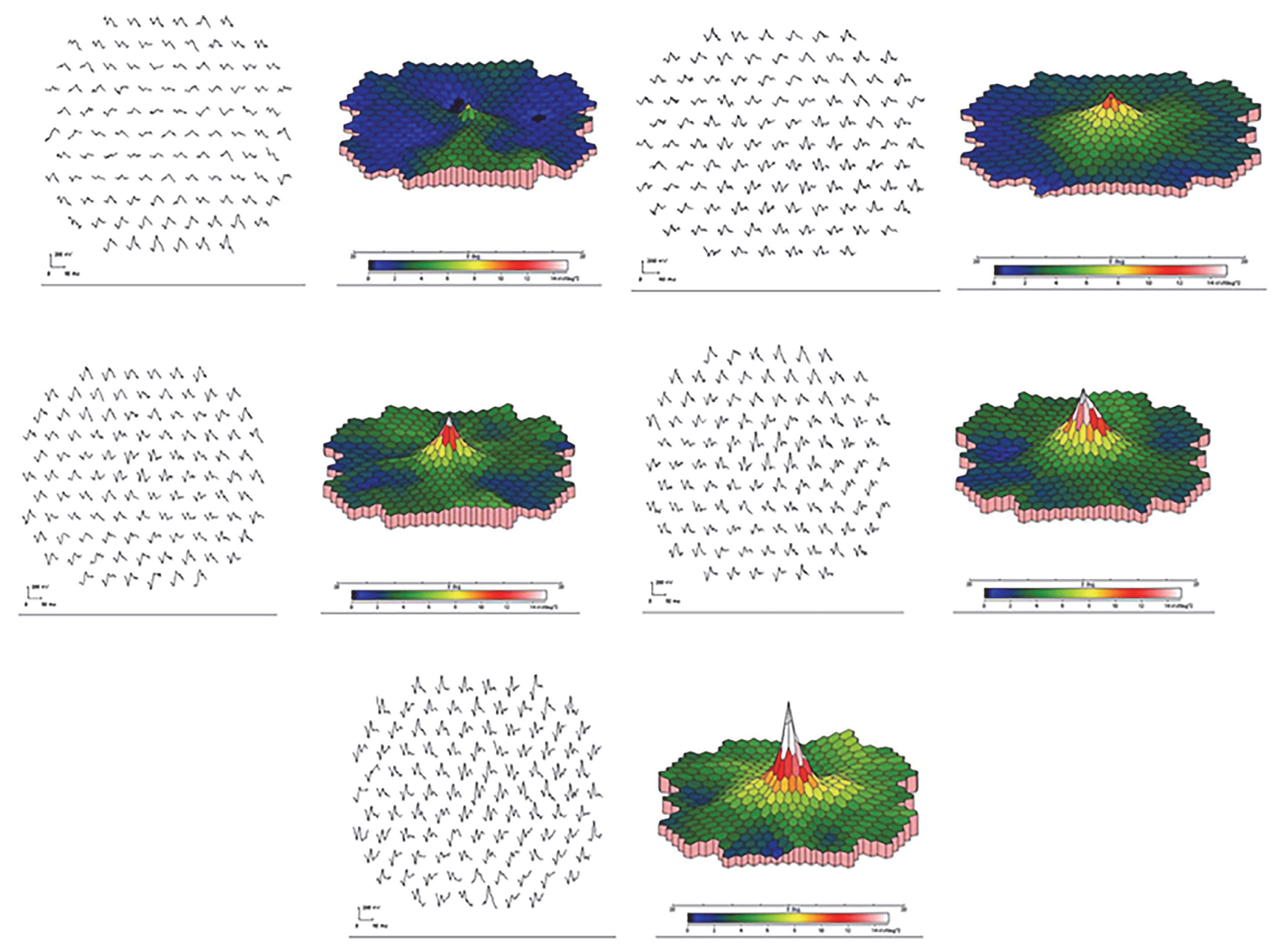

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | ffERG | mfERG | S-cone ERG | VEP | EOG |
|---|---|---|---|---|---|
| APMPPE | Reduction in photopic/scotopic a- and b-waves | Reduction in responses in the affected loci | Reduction | Reduction | |
| AZOOR | Reduction and delay in photopic/scotopic a- and b-waves | Reduction and delay in local responses | |||
| BD | Reduction in photopic/scotopic a- and b-waves | Reduction in responses in the affected loci | |||
| AIR | Reduction and delay in photopic/scotopic a- and b-waves | Reduction in responses, often in central field | Reduction | ||
| AINR | Delay and reduction | Reduction | |||
| BSCR | Reduction in photopic/scotopic a- and b-waves, more obvious in b-wave reduction | ||||
| MEWDS | Reduction in photopic/scotopic a- and b-waves | Reduction in responses in tested field | |||
| VKH | Reduction and delay in photopic/scotopic a- and b-waves | Reduction in responses in the affected loci |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, M.; Kurup, S.K. Application of Ophthalmic Electrophysiology in Inflammatory Disorders of Retina and Optic Nerve. J. Clin. Med. 2024, 13, 3829. https://doi.org/10.3390/jcm13133829
Yu M, Kurup SK. Application of Ophthalmic Electrophysiology in Inflammatory Disorders of Retina and Optic Nerve. Journal of Clinical Medicine. 2024; 13(13):3829. https://doi.org/10.3390/jcm13133829
Chicago/Turabian StyleYu, Minzhong, and Shree K. Kurup. 2024. "Application of Ophthalmic Electrophysiology in Inflammatory Disorders of Retina and Optic Nerve" Journal of Clinical Medicine 13, no. 13: 3829. https://doi.org/10.3390/jcm13133829
APA StyleYu, M., & Kurup, S. K. (2024). Application of Ophthalmic Electrophysiology in Inflammatory Disorders of Retina and Optic Nerve. Journal of Clinical Medicine, 13(13), 3829. https://doi.org/10.3390/jcm13133829