
Diagnostic Modalities in the Detection of Cardiac Amyloidosis
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Recognition of the Disease
3. Echocardiography
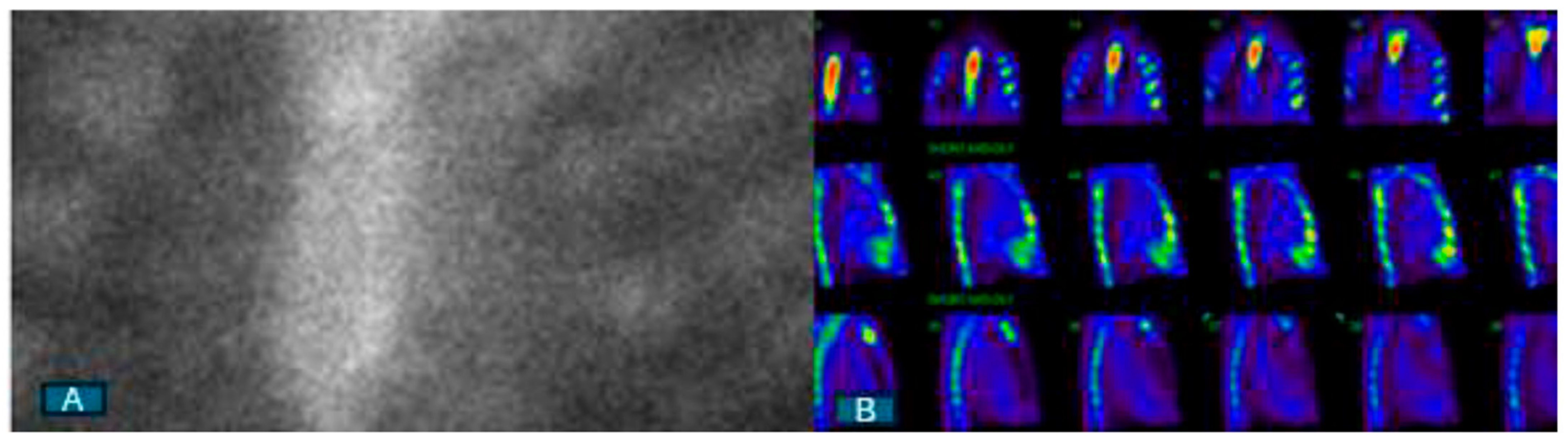
4. Nuclear Scintigraphy with Bone-Avid Tracers
5. CMR
6. Approach to the Diagnosis of CA
7. Biomarker Profile and CA Staging
8. Knowledge Gaps and Directions for Future
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Merlini, G.; Bellotti, V. Molecular Mechanisms of Amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Masri, A.; Bukhari, S.; Eisele, Y.S.; Soman, P. Molecular Imaging of Cardiac Amyloidosis. J. Nucl. Med. 2020, 61, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Pilebro, B.; Suhr, O.B.; Näslund, U.; Westermark, P.; Lindqvist, P.; Sundström, T. (99m)Tc-DPD uptake reflects amyloid fibril composition in hereditary transthyretin amyloidosis. Ups. J. Med. Sci. 2016, 121, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.; Jacobson, D.R.; Tagoe, C.; Alexander, A.; Kitzman, D.W.; Greenberg, B.; Thaneemit-Chen, S.; Lavori, P. Transthyretin V122I in African Americans with congestive heart failure. J. Am. Coll. Cardiol. 2006, 47, 1724–1725. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Linos, A.; Beard, C.M.; Linke, R.P.; Gertz, M.A.; O’Fallon, W.M.; Kurland, L.T. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992, 79, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J. 2017, 38, 2879–2887. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, N.; Rella, V.; Fumagalli, C.; Salerno, S.; Castelletti, S.; Dagradi, F.; Torchio, M.; Marceca, A.; Meda, M.; Gasparini, M.; et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 300, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.; Khan, S.Z.; Ghoweba, M.; Khan, B.; Bashir, Z. Arrhythmias and Device Therapies in Cardiac Amyloidosis. J. Clin. Med. 2024, 13, 1300. [Google Scholar] [CrossRef]
- Ravichandran, S.; Lachmann, H.J.; Wechalekar, A.D. Epidemiologic and Survival Trends in Amyloidosis, 1987–2019. N. Engl. J. Med. 2020, 382, 1567–1568. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Oye, M.; Dhruva, P.; Kandah, F.; Oye, M.; Missov, E. Cardiac amyloid presenting as cardiogenic shock: Case series. Eur. Heart J. Case Rep. 2021, 5, ytab252. [Google Scholar] [CrossRef] [PubMed]
- Dorbala, S.; Vangala, D.; Bruyere, J., Jr.; Quarta, C.; Kruger, J.; Padera, R.; Foster, C.; Hanley, M.; Di Carli, M.F.; Falk, R. Coronary microvascular dysfunction is related to abnormalities in myocardial structure and function in cardiac amyloidosis. JACC Heart Fail. 2014, 2, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.; Kasi, A.; Khan, B. Bradyarrhythmias in Cardiac Amyloidosis and Role of Pacemaker. Curr. Probl. Cardiol. 2023, 48, 101912. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.; Khan, B. Prevalence of ventricular arrhythmias and role of implantable cardioverter-defibrillator in cardiac amyloidosis. J. Cardiol. 2023, 81, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.; Barakat, A.F.; Eisele, Y.S.; Nieves, R.; Jain, S.; Saba, S.; Follansbee, W.P.; Brownell, A.; Soman, P. Prevalence of Atrial Fibrillation and Thromboembolic Risk in Wild-Type Transthyretin Amyloid Cardiomyopathy. Circulation 2021, 143, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C.; Scully, P.R.; Patel, K.P.; Kammerlander, A.A.; Koschutnik, M.; Dona, C.; Wollenweber, T.; Ahmed, N.; Thornton, G.D.; Kelion, A.D.; et al. Prevalence and Outcomes of Concomitant Aortic Stenosis and Cardiac Amyloidosis. J. Am. Coll. Cardiol. 2021, 77, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Milandri, A.; Farioli, A.; Gagliardi, C.; Longhi, S.; Salvi, F.; Curti, S.; Foffi, S.; Caponetti, A.G.; Lorenzini, M.; Ferlini, A.; et al. Carpal tunnel syndrome in cardiac amyloidosis: Implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur. J. Heart Fail. 2020, 22, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Sperry, B.W.; Reyes, B.A.; Ikram, A.; Donnelly, J.P.; Phelan, D.; Jaber, W.A.; Shapiro, D.; Evans, P.J.; Maschke, S.; Kilpatrick, S.E.; et al. Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J. Am. Coll. Cardiol. 2018, 72, 2040–2050. [Google Scholar] [CrossRef]
- Westin, O.; Fosbøl, E.L.; Maurer, M.S.; Leicht, B.P.; Hasbak, P.; Mylin, A.K.; Rørvig, S.; Lindkær, T.H.; Johannesen, H.H.; Gustafsson, F. Screening for Cardiac Amyloidosis 5 to 15 Years After Surgery for Bilateral Carpal Tunnel Syndrome. J. Am. Coll. Cardiol. 2022, 80, 967–977. [Google Scholar] [CrossRef]
- Maurer, M.S.; Smiley, D.; Simsolo, E.; Remotti, F.; Bustamante, A.; Teruya, S.; Helmke, S.; Einstein, A.J.; Lehman, R.; Giles, J.T.; et al. Analysis of lumbar spine stenosis specimens for identification of amyloid. J. Am. Geriatr. Soc. 2022, 70, 3538–3548. [Google Scholar] [CrossRef]
- Geller, H.I.; Singh, A.; Alexander, K.M.; Mirto, T.M.; Falk, R.H. Association Between Ruptured Distal Biceps Tendon and Wild-Type Transthyretin Cardiac Amyloidosis. J. Am. Med. Assoc. 2017, 318, 962–963. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.; Alvarez, J.; Teruya, S.; Castano, A.; Lehman, R.A.; Weidenbaum, M.; Geller, J.A.; Helmke, S.; Maurer, M.S. Hip and knee arthroplasty are common among patients with transthyretin cardiac amyloidosis, occurring years before cardiac amyloid diagnosis: Can we identify affected patients earlier? Amyloid 2017, 24, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S. Cardiac amyloidosis: State-of-the-art review. J. Geriatr. Cardiol. 2023, 20, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Cuddy, S.A.M.; Chetrit, M.; Jankowski, M.; Desai, M.; Falk, R.H.; Weiner, R.B.; Klein, A.L.; Phelan, D.; Grogan, M. Practical Points for Echocardiography in Cardiac Amyloidosis. J. Am. Soc. Echocardiogr. 2022, 35, A31–A40. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Liang, S.; Liu, Z.; Li, Q.; He, W.; Huang, H. Advance of echocardiography in cardiac amyloidosis. Heart Fail. Rev. 2023, 28, 1345–1356. [Google Scholar] [CrossRef]
- Dorbala, S.; Cuddy, S.; Falk, R.H. How to Image Cardiac Amyloidosis: A Practical Approach. JACC Cardiovasc. Imaging 2020, 13, 1368–1383. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Aimo, A.; Barison, A.; Emdin, M.; Porcari, A.; Linhart, A.; Keren, A.; Merlo, M.; Sinagra, G. Restrictive cardiomyopathy: Definition and diagnosis. Eur. Heart J. 2022, 43, 4679–4693. [Google Scholar] [CrossRef]
- Martinez-Naharro, A.; Baksi, A.J.; Hawkins, P.N.; Fontana, M. Diagnostic imaging of cardiac amyloidosis. Nat. Rev. Cardiol. 2020, 17, 413–426. [Google Scholar] [CrossRef]
- Bashir, Z.; Chen, E.W.; Tori, K.; Ghosalkar, D.; Aurigemma, G.P.; Dickey, J.B.; Haines, P. Insight into different phenotypic presentations of heart failure with preserved ejection fraction. Prog. Cardiovasc. Dis. 2023, 79, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.W.J.M.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 2 of 2-Diagnostic Criteria and Appropriate Utilization. Circ. Cardiovasc. Imaging 2021, 14, e000030. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.L.F.; Lim, Y.C.; Evangelista, L.K.M.; Wong, R.C.C.; Chai, P.; Sia, C.H.; Loi, H.Y.; Yeo, T.C.; Lin, W. Utility and pitfalls of the electrocardiogram in the evaluation of cardiac amyloidosis. Ann. Noninvasive Electrocardiol. 2022, 27, e12967. [Google Scholar] [CrossRef] [PubMed]
- Bravo, P.E.; Fujikura, K.; Kijewski, M.F.; Jerosch-Herold, M.; Jacob, S.; El-Sady, M.S.; Sticka, W.; Dubey, S.; Belanger, A.; Park, M.A.; et al. Relative Apical Sparing of Myocardial Longitudinal Strain Is Explained by Regional Differences in Total Amyloid Mass Rather Than the Proportion of Amyloid Deposits. JACC Cardiovasc. Imaging 2019, 12 Pt 1, 1165–1173. [Google Scholar] [CrossRef]
- Quarta, C.C.; Solomon, S.D.; Uraizee, I.; Kruger, J.; Longhi, S.; Ferlito, M.; Gagliardi, C.; Milandri, A.; Rapezzi, C.; Falk, R.H. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation 2014, 129, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Kyrouac, D.; Schiffer, W.; Lennep, B.; Fergestrom, N.; Zhang, K.W.; Gorcsan, J., 3rd; Lenihan, D.J.; Mitchell, J.D. Echocardiographic and clinical predictors of cardiac amyloidosis: Limitations of apical sparing. ESC Heart Fail. 2022, 9, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Minamisawa, M.; Inciardi, R.M.; Claggett, B.; Cuddy, S.A.M.; Quarta, C.C.; Shah, A.M.; Dorbala, S.; Falk, R.H.; Matsushita, K.; Kitzman, D.W.; et al. Left atrial structure and function of the amyloidogenic V122I transthyretin variant in elderly African Americans. Eur. J. Heart Fail. 2021, 23, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Bashir, Z.; Younus, A.; Dhillon, S.; Kasi, A.; Bukhari, S. EXPRESS: Epidemiology, Diagnosis and Management of Cardiac Amyloidosis. J. Investig. Med. 2024, 13, 10815589241261279. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Khan, F.H.; Remme, E.W.; Ohte, N.; García-Izquierdo, E.; Chetrit, M.; Moñivas-Palomero, V.; Mingo-Santos, S.; Andersen, Ø.S.; Gude, E.; et al. Determinants of left atrial reservoir and pump strain and use of atrial strain for evaluation of left ventricular filling pressure. Eur. Heart J. Cardiovasc. Imaging 2021, 23, 61–70. [Google Scholar] [CrossRef]
- Aimo, A.; Fabiani, I.; Giannoni, A.; Mandoli, G.E.; Pastore, M.C.; Vergaro, G.; Spini, V.; Chubuchny, V.; Pasanisi, E.M.; Petersen, C.; et al. Multi-chamber speckle tracking imaging and diagnostic value of left atrial strain in cardiac amyloidosis. Eur. Heart J. Cardiovasc. Imaging 2022, 24, 130–141. [Google Scholar] [CrossRef]
- Bukhari, S.; Khan, S.Z.; Bashir, Z. Atrial Fibrillation, Thromboembolic Risk, and Anticoagulation in Cardiac Amyloidosis: A Review. J. Card. Fail. 2023, 29, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Akintoye, E.; Majid, M.; Klein, A.L.; Hanna, M. Prognostic Utility of Left Atrial Strain to Predict Thrombotic Events and Mortality in Amyloid Cardiomyopathy. JACC Cardiovasc. Imaging 2023, 16, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.; Oliveros, E.; Parekh, H.; Farmakis, D. Epidemiology, Mechanisms, and Management of Atrial Fibrillation in Cardiac Amyloidosis. Curr. Probl. Cardiol. 2023, 48, 101571. [Google Scholar] [CrossRef] [PubMed]
- Moñivas Palomero, V.; Durante-Lopez, A.; Sanabria, M.T.; Cubero, J.S.; González-Mirelis, J.; Lopez-Ibor, J.V.; Navarro Rico, S.M.; Krsnik, I.; Dominguez, F.; Mingo, A.M.; et al. Role of Right Ventricular Strain Measured by Two-Dimensional Echocardiography in the Diagnosis of Cardiac Amyloidosis. J. Am. Soc. Echocardiogr. 2019, 32, 845–853.e1. [Google Scholar] [CrossRef] [PubMed]
- Perugini, E.; Guidalotti, P.L.; Salvi, F.; Cooke, R.M.; Pettinato, C.; Riva, L.; Leone, O.; Farsad, M.; Ciliberti, P.; Bacchi-Reggiani, L.; et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J. Am. Coll. Cardiol. 2005, 46, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, S.; Castaño, A.; Pozniakoff, T.; Deslisle, S.; Latif, F.; Maurer, M.S. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ. Cardiovasc. Imaging 2013, 6, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Masri, A.; Bukhari, S.; Ahmad, S.; Nieves, R.; Eisele, Y.S.; Follansbee, W.; Brownell, A.; Wong, T.C.; Schelbert, E.; Soman, P.; et al. Efficient 1-Hour Technetium-99 m Pyrophosphate Imaging Protocol for the Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Cardiovasc. Imaging 2020, 13, e010249. [Google Scholar] [CrossRef] [PubMed]
- Stats, M.A.; Stone, J.R. Varying levels of small microcalcifications and macrophages in ATTR and AL cardiac amyloidosis: Implications for utilizing nuclear medicine studies to subtype amyloidosis. Cardiovasc. Pathol. 2016, 25, 413–417. [Google Scholar] [CrossRef]
- Musumeci, M.B.; Cappelli, F.; Russo, D.; Tini, G.; Canepa, M.; Milandri, A.; Bonfiglioli, R.; Di Bella, G.; My, F.; Luigetti, M.; et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2020, 13, 1314–1321. [Google Scholar] [CrossRef]
- Maceira, A.M.; Joshi, J.; Prasad, S.K.; Moon, J.C.; Perugini, E.; Harding, I.; Sheppard, M.N.; Poole-Wilson, P.A.; Hawkins, P.N.; Pennell, D.J. Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation 2005, 111, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tian, Z.; Fang, Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: A systematic review and meta-analysis. BMC Cardiovasc. Disord. 2016, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Dungu, J.N.; Valencia, O.; Pinney, J.H.; Gibbs, S.D.; Rowczenio, D.; Gilbertson, J.A.; Lachmann, H.J.; Wechalekar, A.; Gillmore, J.D.; Whelan, C.J.; et al. CMR-Based Differentiation of AL and ATTR Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2014, 7, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, H.; Chacko, B.R.; Irodi, A.; Joseph, E.; Vimala, L.R.; Thomson, V.S. Myocardial nulling pattern in cardiac amyloidosis on time of inversion scout magnetic resonance imaging sequence—A new observation of temporal variability. Indian J. Radiol. Imaging 2018, 28, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Karamitsos, T.D.; Piechnik, S.K.; Banypersad, S.M.; Fontana, M.; Ntusi, N.B.; Ferreira, V.M.; Whelan, C.J.; Myerson, S.G.; Robson, M.D.; Hawkins, P.N.; et al. Noncontrast T1 Mapping for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2013, 6, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Banypersad, S.M.; Fontana, M.; Maestrini, V.; Sado, D.M.; Captur, G.; Petrie, A.; Piechnik, S.K.; Whelan, C.J.; Herrey, A.S.; Gillmore, J.D.; et al. T1 mapping and survival in systemic light-chain amyloidosis. Eur. Heart J. 2015, 36, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Naharro, A.; Kotecha, T.; Norrington, K.; Boldrini, M.; Rezk, T.; Quarta, C.; Treibel, T.A.; Whelan, C.J.; Knight, D.S.; Kellman, P.; et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovasc. Imaging 2019, 12, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Banypersad, S.M.; Sado, D.M.; Flett, A.S.; Gibbs, S.D.; Pinney, J.H.; Maestrini, V.; Cox, A.T.; Fontana, M.; Whelan, C.J.; Wechalekar, A.D.; et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: An equilibrium contrast cardiovascular magnetic resonance study. Circ. Cardiovasc. Imaging 2013, 6, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Nieves, R.A.; Bukhari, S.; Harinstein, M.E. Adding value to myocardial perfusion scintigraphy: A prediction tool to predict adverse cardiac outcomes and risk stratify. J. Nucl. Cardiol. 2021, 28, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Olausson, E.; Wertz, J.; Fridman, Y.; Bering, P.; Maanja, M.; Niklasson, L.; Wong, T.C.; Fukui, M.; Cavalcante, J.L.; Cater, G.; et al. Diffuse myocardial fibrosis associates with incident ventricular arrhythmia in implantable cardioverter defibrillator recipients. medRxiv 2023. [Google Scholar] [CrossRef]
- Takashio, S.; Yamamuro, M.; Izumiya, Y.; Hirakawa, K.; Marume, K.; Yamamoto, M.; Ueda, M.; Yamashita, T.; Ishibashi-Ueda, H.; Yasuda, S.; et al. Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Fail. 2018, 5, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J. Am. Coll. Cardiol. 2016, 68, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, I.Y.; Bukhari, S.; Barakat, A.F.; Pepine, C.J.; Lindley, K.J.; Miller, E.C. American College of Cardiology Cardiovascular Disease in Women Committee. Maternal Stroke: A Call for Action. Circulation 2021, 143, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Damy, T.; Fontana, M.; Hutchinson, M.; Lachmann, H.J.; Martinez-Naharro, A.; Quarta, C.C.; Rezk, T.; Whelan, C.J.; Gonzalez-Lopez, E.; et al. A new staging system for cardiac transthyretin amyloidosis. Eur. Heart J. 2018, 39, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Gertz, M.A.; Kyle, R.A.; Lacy, M.Q.; Burritt, M.F.; Therneau, T.M.; Greipp, P.R.; Witzig, T.E.; Lust, J.A.; Rajkumar, S.V.; et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: A staging system for primary systemic amyloidosis. J. Clin. Oncol. 2004, 22, 3751–3757. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.; Yaghi, S.; Bashir, Z. Stroke in Young Adults. J. Clin. Med. 2023, 12, 4999. [Google Scholar] [CrossRef]
- Kumar, S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Colby, C.; Laumann, K.; Zeldenrust, S.R.; Leung, N.; Dingli, D.; et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol. 2012, 30, 989–995. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukhari, S.; Bashir, Z. Diagnostic Modalities in the Detection of Cardiac Amyloidosis. J. Clin. Med. 2024, 13, 4075. https://doi.org/10.3390/jcm13144075
Bukhari S, Bashir Z. Diagnostic Modalities in the Detection of Cardiac Amyloidosis. Journal of Clinical Medicine. 2024; 13(14):4075. https://doi.org/10.3390/jcm13144075
Chicago/Turabian StyleBukhari, Syed, and Zubair Bashir. 2024. "Diagnostic Modalities in the Detection of Cardiac Amyloidosis" Journal of Clinical Medicine 13, no. 14: 4075. https://doi.org/10.3390/jcm13144075