

Intersecting Pathways: The Role of Metabolic Dysregulation, Gastrointestinal Microbiome, and Inflammation in Acute Ischemic Stroke Pathogenesis and Outcomes

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Inflammation in AIS: Basic Mechanisms and External Factors
2.1. Cellular and Molecular Foundations of Inflammation
2.2. Impact of External Factors: Role of Pathogens, Including Microorganisms and Their Components Like LPS, in Initiating Inflammatory Responses
2.3. Linking Neuroinflammation to AIS
3. Metabolic Dysregulation and Inflammation: Adipokines and Obesity
3.1. Role of Inflammation in Obesity
3.2. Adipokines in Detail
3.3. Direct and Indirect Pathways Linking Obesity to AIS
3.4. Oxidative Stress in Metabolic Dysregulation—The Role of Reactive Oxygen Species in AIS Pathogenesis
4. Microbiota, LPS, and the Neuroinflammatory Cascade
4.1. Obesity-Induced Changes in the Gut Microbiota
4.2. LPS as a Bridge to Neuroinflammation
4.3. The Gut-Brain Axis in AIS
5. Unraveling AIS Mechanisms: Etiology, Neuroinflammation, and Diagnostics
5.1. Influences on AIS Etiology—Beyond Traditional Risk Factors
5.2. Advancements in AIS Diagnostics
6. Addressing AIS: Therapeutic Approaches and Future Directions
6.1. Current Therapeutic Strategies
6.2. Challenges and Opportunities in Therapy Delivery
6.3. Future Research Avenues
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kyu, H.H.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Disability-Adjusted Life-Years (DALYs) for 359 Diseases and Injuries and Healthy Life Expectancy (HALE) for 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1859–1922. [Google Scholar] [CrossRef]
- Soto-Cámara, R.; González-Bernal, J.J.; González-Santos, J.; Aguilar-Parra, J.M.; Trigueros, R.; López-Liria, R. Age-Related Risk Factors at the First Stroke Event. J. Clin. Med. 2020, 9, 2233. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhu, L.; Song, W.; Zhang, M.; Teng, L.; Wu, M. Crosstalk between the Gut and Brain in Ischemic Stroke: Mechanistic Insights and Therapeutic Options. Mediat. Inflamm. 2022, 2022, 6508046. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, L.; Nemeş, S.-A.; Szabo, K.; Teleky, B.-E.; Vodnar, D.-C. Guts Imbalance Imbalances the Brain: A Review of Gut Microbiota Association With Neurological and Psychiatric Disorders. Front. Med. 2022, 9, 813204. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, S.; Zhang, C. The Related Metabolic Diseases and Treatments of Obesity. Healthcare 2022, 10, 1616. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpour-Niazi, S.; Afaghi, S.; Hadaegh, P.; Mahdavi, M.; Farhadnejad, H.; Tohidi, M.; Mirmiran, P.; Azizi, F.; Hadaegh, F. The Association between Metabolic Syndrome and Insulin Resistance with Risk of Cardiovascular Events in Different States of Cardiovascular Health Status. J. Diabetes Investig. 2024, 15, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, J.H.; Lee, Y.J. The Role of Adipokines in Tumor Progression and Its Association with Obesity. Biomedicines 2024, 12, 97. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Martín-Rodríguez, A.; Martínez-Guardado, I.; Navarro-Jiménez, E.; Laborde-Cárdenas, C.C.; Tornero-Aguilera, J.F. The Role of Adipokines in Health and Disease. Biomedicines 2023, 11, 1290. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the Gut Microbiota in Nutrition and Health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef]
- Vetrani, C.; Di Nisio, A.; Paschou, S.A.; Barrea, L.; Muscogiuri, G.; Graziadio, C.; Savastano, S.; Colao, A. From Gut Microbiota through Low-Grade Inflammation to Obesity: Key Players and Potential Targets. Nutrients 2022, 14, 2103. [Google Scholar] [CrossRef] [PubMed]
- Kandpal, M.; Indari, O.; Baral, B.; Jakhmola, S.; Tiwari, D.; Bhandari, V.; Pandey, R.K.; Bala, K.; Sonawane, A.; Jha, H.C. Dysbiosis of Gut Microbiota from the Perspective of the Gut–Brain Axis: Role in the Provocation of Neurological Disorders. Metabolites 2022, 12, 1064. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Nieß, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The Role of Gut-Derived Lipopolysaccharides and the Intestinal Barrier in Fatty Liver Diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zadeh, M.; Yang, C.; Candelario-Jalil, E.; Mohamadzadeh, M. Ischemic Stroke Impacts the Gut Microbiome, Ileal Epithelial and Immune Homeostasis. iScience 2022, 25, 105437. [Google Scholar] [CrossRef] [PubMed]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and Cardiovascular Disease: From Mechanisms to Therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hussain, B.; Chang, J. Peripheral Inflammation and Blood–Brain Barrier Disruption: Effects and Mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Vandresen-Filho, S.; Martins, W.C.; Bertoldo, D.B.; Mancini, G.; De Bem, A.F.; Tasca, C.I. Cerebral Cortex, Hippocampus, Striatum and Cerebellum Show Differential Susceptibility to Quinolinic Acid-Induced Oxidative Stress. Neurol. Sci. 2015, 36, 1449–1456. [Google Scholar] [CrossRef]
- Sun, Y.; Koyama, Y.; Shimada, S. Inflammation From Peripheral Organs to the Brain: How Does Systemic Inflammation Cause Neuroinflammation? Front. Aging Neurosci. 2022, 14, 903455. [Google Scholar] [CrossRef]
- Gąssowska-Dobrowolska, M.; Chlubek, M.; Kolasa, A.; Tomasiak, P.; Korbecki, J.; Skowrońska, K.; Tarnowski, M.; Masztalewicz, M.; Baranowska-Bosiacka, I. Microglia and Astroglia—The Potential Role in Neuroinflammation Induced by Pre- and Neonatal Exposure to Lead (Pb). Int. J. Mol. Sci. 2023, 24, 9903. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhang, C.; Chen, A.; Wang, H.; Zhou, Y.; Li, Y.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef] [PubMed]
- Roe, K. An Inflammation Classification System Using Cytokine Parameters. Scand. J. Immunol. 2021, 93, e12970. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Mes, L.; Newling, M.; den Dunnen, J.; Hoepel, W. Physiological and Pathological Inflammation Induced by Antibodies and Pentraxins. Cells 2021, 10, 1175. [Google Scholar] [CrossRef] [PubMed]
- Germolec, D.R.; Shipkowski, K.A.; Frawley, R.P.; Evans, E. Markers of Inflammation. Methods Mol. Biol. 2018, 1803, 57–79. [Google Scholar] [PubMed]
- Varela, M.L.; Mogildea, M.; Moreno, I.; Lopes, A. Acute Inflammation and Metabolism. Inflammation 2018, 41, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef] [PubMed]
- Millán Solano, M.V.; Salinas Lara, C.; Sánchez-Garibay, C.; Soto-Rojas, L.O.; Escobedo-Ávila, I.; Tena-Suck, M.L.; Ortíz-Butrón, R.; Choreño-Parra, J.A.; Romero-López, J.P.; Meléndez Camargo, M.E. Effect of Systemic Inflammation in the CNS: A Silent History of Neuronal Damage. Int. J. Mol. Sci. 2023, 24, 11902. [Google Scholar] [CrossRef] [PubMed]
- Simats, A.; Liesz, A. Systemic Inflammation after Stroke: Implications for Post-stroke Comorbidities. EMBO Mol. Med. 2022, 14, e16269. [Google Scholar] [CrossRef]
- Liu, C.; Chu, D.; Kalantar-Zadeh, K.; George, J.; Young, H.A.; Liu, G. Cytokines: From Clinical Significance to Quantification. Adv. Sci. 2021, 8, e2004433. [Google Scholar] [CrossRef]
- Wautier, J.-L.; Wautier, M.-P. Pro- and Anti-Inflammatory Prostaglandins and Cytokines in Humans: A Mini Review. Int. J. Mol. Sci. 2023, 24, 9647. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The Pro-Inflammatory Cytokines in COVID-19 Pathogenesis: What Goes Wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef]
- Levin, S.G.; Pershina, E.V.; Bugaev-Makarovskiy, N.A.; Chernomorets, I.Y.; Konakov, M.V.; Arkhipov, V.I. Why Do Levels Of Anti-Inflammatory Cytokines Increase During Memory Acquisition? Neuroscience 2021, 473, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Hadrup, N.; Zhernovkov, V.; Jacobsen, N.R.; Voss, C.; Strunz, M.; Ansari, M.; Schiller, H.B.; Halappanavar, S.; Poulsen, S.S.; Kholodenko, B.; et al. Acute Phase Response as a Biological Mechanism-of-Action of (Nano)Particle-Induced Cardiovascular Disease. Small 2020, 16, e1907476. [Google Scholar] [CrossRef]
- Wang, S.; Wu, P.; Chen, Y.; Chai, Y. Ambiguous Roles and Potential Therapeutic Strategies of Innate Lymphoid Cells in Different Types of Tumor (Review). Oncol. Lett. 2020, 20, 1513–1525. [Google Scholar] [CrossRef]
- Kiaie, N.; Gorabi, A.M.; Loveless, R.; Teng, Y.; Jamialahmadi, T.; Sahebkar, A. The Regenerative Potential of Glial Progenitor Cells and Reactive Astrocytes in CNS Injuries. Neurosci. Biobehav. Rev. 2022, 140, 104794. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.-Q.; Ma, X.-T.; Hu, Z.-W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.-J.; Tian, D.-S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Planas, A.M. Role of Microglia in Stroke. Glia 2024, 72, 1016–1053. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Darwish, S.F.; Elbadry, A.M.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The Dual Face of Microglia (M1/M2) as a Potential Target in the Protective Effect of Nutraceuticals against Neurodegenerative Diseases. Front. Aging 2023, 4, 1231706. [Google Scholar] [CrossRef]
- Song, S.; Huang, H.; Guan, X.; Fiesler, V.; Bhuiyan, M.I.H.; Liu, R.; Jalali, S.; Hasan, M.N.; Tai, A.K.; Chattopadhyay, A.; et al. Activation of Endothelial Wnt/β-Catenin Signaling by Protective Astrocytes Repairs BBB Damage in Ischemic Stroke. Prog. Neurobiol. 2021, 199, 101963. [Google Scholar] [CrossRef]
- Kumar, A.; Fontana, I.C.; Nordberg, A. Reactive Astrogliosis: A Friend or Foe in the Pathogenesis of Alzheimer’s Disease. J. Neurochem. 2023, 164, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Chan, K.L.; Poller, W.C.; Swirski, F.K.; Russo, S.J. Central Regulation of Stress-Evoked Peripheral Immune Responses. Nat. Rev. Neurosci. 2023, 24, 591–604. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The Movers and Shapers in Immune Privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Herz, J.; Bendix, I.; Felderhoff-Müser, U. Peripheral Immune Cells and Perinatal Brain Injury: A Double-Edged Sword? Pediatr. Res. 2022, 91, 392–403. [Google Scholar] [CrossRef]
- Denorme, F.; Portier, I.; Rustad, J.L.; Cody, M.J.; de Araujo, C.V.; Hoki, C.; Alexander, M.D.; Grandhi, R.; Dyer, M.R.; Neal, M.D.; et al. Neutrophil Extracellular Traps Regulate Ischemic Stroke Brain Injury. J. Clin. Investig. 2022, 132, e154225. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting Neutrophils in Ischemic Stroke: Translational Insights from Experimental Studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef]
- Yu, F.; Wang, Y.; Stetler, A.R.; Leak, R.K.; Hu, X.; Chen, J. Phagocytic Microglia and Macrophages in Brain Injury and Repair. CNS Neurosci. Ther. 2022, 28, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Ohyagi, M.; Ito, M. T Cells in the Brain Inflammation. Adv. Immunol. 2023, 157, 29–58. [Google Scholar] [PubMed]
- Liston, A.; Dooley, J.; Yshii, L. Brain-Resident Regulatory T Cells and Their Role in Health and Disease. Immunol. Lett. 2022, 248, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Feng, B.; Zhang, K.; Guo, Y.; Liu, S.; Wu, Y.; Li, X.; Zhao, M. Excessive Astrocyte-Derived Neurotrophin-3 Contributes to the Abnormal Neuronal Dendritic Development in a Mouse Model of Fragile X Syndrome. PLoS Genet. 2012, 8, e1003172. [Google Scholar] [CrossRef]
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. NADPH Oxidases in Oxidant Production by Microglia: Activating Receptors, Pharmacology and Association with Disease. Br. J. Pharmacol. 2017, 174, 1733–1749. [Google Scholar] [CrossRef]
- Larochelle, J.; Tishko, R.J.; Yang, C.; Ge, Y.; Phan, L.T.; Gunraj, R.E.; Stansbury, S.M.; Liu, L.; Mohamadzadeh, M.; Khoshbouei, H.; et al. Receptor-Interacting Protein Kinase 2 (RIPK2) Profoundly Contributes to Post-Stroke Neuroinflammation and Behavioral Deficits with Microglia as Unique Perpetrators. J. Neuroinflammation 2023, 20, 221. [Google Scholar] [CrossRef]
- Shi, K.; Tian, D.-C.; Li, Z.-G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.-D. Global Brain Inflammation in Stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release Mechanisms of Major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Thundyil, J.; Lim, K.-L. DAMPs and Neurodegeneration. Ageing Res. Rev. 2015, 24, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Aal-Aaboda, M.; Abu Raghif, A.R.; Almudhafer, R.H.; Hadi, N.R. Lipopolysaccharide from Rhodobacter Spheroids Modulate Toll-like Receptors Expression and Tissue Damage in an Animal Model of Bilateral Renal Ischemic Reperfusion Injury. J. Med. Life 2022, 15, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, Y.; Luo, Y.; Du, Y.; Zhang, X.; Fu, J. Curcumin Inhibits LPS-Induced Neuroinflammation by Promoting Microglial M2 Polarization via TREM2/ TLR4/ NF-ΚB Pathways in BV2 Cells. Mol. Immunol. 2019, 116, 29–37. [Google Scholar] [CrossRef]
- Gesuete, R.; Kohama, S.G.; Stenzel-Poore, M.P. Toll-Like Receptors and Ischemic Brain Injury. J. Neuropathol. Exp. Neurol. 2014, 73, 378–386. [Google Scholar] [CrossRef]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger Signals in Stroke and Their Role on Microglia Activation after Ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 175628641877425. [Google Scholar] [CrossRef]
- Li, M.; Liu, J.; Bi, Y.; Chen, J.; Zhao, L. Potential Medications or Compounds Acting on Toll-like Receptors in Cerebral Ischemia. Curr. Neuropharmacol. 2018, 16, 160–175. [Google Scholar] [CrossRef]
- Stanzione, R.; Forte, M.; Cotugno, M.; Bianchi, F.; Marchitti, S.; Rubattu, S. Role of DAMPs and of Leukocytes Infiltration in Ischemic Stroke: Insights from Animal Models and Translation to the Human Disease. Cell. Mol. Neurobiol. 2022, 42, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Saberi, A.; Akhondzadeh, S.; Kazemi, S.; Kazemi, S. Infectious Agents and Stroke: A Systematic Review. Basic Clin. Neurosci. J. 2021, 12, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Keikha, M.; Karbalaei, M. Potential Association between Bacterial Infections and Ischemic Stroke Based on Fifty Case-Control Studies: A Systematic Review and Meta-Analysis. New Microbes New Infect. 2022, 47, 100980. [Google Scholar] [CrossRef] [PubMed]
- Hasan, Z.N. Association of Chlamydia Pneumoniae Serology and Ischemic Stroke. South. Med. J. 2011, 104, 319–321. [Google Scholar] [CrossRef]
- Freiherr Von Seckendorff, A.; Nomenjanahary, M.S.; Labreuche, J.; Ollivier, V.; Di Meglio, L.; Dupont, S.; Hamdani, M.; Brikci-Nigassa, N.; Brun, A.; Boursin, P.; et al. Periodontitis in Ischemic Stroke: Impact of Porphyromonas Gingivalis on Thrombus Composition and Ischemic Stroke Outcomes. Res. Pract. Thromb. Haemost. 2024, 8, 102313. [Google Scholar] [CrossRef]
- Hakoupian, M.; Ferino, E.; Jickling, G.C.; Amini, H.; Stamova, B.; Ander, B.P.; Alomar, N.; Sharp, F.R.; Zhan, X. Bacterial Lipopolysaccharide Is Associated with Stroke. Sci. Rep. 2021, 11, 6570. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983–996.e6. [Google Scholar] [CrossRef] [PubMed]
- Forbes, H.J.; Williamson, E.; Benjamin, L.; Breuer, J.; Brown, M.M.; Langan, S.M.; Minassian, C.; Smeeth, L.; Thomas, S.L.; Warren-Gash, C. Association of Herpesviruses and Stroke: Systematic Review and Meta-Analysis. PLoS ONE 2018, 13, e0206163. [Google Scholar] [CrossRef]
- Amlie-Lefond, C.; Gilden, D. Varicella Zoster Virus: A Common Cause of Stroke in Children and Adults. J. Stroke Cerebrovasc. Dis. 2016, 25, 1561–1569. [Google Scholar] [CrossRef]
- Urbanek, C.; Palm, F.; Grau, A. Influenza and Stroke Risk: A Key Target Not to Be Missed? Infect. Disord.—Drug Targets 2010, 10, 122–131. [Google Scholar] [CrossRef]
- Che Mohd Nassir, C.M.N.; Zolkefley, M.K.I.; Ramli, M.D.; Norman, H.H.; Abdul Hamid, H.; Mustapha, M. Neuroinflammation and COVID-19 Ischemic Stroke Recovery—Evolving Evidence for the Mediating Roles of the ACE2/Angiotensin-(1–7)/Mas Receptor Axis and NLRP3 Inflammasome. Int. J. Mol. Sci. 2022, 23, 3085. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, A.G.F.; Rodrigues, W.F.; Farnesi-de-Assunção, T.S.; Borges, A.V.B.E.; Obata, M.M.S.; Neto, J.R. do C.; da Silva, D.A.A.; Andrade-Silva, L.E.; Desidério, C.S.; Costa-Madeira, J.C.; et al. Inflammatory Response and Activation of Coagulation after COVID-19 Infection. Viruses 2023, 15, 938. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.P.; Kalata, N.; Joekes, E.C.; Kampondeni, S.; Benjamin, L.A.; Harrison, T.S.; Lalloo, D.G.; Heyderman, R.S. Ischemic Stroke as a Complication of Cryptococcal Meningitis and Immune Reconstitution Inflammatory Syndrome: A Case Report. BMC Infect. Dis. 2018, 18, 520. [Google Scholar] [CrossRef] [PubMed]
- Tarhan, B.; Mehkri, Y.; De Prey, J.; Hu, C.; Tuna, I.S.; Shuhaiber, H. Cryptococcosis Presenting as Cerebrovascular Disease. Cureus 2021, 13, e19442. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, V.; Mushi, V.; Ngasala, B.; Kihwele, J.; Sabas, D.; Rocchi, L. Stroke in Patients with Schistosomiasis: Review of Cases in Literature. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vizuete, A.F.K.; Fróes, F.; Seady, M.; Zanotto, C.; Bobermin, L.D.; Roginski, A.C.; Wajner, M.; Quincozes-Santos, A.; Gonçalves, C.A. Early Effects of LPS-Induced Neuroinflammation on the Rat Hippocampal Glycolytic Pathway. J. Neuroinflammation 2022, 19, 255. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.-O. Recognition of Lipopolysaccharide Pattern by TLR4 Complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed]
- Mbongue, J.C.; Vanterpool, E.; Firek, A.; Langridge, W.H.R. Lipopolysaccharide-Induced Immunological Tolerance in Monocyte-Derived Dendritic Cells. Immuno 2022, 2, 482–500. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 Trafficking and Its Influence on LPS-Induced pro-Inflammatory Signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Lai, J.; Liu, Y.; Liu, C.; Qi, M.; Liu, R.; Zhu, X.; Zhou, Q.; Chen, Y.; Guo, A.; Hu, C. Indirubin Inhibits LPS-Induced Inflammation via TLR4 Abrogation Mediated by the NF-KB and MAPK Signaling Pathways. Inflammation 2017, 40, 1–12. [Google Scholar] [CrossRef]
- Murch, O.; Collin, M.; Hinds, C.J.; Thiemermann, C. Lipoproteins in Inflammation and Sepsis. I. Basic Science. Intensive Care Med. 2007, 33, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.; Karim, N.; Wasay, M.; Venketasubramanian, N. Emerging Stroke Risk Factors: A Focus on Infectious and Environmental Determinants. J. Cardiovasc. Dev. Dis. 2024, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Spence, J. Nutrition and Risk of Stroke. Nutrients 2019, 11, 647. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Miller, M.R.; Shah, A.S.V. Air Pollution and Stroke. J. Stroke 2018, 20, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef]
- Sureda, A.; Bibiloni, M.; Julibert, A.; Bouzas, C.; Argelich, E.; Llompart, I.; Pons, A.; Tur, J. Adherence to the Mediterranean Diet and Inflammatory Markers. Nutrients 2018, 10, 62. [Google Scholar] [CrossRef]
- Kumar, M.; Pal, N.; Sharma, P.; Kumawat, M.; Sarma, D.K.; Nabi, B.; Verma, V.; Tiwari, R.R.; Shubham, S.; Arjmandi, B.; et al. Omega-3 Fatty Acids and Their Interaction with the Gut Microbiome in the Prevention and Amelioration of Type-2 Diabetes. Nutrients 2022, 14, 1723. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic Stroke. Nat. Rev. Dis. Prim. 2019, 5, 70. [Google Scholar] [CrossRef]
- Yousufuddin, M.; Young, N. Aging and Ischemic Stroke. Aging 2019, 11, 2542–2544. [Google Scholar] [CrossRef]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, Regional, and National Burden of Stroke and Its Risk Factors, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Lavallée, P.C.; Labreuche, J.; Albers, G.W.; Bornstein, N.M.; Canhão, P.; Caplan, L.R.; Donnan, G.A.; Ferro, J.M.; Hennerici, M.G.; et al. One-Year Risk of Stroke after Transient Ischemic Attack or Minor Stroke. N. Engl. J. Med. 2016, 374, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Donnan, G.A. Secondary Prevention after Ischemic Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2012, 366, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Kernan, W.N.; Ovbiagele, B.; Black, H.R.; Bravata, D.M.; Chimowitz, M.I.; Ezekowitz, M.D.; Fang, M.C.; Fisher, M.; Furie, K.L.; Heck, D.V.; et al. Guidelines for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack. Stroke 2014, 45, 2160–2236. [Google Scholar] [CrossRef] [PubMed]
- Bendorius, M.; Po, C.; Muller, S.; Jeltsch-David, H. From Systemic Inflammation to Neuroinflammation: The Case of Neurolupus. Int. J. Mol. Sci. 2018, 19, 3588. [Google Scholar] [CrossRef]
- Mou, Y.; Du, Y.; Zhou, L.; Yue, J.; Hu, X.; Liu, Y.; Chen, S.; Lin, X.; Zhang, G.; Xiao, H.; et al. Gut Microbiota Interact With the Brain Through Systemic Chronic Inflammation: Implications on Neuroinflammation, Neurodegeneration, and Aging. Front. Immunol. 2022, 13, 796288. [Google Scholar] [CrossRef]
- Yang, J.; Ran, M.; Li, H.; Lin, Y.; Ma, K.; Yang, Y.; Fu, X.; Yang, S. New Insight into Neurological Degeneration: Inflammatory Cytokines and Blood–Brain Barrier. Front. Mol. Neurosci. 2022, 15, 1013933. [Google Scholar] [CrossRef]
- Brandl, S.; Reindl, M. Blood–Brain Barrier Breakdown in Neuroinflammation: Current In Vitro Models. Int. J. Mol. Sci. 2023, 24, 12699. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Li, M.; Gao, F.; Wei, P.; Wang, J. Construction and Imaging of a Neurovascular Unit Model. Neural Regen. Res. 2022, 17, 1685. [Google Scholar] [CrossRef]
- Schaeffer, S.; Iadecola, C. Revisiting the Neurovascular Unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Smith, B.C.; Tinkey, R.A.; Shaw, B.C.; Williams, J.L. Targetability of the Neurovascular Unit in Inflammatory Diseases of the Central Nervous System. Immunol. Rev. 2022, 311, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Bao, T.; Yang, K.; Zhu, X.; Wang, S.; Xiang, W.; Ge, A.; Zeng, L.; Ge, J. The Mechanism of Microglia-Mediated Immune Inflammation in Ischemic Stroke and the Role of Natural Botanical Components in Regulating Microglia: A Review. Front. Immunol. 2023, 13, 1047550. [Google Scholar] [CrossRef] [PubMed]
- Maida, C.D.; Norrito, R.L.; Daidone, M.; Tuttolomondo, A.; Pinto, A. Neuroinflammatory Mechanisms in Ischemic Stroke: Focus on Cardioembolic Stroke, Background, and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 6454. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Coria, H.; Arrieta-Cruz, I.; Cruz, M.-E.; López-Valdés, H. Physiopathology of Ischemic Stroke and Its Modulation Using Memantine: Evidence from Preclinical Stroke. Neural Regen. Res. 2021, 16, 433. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic Cascades in Ischemic Stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Kong, R.; Zhang, L.; Zhang, J. Mitochondria in Traumatic Brain Injury and Mitochondrial-targeted Multipotential Therapeutic Strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chu, J.-M.-T.; Wong, G.-T.-C. Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction. Biomolecules 2022, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Naik, U.P.; Naik, M.U.; Yadav, S.K.; Chaurasia, R.N.; Dash, D. Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications. Biomolecules 2023, 13, 1609. [Google Scholar] [CrossRef]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, Calcium and Mitochondria: A Triad in Synaptic Neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Olufunmilayo, E.O.; Gerke-Duncan, M.B.; Holsinger, R.M.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants 2023, 12, 517. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, H.; Li, H.; Xie, F.; Zhang, J. Research Progress of Neuroinflammation-Related Cells in Traumatic Brain Injury: A Review. Medicine 2023, 102, e34009. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Zhong, L.; Jin, X.; Cui, R.; Yang, W.; Gao, S.; Lv, J.; Li, B.; Liu, T. Effect of Inflammation on the Process of Stroke Rehabilitation and Poststroke Depression. Front. Psychiatry 2019, 10, 184. [Google Scholar] [CrossRef]
- Stuckey, S.M.; Ong, L.K.; Collins-Praino, L.E.; Turner, R.J. Neuroinflammation as a Key Driver of Secondary Neurodegeneration Following Stroke? Int. J. Mol. Sci. 2021, 22, 13101. [Google Scholar] [CrossRef]
- Shao, F.; Wang, X.; Wu, H.; Wu, Q.; Zhang, J. Microglia and Neuroinflammation: Crucial Pathological Mechanisms in Traumatic Brain Injury-Induced Neurodegeneration. Front. Aging Neurosci. 2022, 14, 825086. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leak, R.K.; Cao, G. Microglia-Mediated Neuroinflammation and Neuroplasticity after Stroke. Front. Cell. Neurosci. 2022, 16, 980722. [Google Scholar] [CrossRef]
- Theus, M.H. Neuroinflammation and Acquired Traumatic CNS Injury: A Mini Review. Front. Neurol. 2024, 15, 1334847. [Google Scholar] [CrossRef]
- Perdomo, C.M.; Cohen, R.V.; Sumithran, P.; Clément, K.; Frühbeck, G. Contemporary Medical, Device, and Surgical Therapies for Obesity in Adults. Lancet 2023, 401, 1116–1130. [Google Scholar] [CrossRef]
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global Burden of 369 Diseases and Injuries in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Scuteri, A.; Laurent, S.; Cucca, F.; Cockcroft, J.; Cunha, P.G.; Mañas, L.R.; Raso, F.U.M.; Muiesan, M.L.; Ryliškytė, L.; Rietzschel, E.; et al. Metabolic Syndrome across Europe: Different Clusters of Risk Factors. Eur. J. Prev. Cardiol. 2015, 22, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Elagizi, A.; Kachur, S.; Lavie, C.J.; Carbone, S.; Pandey, A.; Ortega, F.B.; Milani, R.V. An Overview and Update on Obesity and the Obesity Paradox in Cardiovascular Diseases. Prog. Cardiovasc. Dis. 2018, 61, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Tutor, A.W.; Lavie, C.J.; Kachur, S.; Milani, R.V.; Ventura, H.O. Updates on Obesity and the Obesity Paradox in Cardiovascular Diseases. Prog. Cardiovasc. Dis. 2023, 78, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose Tissue Inflammation and Metabolic Dysfunction in Obesity. Am. J. Physiol. Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Cobos-Palacios, L.; Ruiz-Moreno, M.I.; Vilches-Perez, A.; Vargas-Candela, A.; Muñoz-Úbeda, M.; Benítez Porres, J.; Navarro-Sanz, A.; Lopez-Carmona, M.D.; Sanz-Canovas, J.; Perez-Belmonte, L.M.; et al. Metabolically Healthy Obesity: Inflammatory Biomarkers and Adipokines in Elderly Population. PLoS ONE 2022, 17, e0265362. [Google Scholar] [CrossRef] [PubMed]
- Kanmani, S.; Kwon, M.; Shin, M.-K.; Kim, M.K. Association of C-Reactive Protein with Risk of Developing Type 2 Diabetes Mellitus, and Role of Obesity and Hypertension: A Large Population-Based Korean Cohort Study. Sci. Rep. 2019, 9, 4573. [Google Scholar] [CrossRef] [PubMed]
- Kern, L.; Mittenbühler, M.; Vesting, A.; Ostermann, A.; Wunderlich, C.; Wunderlich, F. Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation—Driven Liver and Colorectal Cancers. Cancers 2018, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.; Choi, M.-S. Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed]
- Kirichenko, T.V.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Varaeva, Y.R.; Starodubova, A.V. The Role of Adipokines in Inflammatory Mechanisms of Obesity. Int. J. Mol. Sci. 2022, 23, 14982. [Google Scholar] [CrossRef]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef]
- Taylor, C.T.; Colgan, S.P. Regulation of Immunity and Inflammation by Hypoxia in Immunological Niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.; Sarapultsev, A. Atherosclerosis and Inflammation: Insights from the Theory of General Pathological Processes. Int. J. Mol. Sci. 2023, 24, 7910. [Google Scholar] [CrossRef]
- Sprick, J.D.; Mallet, R.T.; Przyklenk, K.; Rickards, C.A. Ischaemic and Hypoxic Conditioning: Potential for Protection of Vital Organs. Exp. Physiol. 2019, 104, 278–294. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, J.; Gu, Y.; Ji, X.; Nan, G. Intermittent Hypoxia Conditioning as a Potential Prevention and Treatment Strategy for Ischemic Stroke: Current Evidence and Future Directions. Front. Neurosci. 2022, 16, 1067411. [Google Scholar] [CrossRef]
- Recinella, L.; Orlando, G.; Ferrante, C.; Chiavaroli, A.; Brunetti, L.; Leone, S. Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases. Front. Physiol. 2020, 11, 578966. [Google Scholar] [CrossRef]
- Mechanick, J.I.; Zhao, S.; Garvey, W.T. Leptin, An Adipokine With Central Importance in the Global Obesity Problem. Glob. Heart 2018, 13, 113. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef]
- Nyangasa, M.A.; Buck, C.; Kelm, S.; Sheikh, M.A.; Günther, K.; Hebestreit, A. The Association between Leptin and Inflammatory Markers with Obesity Indices in Zanzibari Children, Adolescents, and Adults. Obes. Sci. Pract. 2021, 7, 71–81. [Google Scholar] [CrossRef]
- La Cava, A. Leptin in Inflammation and Autoimmunity. Cytokine 2017, 98, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C.; Akanji, A.O. Leptin, Obesity, and Hypertension: A Review of Pathogenetic Mechanisms. Metab. Syndr. Relat. Disord. 2020, 18, 399–405. [Google Scholar] [CrossRef]
- Villarreal-Molina, M.T.; Antuna-Puente, B. Adiponectin: Anti-Inflammatory and Cardioprotective Effects. Biochimie 2012, 94, 2143–2149. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Howard, A.G.; Blanco, E.; Burrows, R.; Correa-Burrows, P.; Memili, A.; Albala, C.; Santos, J.L.; Angel, B.; Lozoff, B.; et al. Dynamic Relationships between Body Fat and Circulating Adipokine Levels from Adolescence to Young Adulthood: The Santiago Longitudinal Study. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Arefhosseini, S.R.; Ebrahimi-Mamaeghani, M.; Fallah, P.; Bazi, Z. Adiponectin as a Potential Biomarker of Vascular Disease. Vasc. Health Risk Manag. 2015, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Qiu, S.; Yang, G.; Wu, Q. Adiponectin and Metabolic Cardiovascular Diseases: Therapeutic Opportunities and Challenges. Genes Dis. 2023, 10, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Ambroszkiewicz, J.; Chełchowska, M.; Rowicka, G.; Klemarczyk, W.; Strucińska, M.; Gajewska, J. Anti-Inflammatory and Pro-Inflammatory Adipokine Profiles in Children on Vegetarian and Omnivorous Diets. Nutrients 2018, 10, 1241. [Google Scholar] [CrossRef]
- Yawoot, N.; Govitrapong, P.; Tocharus, C.; Tocharus, J. Ischemic Stroke, Obesity, and the Anti-inflammatory Role of Melatonin. BioFactors 2021, 47, 41–58. [Google Scholar] [CrossRef]
- Guzik, A.; Bushnell, C. Stroke Epidemiology and Risk Factor Management. Contin. Lifelong Learn. Neurol. 2017, 23, 15–39. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, E984–E1010. [Google Scholar] [CrossRef]
- Dorrance, A.; Matin, N.; Pires, P. The Effects of Obesity on the Cerebral Vasculature. Curr. Vasc. Pharmacol. 2014, 12, 462–472. [Google Scholar] [CrossRef]
- Li, H.; Khan, S.; Siddique, R.; Bai, Q.; Liu, Y.; Zhang, R.; Zhang, Y.; Yong, V.W.; Xue, M. Obesity in Acute Ischaemic Stroke Patients Treated with Intravenous Thrombolysis Therapy. Neurol. Res. 2023, 45, 796–803. [Google Scholar] [CrossRef]
- Ramírez-Carreto, R.J.; Rodríguez-Cortés, Y.M.; Torres-Guerrero, H.; Chavarría, A. Possible Implications of Obesity-Primed Microglia That Could Contribute to Stroke-Associated Damage. Cell. Mol. Neurobiol. 2023, 43, 2473–2490. [Google Scholar] [CrossRef]
- Letra, L.; Sena, C. Cerebrovascular Disease: Consequences of Obesity-Induced Endothelial Dysfunction. In Advances in Neurobiology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 19, pp. 163–189. ISBN 9783319632605. [Google Scholar]
- Forlivesi, S.; Cappellari, M.; Bonetti, B. Obesity Paradox and Stroke: A Narrative Review. Eat. Weight Disord.—Stud. Anorex. Bulim. Obes. 2021, 26, 417–423. [Google Scholar] [CrossRef]
- Quiñones-Ossa, G.A.; Lobo, C.; Garcia-Ballestas, E.; Florez, W.A.; Moscote-Salazar, L.R.; Agrawal, A. Obesity and Stroke: Does the Paradox Apply for Stroke? Neurointervention 2021, 16, 9–19. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, W.J.; Khera, A.V.; Kim, J.Y.; Yon, D.K.; Lee, S.W.; Shin, J.I.; Won, H.-H. Association between Adiposity and Cardiovascular Outcomes: An Umbrella Review and Meta-Analysis of Observational and Mendelian Randomization Studies. Eur. Heart J. 2021, 42, 3388–3403. [Google Scholar] [CrossRef]
- da Silva, C.L.; Sousa, T.M.M.; de Sousa Junior, J.B.; Nakano, E.Y. Nutritional Factors Associated with Mortality in Hospitalized Patients with COVID-19. Clin. Nutr. Open Sci. 2022, 45, 17–26. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, S.; Leng, S.X. Chronic Low-Grade Inflammatory Phenotype (CLIP) and Senescent Immune Dysregulation. Clin. Ther. 2019, 41, 400–409. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive Oxygen Species, Toxicity, Oxidative Stress, and Antioxidants: Chronic Diseases and Aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Khalil, A.A.; Awadallah, S.; Khan, S.A.; Abu-Izneid, T.; Kamran, M.; Hemeg, H.A.; Mubarak, M.S.; Khalid, A.; Wilairatana, P. Reactive Oxygen Species in Biological Systems: Pathways, Associated Diseases, and Potential Inhibitors—A Review. Food Sci. Nutr. 2024, 12, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- Obeagu, E.I.; Igwe, M.C.; Obeagu, G.U. Oxidative Stress’s Impact on Red Blood Cells: Unveiling Implications for Health and Disease. Medicine 2024, 103, e37360. [Google Scholar] [CrossRef]
- Singer, R.E.; Moss, K.; Kim, S.J.; Beck, J.D.; Offenbacher, S. Oxidative Stress and IgG Antibody Modify Periodontitis-CRP Association. J. Dent. Res. 2015, 94, 1698–1705. [Google Scholar] [CrossRef]
- Endale, H.T.; Tesfaye, W.; Mengstie, T.A. ROS Induced Lipid Peroxidation and Their Role in Ferroptosis. Front. Cell Dev. Biol. 2023, 11, 1226044. [Google Scholar] [CrossRef]
- Papaccio, F.; D′Arino, A.; Caputo, S.; Bellei, B. Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants 2022, 11, 1121. [Google Scholar] [CrossRef]
- Liemburg-Apers, D.C.; Willems, P.H.G.M.; Koopman, W.J.H.; Grefte, S. Interactions between Mitochondrial Reactive Oxygen Species and Cellular Glucose Metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Rampelotto, P.H.; Giannakos, N.R.O.; Mena Canata, D.A.; Pereira, F.D.; Hackenhaar, F.S.; Pereira, M.J.R.; Benfato, M.S. Oxidative Stress and Antioxidant Defense in the Brain of Bat Species with Different Feeding Habits. Int. J. Mol. Sci. 2023, 24, 12162. [Google Scholar] [CrossRef]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative Stress Increases Blood–Brain Barrier Permeability and Induces Alterations in Occludin during Hypoxia–Reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef]
- Lehner, C.; Gehwolf, R.; Tempfer, H.; Krizbai, I.; Hennig, B.; Bauer, H.-C.; Bauer, H. Oxidative Stress and Blood–Brain Barrier Dysfunction Under Particular Consideration of Matrix Metalloproteinases. Antioxid. Redox Signal. 2011, 15, 1305–1323. [Google Scholar] [CrossRef]
- Costea, L.; Mészáros, Á.; Bauer, H.; Bauer, H.-C.; Traweger, A.; Wilhelm, I.; Farkas, A.E.; Krizbai, I.A. The Blood–Brain Barrier and Its Intercellular Junctions in Age-Related Brain Disorders. Int. J. Mol. Sci. 2019, 20, 5472. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The Human Gut Microbiota: Metabolism and Perspective in Obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Ryan, P.M.; Cryan, J.F.; Dinan, T.G.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Gut Microbiota, Obesity and Diabetes. Postgrad. Med. J. 2016, 92, 286–300. [Google Scholar] [CrossRef]
- Geng, J.; Ni, Q.; Sun, W.; Li, L.; Feng, X. The Links between Gut Microbiota and Obesity and Obesity Related Diseases. Biomed. Pharmacother. 2022, 147, 112678. [Google Scholar] [CrossRef]
- Gérard, P. Gut Microbiota and Obesity. Cell. Mol. Life Sci. 2016, 73, 147–162. [Google Scholar] [CrossRef]
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, Inflammation, and the Gut Microbiota. Lancet Diabetes Endocrinol. 2015, 3, 207–215. [Google Scholar] [CrossRef]
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut Microbiota and Obesity: A Role for Probiotics. Nutrients 2019, 11, 2690. [Google Scholar] [CrossRef]
- Aragón-Vela, J.; Solis-Urra, P.; Ruiz-Ojeda, F.J.; Álvarez-Mercado, A.I.; Olivares-Arancibia, J.; Plaza-Diaz, J. Impact of Exercise on Gut Microbiota in Obesity. Nutrients 2021, 13, 3999. [Google Scholar] [CrossRef]
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef]
- Liu, B.-N.; Liu, X.-T.; Liang, Z.-H.; Wang, J.-H. Gut Microbiota in Obesity. World J. Gastroenterol. 2021, 27, 3837–3850. [Google Scholar] [CrossRef]
- Sergeev, I.N.; Aljutaily, T.; Walton, G.; Huarte, E. Effects of Synbiotic Supplement on Human Gut Microbiota, Body Composition and Weight Loss in Obesity. Nutrients 2020, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Warmbrunn, M.V.; Nieuwdorp, M.; Clément, K. Metabolism and Metabolic Disorders and the Microbiome: The Intestinal Microbiota Associated With Obesity, Lipid Metabolism, and Metabolic Health—Pathophysiology and Therapeutic Strategies. Gastroenterology 2021, 160, 573–599. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 1–29. [Google Scholar] [CrossRef]
- Wacleche, V.; Tremblay, C.; Routy, J.-P.; Ancuta, P. The Biology of Monocytes and Dendritic Cells: Contribution to HIV Pathogenesis. Viruses 2018, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Mahla, R.S. Sweeten PAMPs: Role of Sugar Complexed PAMPs in Innate Immunity and Vaccine Biology. Front. Immunol. 2013, 4, 248. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like Receptors in Immunity and Inflammatory Diseases: Past, Present, and Future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Skrzypczak-Wiercioch, A.; Sałat, K. Lipopolysaccharide-Induced Model of Neuroinflammation: Mechanisms of Action, Research Application and Future Directions for Its Use. Molecules 2022, 27, 5481. [Google Scholar] [CrossRef]
- Nijland, R.; Hofland, T.; Van Strijp, J. Recognition of LPS by TLR4: Potential for Anti-Inflammatory Therapies. Mar. Drugs 2014, 12, 4260–4273. [Google Scholar] [CrossRef]
- Peñaloza, H.F.; Noguera, L.P.; Riedel, C.A.; Bueno, S.M. Expanding the Current Knowledge About the Role of Interleukin-10 to Major Concerning Bacteria. Front. Microbiol. 2018, 9, 2047. [Google Scholar] [CrossRef]
- Peng, X.; Luo, Z.; He, S.; Zhang, L.; Li, Y. Blood-Brain Barrier Disruption by Lipopolysaccharide and Sepsis-Associated Encephalopathy. Front. Cell. Infect. Microbiol. 2021, 11, 768108. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lin, F.; Ren, M.; Liu, X.; Xie, W.; Zhang, A.; Qian, M.; Mo, Y.; Wang, J.; Lv, Y. The PICK1/TLR4 Complex on Microglia Is Involved in the Regulation of LPS-Induced Sepsis-Associated Encephalopathy. Int. Immunopharmacol. 2021, 100, 108116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, P.; Li, Y.; Zhao, Z.; Wu, X.; Zhang, L.; Feng, J.; Hong, J.-S. Early-Released Interleukin-10 Significantly Inhibits Lipopolysaccharide-Elicited Neuroinflammation In Vitro. Cells 2021, 10, 2173. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation Induced by Lipopolysaccharide Causes Cognitive Impairment in Mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, M.; Tousif, A.H.; Sonali, S.; Vichitra, C.; Sunanda, T.; Praveenraj, S.S.; Ray, B.; Gorantla, V.R.; Rungratanawanich, W.; Mahalakshmi, A.M.; et al. Role of Endogenous Lipopolysaccharides in Neurological Disorders. Cells 2022, 11, 4038. [Google Scholar] [CrossRef]
- Vandenbark, A.A.; Offner, H.; Matejuk, S.; Matejuk, A. Microglia and Astrocyte Involvement in Neurodegeneration and Brain Cancer. J. Neuroinflammation 2021, 18, 298. [Google Scholar] [CrossRef]
- Wu, H.-J.; Wu, E. The Role of Gut Microbiota in Immune Homeostasis and Autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the Gut Microbiota in Immunity and Inflammatory Disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, X.; Ye, Y.; Wang, F.; Chen, F.; Zheng, C. The Role of Microbiota in Infant Health: From Early Life to Adulthood. Front. Immunol. 2021, 12, 708472. [Google Scholar] [CrossRef] [PubMed]
- Coscia, A.; Bardanzellu, F.; Caboni, E.; Fanos, V.; Peroni, D.G. When a Neonate Is Born, So Is a Microbiota. Life 2021, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, L.; Jin, B.; Xu, X.; Zuo, X.; Li, Y.; Li, Z. The Effects of Delivery Mode on the Gut Microbiota and Health: State of Art. Front. Microbiol. 2021, 12, 724449. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Blacher, E.; Elinav, E.; Pettersson, S. Our Gut Microbiome: The Evolving Inner Self. Cell 2017, 171, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Oziom, J.; Budrewicz, S. Rola Mikrobioty Jelitowej w Patogenezie i Przebiegu Wybranych Schorzeń Układu Nerwowego. Pol. Przegląd Neurol. 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Lee, S.H. Intestinal Permeability Regulation by Tight Junction: Implication on Inflammatory Bowel Diseases. Intest. Res. 2015, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.-D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal Permeability—A New Target for Disease Prevention and Therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef]
- Toribio-Mateas, M. Harnessing the Power of Microbiome Assessment Tools as Part of Neuroprotective Nutrition and Lifestyle Medicine Interventions. Microorganisms 2018, 6, 35. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.; Hu, B. Brain–Gut Axis after Stroke. Brain Circ. 2018, 4, 165. [Google Scholar] [CrossRef]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Battaglini, D.; Pimentel-Coelho, P.M.; Robba, C.; dos Santos, C.C.; Cruz, F.F.; Pelosi, P.; Rocco, P.R.M. Gut Microbiota in Acute Ischemic Stroke: From Pathophysiology to Therapeutic Implications. Front. Neurol. 2020, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Amlerova, J.; Šroubek, J.; Angelucci, F.; Hort, J. Evidences for a Role of Gut Microbiota in Pathogenesis and Management of Epilepsy. Int. J. Mol. Sci. 2021, 22, 5576. [Google Scholar] [CrossRef]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The Gut Microbiome in Human Neurological Disease: A Review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Peh, A.; O’Donnell, J.A.; Broughton, B.R.S.; Marques, F.Z. Gut Microbiota and Their Metabolites in Stroke: A Double-Edged Sword. Stroke 2022, 53, 1788–1801. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Liesirova, K.; Broeg-Morvay, A.; Meisterernst, J.; Schlager, M.; Mono, M.-L.; El-Koussy, M.; Kägi, G.; Jung, S.; Sarikaya, H. Dysphagia in Acute Stroke: Incidence, Burden and Impact on Clinical Outcome. PLoS ONE 2016, 11, e0148424. [Google Scholar] [CrossRef]
- Larsson, S.C. Dietary Approaches for Stroke Prevention. Stroke 2017, 48, 2905–2911. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Shiba, T.; Tohara, H.; Yamaguchi, K.; Hara, K.; Nakagawa, K.; Komatsu, K.; Watanabe, K.; Ohsugi, Y.; Maekawa, S.; et al. Re-Initiation of Oral Food Intake Following Enteral Nutrition Alters Oral and Gut Microbiota Communities. Front. Cell. Infect. Microbiol. 2019, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, K.; Tanaka, R.; Urabe, T.; Ueno, Y.; Yamashiro, Y.; Nomoto, K.; Takahashi, T.; Tsuji, H.; Asahara, T.; Hattori, N. Gut Dysbiosis Is Associated with Metabolism and Systemic Inflammation in Patients with Ischemic Stroke. PLoS ONE 2017, 12, e0171521. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host Microbiota Constantly Control Maturation and Function of Microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Tana, C.; Umesaki, Y.; Imaoka, A.; Handa, T.; Kanazawa, M.; Fukudo, S. Altered Profiles of Intestinal Microbiota and Organic Acids May Be the Origin of Symptoms in Irritable Bowel Syndrome. Neurogastroenterol. Motil. 2009, 22, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal Microbiota Metabolism of L-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Giugliano, G.; Gargiulo, G.; Franzone, A.; Trimarco, B.; Esposito, G.; Perrino, C. Gut Microbe-Generated Metabolite Trimethylamine-N-Oxide as Cardiovascular Risk Biomarker: A Systematic Review and Dose-Response Meta-Analysis. Eur. Heart J. 2017, 38, 2948–2956. [Google Scholar] [CrossRef]
- Zhang, S.R.; Phan, T.G.; Sobey, C.G. Targeting the Immune System for Ischemic Stroke. Trends Pharmacol. Sci. 2021, 42, 96–105. [Google Scholar] [CrossRef]
- Hussain, A.; Lee, M.; Rana, J.; Virani, S.S. Epidemiology and Risk Factors for Stroke in Young Individuals: Implications for Prevention. Curr. Opin. Cardiol. 2021, 36, 565–571. [Google Scholar] [CrossRef]
- Shi, Y.; Guo, L.; Chen, Y.; Xie, Q.; Yan, Z.; Liu, Y.; Kang, J.; Li, S. Risk Factors for Ischemic Stroke: Differences between Cerebral Small Vessel and Large Artery Atherosclerosis Aetiologies. Folia Neuropathol. 2021, 59, 378–385. [Google Scholar] [CrossRef]
- Adams, H.P.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E. Classification of Subtype of Acute Ischemic Stroke. Definitions for Use in a Multicenter Clinical Trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef]
- Markus, H.S.; de Leeuw, F.E. Cerebral Small Vessel Disease: Recent Advances and Future Directions. Int. J. Stroke 2023, 18, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassègue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-Induced Production of Mitochondrial Superoxide in Angiotensin II-Mediated Endothelial Oxidative Stress and Hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Poredos, P.; Poredos, A.V.; Gregoric, I. Endothelial Dysfunction and Its Clinical Implications. Angiology 2021, 72, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V. Monocyte Recruitment and Foam Cell Formation in Atherosclerosis. Micron 2006, 37, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Zwaka, T.P.; Hombach, V.; Torzewski, J. C-Reactive Protein–Mediated Low Density Lipoprotein Uptake by Macrophages. Circulation 2001, 103, 1194–1197. [Google Scholar] [CrossRef]
- Miller, Y.I.; Shyy, J.Y.-J. Context-Dependent Role of Oxidized Lipids and Lipoproteins in Inflammation. Trends Endocrinol. Metab. 2017, 28, 143–152. [Google Scholar] [CrossRef]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. The Role of Inflammasomes in Human Diseases and Their Potential as Therapeutic Targets. Signal Transduct. Target. Ther. 2024, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Carmo, H.R.P.; Bonilha, I.; Barreto, J.; Tognolini, M.; Zanotti, I.; Sposito, A.C. High-Density Lipoproteins at the Interface between the NLRP3 Inflammasome and Myocardial Infarction. Int. J. Mol. Sci. 2024, 25, 1290. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of Inflammation: The Beginning Programs the End. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Kretzer, C.; Jordan, P.M.; Bilancia, R.; Rossi, A.; Gür Maz, T.; Banoglu, E.; Schubert, U.S.; Werz, O. Shifting the Biosynthesis of Leukotrienes Toward Specialized Pro-Resolving Mediators by the 5-Lipoxygenase-Activating Protein (FLAP) Antagonist BRP-201. J. Inflamm. Res. 2022, 15, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Gabrielsen, A.; Agardh, H.E.; Wan, M.; Wetterholm, A.; Wong, C.-H.; Hedin, U.; Swedenborg, J.; Hansson, G.K.; Samuelsson, B.; et al. Expression of 5-Lipoxygenase and Leukotriene A 4 Hydrolase in Human Atherosclerotic Lesions Correlates with Symptoms of Plaque Instability. Proc. Natl. Acad. Sci. USA 2006, 103, 8161–8166. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.K.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-Triggered Lipoxin and Resolvin E1 Modulate Vascular Smooth Muscle Phenotype and Correlate with Peripheral Atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef]
- Watson, T.; Shantsila, E.; Lip, G.Y. Mechanisms of Thrombogenesis in Atrial Fibrillation: Virchow’s Triad Revisited. Lancet 2009, 373, 155–166. [Google Scholar] [CrossRef]
- Ding, W.Y.; Gupta, D.; Lip, G.Y.H. Atrial Fibrillation and the Prothrombotic State: Revisiting Virchow’s Triad in 2020. Heart 2020, 106, 1463–1468. [Google Scholar] [CrossRef]
- Marin, F. Plasma von Willebrand Factor, Soluble Thrombomodulin, and Fibrin D-Dimer Concentrations in Acute Onset Non-Rheumatic Atrial Fibrillation. Heart 2004, 90, 1162–1166. [Google Scholar] [CrossRef]
- Akar, J.G.; Jeske, W.; Wilber, D.J. Acute Onset Human Atrial Fibrillation Is Associated With Local Cardiac Platelet Activation and Endothelial Dysfunction. J. Am. Coll. Cardiol. 2008, 51, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Rafaqat, S.; Gluscevic, S.; Patoulias, D.; Sharif, S.; Klisic, A. The Association between Coagulation and Atrial Fibrillation. Biomedicines 2024, 12, 274. [Google Scholar] [CrossRef] [PubMed]
- Makowski, M.; Smorag, I.; Makowska, J.; Bissinger, A.; Grycewicz, T.; Paśnik, J.; Kidawa, M.; Lubiński, A.; Zielińska, M.; Baj, Z. Platelet Reactivity and Mean Platelet Volume as Risk Markers of Thrombogenesis in Atrial Fibrillation. Int. J. Cardiol. 2017, 235, 1–5. [Google Scholar] [CrossRef]
- Nagao, T.; Hamamoto, M.; Kanda, A.; Tsuganesawa, T.; Ueda, M.; Kobayashi, K.; Miyazaki, T.; Terashi, A. Platelet Activation Is Not Involved in Acceleration of the Coagulation System in Acute Cardioembolic Stroke With Nonvalvular Atrial Fibrillation. Stroke 1995, 26, 1365–1368. [Google Scholar] [CrossRef]
- Stroke Prevention in Atrial Fibrillation Investigators. The Stroke Prevention in Atrial Fibrillation III Study: Rationale, Design, and Patient Features. J. Stroke Cerebrovasc. Dis. 1997, 6, 341–353. [Google Scholar] [CrossRef]
- Aviles, R.J.; Martin, D.O.; Apperson-Hansen, C.; Houghtaling, P.L.; Rautaharju, P.; Kronmal, R.A.; Tracy, R.P.; Van Wagoner, D.R.; Psaty, B.M.; Lauer, M.S.; et al. Inflammation as a Risk Factor for Atrial Fibrillation. Circulation 2003, 108, 3006–3010. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.; Kwan, G.F.; Benjamin, E.J. Global Epidemiology of Atrial Fibrillation. Nat. Rev. Cardiol. 2014, 11, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Pujadas Capmany, R.; Arboix, A.; Casañas-Muñoz, R.; Anguera-Ferrando, N. Specific Cardiac Disorders in 402 Consecutive Patients with Ischaemic Cardioembolic Stroke. Int. J. Cardiol. 2004, 95, 129–134. [Google Scholar] [CrossRef]
- Alkhouli, M.; Alqahtani, F.; Aljohani, S.; Alvi, M.; Holmes, D.R. Burden of Atrial Fibrillation–Associated Ischemic Stroke in the United States. JACC Clin. Electrophysiol. 2018, 4, 618–625. [Google Scholar] [CrossRef]
- Sacco, R.L.; Ellenberg, J.H.; Mohr, J.P.; Tatemichi, T.K.; Hier, D.B.; Price, T.R.; Wolf, P.A. Infarcts of Undetermined Cause: The NINCDS Stroke Data Bank. Ann. Neurol. 1989, 25, 382–390. [Google Scholar] [CrossRef]
- Li, L.; Yiin, G.S.; Geraghty, O.C.; Schulz, U.G.; Kuker, W.; Mehta, Z.; Rothwell, P.M. Incidence, Outcome, Risk Factors, and Long-Term Prognosis of Cryptogenic Transient Ischaemic Attack and Ischaemic Stroke: A Population-Based Study. Lancet Neurol. 2015, 14, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Jabaudon, D.; Sztajzel, J.; Sievert, K.; Landis, T.; Sztajzel, R. Usefulness of Ambulatory 7-Day ECG Monitoring for the Detection of Atrial Fibrillation and Flutter After Acute Stroke and Transient Ischemic Attack. Stroke 2004, 35, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Kaura, A.; Sztriha, L.; Chan, F.K.; Aeron-Thomas, J.; Gall, N.; Piechowski-Jozwiak, B.; Teo, J.T. Early Prolonged Ambulatory Cardiac Monitoring in Stroke (EPACS): An Open-Label Randomised Controlled Trial. Eur. J. Med. Res. 2019, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Dernellis, J. Relationship between C-Reactive Protein Concentrations during Glucocorticoid Therapy and Recurrent Atrial Fibrillation. Eur. Heart J. 2004, 25, 1100–1107. [Google Scholar] [CrossRef]
- Singh, A.; Bonnell, G.; De Prey, J.; Buchwald, N.; Eskander, K.; Kincaid, K.J.; Wilson, C.A. Small-Vessel Disease in the Brain. Am. Hear. J. Plus Cardiol. Res. Pract. 2023, 27, 100277. [Google Scholar] [CrossRef] [PubMed]
- Marini, S.; Anderson, C.D.; Rosand, J. Genetics of Cerebral Small Vessel Disease. Stroke 2020, 51, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.S.; van Der Flier, W.M.; Smith, E.E.; Bath, P.; Biessels, G.J.; Briceno, E.; Brodtman, A.; Chabriat, H.; Chen, C.; de Leeuw, F.-E.; et al. Framework for Clinical Trials in Cerebral Small Vessel Disease (FINESSE). JAMA Neurol. 2022, 79, 1187. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging Standards for Research into Small Vessel Disease and Its Contribution to Ageing and Neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef] [PubMed]
- Duering, M.; Biessels, G.J.; Brodtmann, A.; Chen, C.; Cordonnier, C.; de Leeuw, F.-E.; Debette, S.; Frayne, R.; Jouvent, E.; Rost, N.S.; et al. Neuroimaging Standards for Research into Small Vessel Disease—Advances since 2013. Lancet Neurol. 2023, 22, 602–618. [Google Scholar] [CrossRef]
- Shi, Y.; Thrippleton, M.J.; Marshall, I.; Wardlaw, J.M. Intracranial Pulsatility in Patients with Cerebral Small Vessel Disease: A Systematic Review. Clin. Sci. 2018, 132, 157–171. [Google Scholar] [CrossRef]
- Shi, Y.; Thrippleton, M.J.; Blair, G.W.; Dickie, D.A.; Marshall, I.; Hamilton, I.; Doubal, F.N.; Chappell, F.; Wardlaw, J.M. Small Vessel Disease Is Associated with Altered Cerebrovascular Pulsatility but Not Resting Cerebral Blood Flow. J. Cereb. Blood Flow Metab. 2020, 40, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-J.; Jung, K.-H.; Ryu, Y.J.; Lee, K.-J.; Kim, J.-M.; Lee, S.-T.; Chu, K.; Kim, M.; Lee, S.K.; Roh, J.-K. Progression of Cerebral White Matter Hyperintensities and the Associated Sonographic Index. Radiology 2017, 284, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Altmann, M.; Thommessen, B.; Rønning, O.M.; Benth, J.Š.; Reichenbach, A.S.; Fure, B. Middle Cerebral Artery Pulsatility Index Is Associated with Cognitive Impairment in Lacunar Stroke. J. Neuroimaging 2016, 26, 431–435. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Small Vessel Disease: Mechanisms and Clinical Implications. Lancet Neurol. 2019, 18, 684–696. [Google Scholar] [CrossRef]
- Seo, S.-U.; Kamada, N.; Muñoz-Planillo, R.; Kim, Y.-G.; Kim, D.; Koizumi, Y.; Hasegawa, M.; Himpsl, S.D.; Browne, H.P.; Lawley, T.D.; et al. Distinct Commensals Induce Interleukin-1β via NLRP3 Inflammasome in Inflammatory Monocytes to Promote Intestinal Inflammation in Response to Injury. Immunity 2015, 42, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Kochumon, S.; Al Madhoun, A.; Al-Rashed, F.; Thomas, R.; Sindhu, S.; Al-Ozairi, E.; Al-Mulla, F.; Ahmad, R. Elevated Adipose Tissue Associated IL-2 Expression in Obesity Correlates with Metabolic Inflammation and Insulin Resistance. Sci. Rep. 2020, 10, 16364. [Google Scholar] [CrossRef] [PubMed]
- Tchitchek, N.; Nguekap Tchoumba, O.; Pires, G.; Dandou, S.; Campagne, J.; Churlaud, G.; Fourcade, G.; Hoffmann, T.W.; Strozzi, F.; Gaal, C.; et al. Low-Dose IL-2 Shapes a Tolerogenic Gut Microbiota That Improves Autoimmunity and Gut Inflammation. JCI Insight 2022, 7, e159406. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, S.; He, Q.; Zhang, D.; Chang, J. The Role of Immune Cells in Post-Stroke Angiogenesis and Neuronal Remodeling: The Known and the Unknown. Front. Immunol. 2021, 12, 784098. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Lee, S.-H.; Hong, S.-J. Antibiotics-Induced Dysbiosis of Intestinal Microbiota Aggravates Atopic Dermatitis in Mice by Altered Short-Chain Fatty Acids. Allergy. Asthma Immunol. Res. 2020, 12, 137. [Google Scholar] [CrossRef]
- Corriere, T.; Di Marca, S.; Cataudella, E.; Pulvirenti, A.; Alaimo, S.; Stancanelli, B.; Malatino, L. Neutrophil-to-Lymphocyte Ratio Is a Strong Predictor of Atherosclerotic Carotid Plaques in Older Adults. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 23–27. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-Reactive Protein Concentration and Risk of Coronary Heart Disease, Stroke, and Mortality: An Individual Participant Meta-Analysis. Lancet 2010, 375, 132–140. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, S.; Li, Y.; Sun, Y.; Xiong, X.; Hu, X.; Chen, J.; Qiu, S. Interleukins and Ischemic Stroke. Front. Immunol. 2022, 13, 828447. [Google Scholar] [CrossRef]
- Levast, B.; Li, Z.; Madrenas, J. The Role of IL-10 in Microbiome-Associated Immune Modulation and Disease Tolerance. Cytokine 2015, 75, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, L.A.; Shen, W.-J.; Joyce-Shaikh, B.; Pyatnova, E.A.; Richards, A.G.; Thom, C.; Andrade, S.M.; Cua, D.J.; Kraemer, F.B.; Butcher, E.C. IL-17 Regulates Adipogenesis, Glucose Homeostasis, and Obesity. J. Immunol. 2010, 185, 6947–6959. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Monin, L.; Castillo, P.; Elsegeiny, W.; Horne, W.; Eddens, T.; Vikram, A.; Good, M.; Schoenborn, A.A.; Bibby, K.; et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 2016, 44, 659–671. [Google Scholar] [CrossRef]
- Trøseid, M.; Seljeflot, I.; Arnesen, H. The Role of Interleukin-18 in the Metabolic Syndrome. Cardiovasc. Diabetol. 2010, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, H.; He, J.; Xiong, X. The Role of the Gut Microbiota in the Development of Ischemic Stroke. Front. Immunol. 2022, 13, 845243. [Google Scholar] [CrossRef]
- Winek, K.; Dirnagl, U.; Meisel, A. The Gut Microbiome as Therapeutic Target in Central Nervous System Diseases: Implications for Stroke. Neurotherapeutics 2016, 13, 762–774. [Google Scholar] [CrossRef]
- Kim, C.-S.; Park, H.-S.; Kawada, T.; Kim, J.-H.; Lim, D.; Hubbard, N.E.; Kwon, B.-S.; Erickson, K.L.; Yu, R. Circulating Levels of MCP-1 and IL-8 Are Elevated in Human Obese Subjects and Associated with Obesity-Related Parameters. Int. J. Obes. 2006, 30, 1347–1355. [Google Scholar] [CrossRef]
- Duan, W.-L.; Wang, X.-J.; Ma, Y.-P.; Sheng, Z.-M.; Dong, H.; Zhang, L.-Y.; Zhang, B.-G.; He, M.-T. Therapeutic Strategies Targeting the NLRP3-mediated Inflammatory Response and Pyroptosis in Cerebral Ischemia/Reperfusion Injury (Review). Mol. Med. Rep. 2024, 29, 46. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, H.; Dong, L.; Sun, S.; Li, L. MiRNA-20b Inhibits Cerebral Ischemia-Induced Inflammation through Targeting NLRP3. Int. J. Mol. Med. 2018, 43, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, Y. Implications of Gut Microbiome on Coronary Artery Disease. Cardiovasc. Diagn. Ther. 2020, 10, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.W.-M.; Gohel, M.-D.I.; Ng, C.-F.; Zeppieri, M.; Santos, J.; Fonseca, I.; Kang, J.-S.; Park, J.-H.; Bukhvostov, A.A.; Dvornikov, A.S.; et al. Current Topics in Medicine and Medical Research Vol. 1; Qureshi, D.N.A., Ed.; (A Part of Sciencedomain International); Book Publisher International: Hooghly, West Bengal, India, 2020; ISBN 9789390149599. Available online: https://bp.bookpi.org/index.php/bpi/catalog/book/195 (accessed on 15 May 2024).
- Rashid, H.; Hossain, B.; Siddiqua, T.; Kabir, M.; Noor, Z.; Ahmed, M.; Haque, R. Fecal MicroRNAs as Potential Biomarkers for Screening and Diagnosis of Intestinal Diseases. Front. Mol. Biosci. 2020, 7, 181. [Google Scholar] [CrossRef]
- Raitoharju, E.; Lyytikäinen, L.-P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kähönen, M.; Karhunen, P.J.; et al. MiR-21, MiR-210, MiR-34a, and MiR-146a/b Are up-Regulated in Human Atherosclerotic Plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Vilar, R.; Fish, R.J.; Casini, A.; Neerman-Arbez, M. Fibrin(Ogen) in Human Disease: Both Friend and Foe. Haematologica 2020, 105, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, M.; Kaczorowska, B.; Baj, Z. Fibrinogen Concentrations in Ischaemic Stroke Patients with Metabolic Disorders. Neurol. Neurochir. Pol. 2020, 54, 259–264. [Google Scholar] [CrossRef]
- Ricciardi, R.M.; Cipollone, A.; D’Ardes, D.; Di Giacomo, D.; Pignatelli, P.; Cipollone, F.; Curia, M.C.; Magni, P.; Bucci, M. Risk Factors and Immunoinflammatory Mechanisms Leading to Atherosclerosis: Focus on the Role of Oral Microbiota Dysbiosis. Microorganisms 2023, 11, 1479. [Google Scholar] [CrossRef] [PubMed]
- Geurts, L.; Biessels, G.J.; Luijten, P.; Zwanenburg, J. Better and Faster Velocity Pulsatility Assessment in Cerebral White Matter Perforating Arteries with 7T Quantitative Flow MRI through Improved Slice Profile, Acquisition Scheme, and Postprocessing. Magn. Reson. Med. 2018, 79, 1473–1482. [Google Scholar] [CrossRef]
- Geurts, L.J.; Zwanenburg, J.J.M.; Klijn, C.J.M.; Luijten, P.R.; Biessels, G.J. Higher Pulsatility in Cerebral Perforating Arteries in Patients With Small Vessel Disease Related Stroke, a 7T MRI Study. Stroke 2019, 50, 62–68. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional Morphology of the Blood–Brain Barrier in Health and Disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood–Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672. [Google Scholar] [CrossRef]
- Mokrov, G.V.; Deeva, O.A.; Gudasheva, T.A. The Ligands of Translocator Protein: Design and Biological Properties. Curr. Pharm. Des. 2021, 27, 217–237. [Google Scholar] [CrossRef]
- Chaney, A.; Cropper, H.C.; Johnson, E.M.; Lechtenberg, K.J.; Peterson, T.C.; Stevens, M.Y.; Buckwalter, M.S.; James, M.L. 11 C-DPA-713 Versus 18 F-GE-180: A Preclinical Comparison of Translocator Protein 18 KDa PET Tracers to Visualize Acute and Chronic Neuroinflammation in a Mouse Model of Ischemic Stroke. J. Nucl. Med. 2019, 60, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, N.; Lavisse, S.; Roost, P.; Gubinelli, F.; Hillmer, A.; Boutin, H. TSPO Imaging in Animal Models of Brain Diseases. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 77–109. [Google Scholar] [CrossRef]
- Schaechter, J.D.; Hightower, B.G.; Kim, M.; Loggia, M.L. A Pilot [11C]PBR28 PET/MRI Study of Neuroinflammation and Neurodegeneration in Chronic Stroke Patients. Brain, Behav. Immun.—Heal. 2021, 17, 100336. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Rodichkin, A.N.; McGlothan, J.L.; Acanda De La Rocha, A.M.; Azzam, D.J. Imaging Neuroinflammation with TSPO: A New Perspective on the Cellular Sources and Subcellular Localization. Pharmacol. Ther. 2022, 234, 108048. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Marks, J.D.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Characterization of the 18 KDa Translocator Protein (TSPO) Expression in Post-mortem Normal and Alzheimer’s Disease Brains. Brain Pathol. 2020, 30, 151–164. [Google Scholar] [CrossRef]
- Horti, A.G.; Naik, R.; Foss, C.A.; Minn, I.; Misheneva, V.; Du, Y.; Wang, Y.; Mathews, W.B.; Wu, Y.; Hall, A.; et al. PET Imaging of Microglia by Targeting Macrophage Colony-Stimulating Factor 1 Receptor (CSF1R). Proc. Natl. Acad. Sci. USA 2019, 116, 1686–1691. [Google Scholar] [CrossRef]
- Backes, H.; Walberer, M.; Ladwig, A.; Rueger, M.A.; Neumaier, B.; Endepols, H.; Hoehn, M.; Fink, G.R.; Schroeter, M.; Graf, R. Glucose Consumption of Inflammatory Cells Masks Metabolic Deficits in the Brain. Neuroimage 2016, 128, 54–62. [Google Scholar] [CrossRef]
- Colás, L.; Domercq, M.; Ramos-Cabrer, P.; Palma, A.; Gómez-Vallejo, V.; Padro, D.; Plaza-García, S.; Pulagam, K.R.; Higuchi, M.; Matute, C.; et al. In Vivo Imaging of A7 Nicotinic Receptors as a Novel Method to Monitor Neuroinflammation after Cerebral Ischemia. Glia 2018, 66, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Joya, A.; Ardaya, M.; Montilla, A.; Garbizu, M.; Plaza-García, S.; Gómez-Vallejo, V.; Padro, D.; Gutiérrez, J.J.; Rios, X.; Ramos-Cabrer, P.; et al. In Vivo Multimodal Imaging of Adenosine A 1 Receptors in Neuroinflammation after Experimental Stroke. Theranostics 2021, 11, 410–425. [Google Scholar] [CrossRef]
- Gauberti, M.; Fournier, A.P.; Docagne, F.; Vivien, D.; Martinez de Lizarrondo, S. Molecular Magnetic Resonance Imaging of Endothelial Activation in the Central Nervous System. Theranostics 2018, 8, 1195–1212. [Google Scholar] [CrossRef]
- Tokgoz, S.; Kayrak, M.; Akpinar, Z.; Seyithanoğlu, A.; Güney, F.; Yürüten, B. Neutrophil Lymphocyte Ratio as a Predictor of Stroke. J. Stroke Cerebrovasc. Dis. 2013, 22, 1169–1174. [Google Scholar] [CrossRef]
- Prentice, R.L.; Szatrowski, T.P.; Kato, H.; Mason, M.W. Leukocyte Counts and Cerebrovascular Disease. J. Chronic Dis. 1982, 35, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Zia, E.; Melander, O.; Björkbacka, H.; Hedblad, B.; Engström, G. Total and Differential Leucocyte Counts in Relation to Incidence of Stroke Subtypes and Mortality: A Prospective Cohort Study. J. Intern. Med. 2012, 272, 298–304. [Google Scholar] [CrossRef]
- Baetta, R.; Corsini, A. Role of Polymorphonuclear Neutrophils in Atherosclerosis: Current State and Future Perspectives. Atherosclerosis 2010, 210, 1–13. [Google Scholar] [CrossRef]
- Nasr, N.; Ruidavets, J.B.; Arnal, J.F.; Sie, P.; Larrue, V. Association of Neutrophil Count with Microembolization in Patients with Symptomatic Carotid Artery Stenosis. Atherosclerosis 2009, 207, 519–523. [Google Scholar] [CrossRef]
- Furlan, J.C.; Vergouwen, M.D.I.; Fang, J.; Silver, F.L. White Blood Cell Count Is an Independent Predictor of Outcomes after Acute Ischaemic Stroke. Eur. J. Neurol. 2014, 21, 215–222. [Google Scholar] [CrossRef]
- Lin, K.; Fan, F.; Cai, M.; Yu, Y.; Fu, C.; Ding, L.; Sun, Y.; Sun, J.; Shi, Y.; Dong, Z.; et al. Systemic Immune Inflammation Index and System Inflammation Response Index Are Potential Biomarkers of Atrial Fibrillation among the Patients Presenting with Ischemic Stroke. Eur. J. Med. Res. 2022, 27, 106. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Yang, Y.; Chen, Z.; Yang, L.; Diao, S.; Huang, S.; Wang, Y.; Wang, Y.; Sun, B.; Yuan, X.; et al. Neutrophil to Lymphocyte Ratio Predicts Outcome of Stroke by Cervicocranial Arterial Dissection. Front. Med. 2020, 7, 598055. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Chen, R.; Liu, L.; Liu, X.; Hou, J.; Liao, J.; Zhang, P.; Huang, J.; Lu, L.; Chen, L.; et al. Systemic Immune-Inflammation Index and Incident Cardiovascular Diseases among Middle-Aged and Elderly Chinese Adults: The Dongfeng-Tongji Cohort Study. Atherosclerosis 2021, 323, 20–29. [Google Scholar] [CrossRef]
- Chen, L.; Pandey, S.; Shen, R.; Xu, Y.; Zhang, Q. Increased Systemic Immune-Inflammation Index Is Associated With Delayed Cerebral Ischemia in Aneurysmal Subarachnoid Hemorrhage Patients. Front. Neurol. 2021, 12, 745175. [Google Scholar] [CrossRef] [PubMed]
- Strindhall, J.; Nilsson, B.-O.; Löfgren, S.; Ernerudh, J.; Pawelec, G.; Johansson, B.; Wikby, A. No Immune Risk Profile among Individuals Who Reach 100 Years of Age: Findings from the Swedish NONA Immune Longitudinal Study. Exp. Gerontol. 2007, 42, 753–761. [Google Scholar] [CrossRef]
- Gkrania-Klotsas, E.; Langenberg, C.; Sharp, S.J.; Luben, R.; Khaw, K.-T.; Wareham, N.J. Seropositivity and Higher Immunoglobulin G Antibody Levels Against Cytomegalovirus Are Associated With Mortality in the Population-Based European Prospective Investigation of Cancer–Norfolk Cohort. Clin. Infect. Dis. 2013, 56, 1421–1427. [Google Scholar] [CrossRef]
- Mathei, C.; Adriaensen, W.; Vaes, B.; Van Pottelbergh, G.; Wallemacq, P.; Degryse, J. No Relation between CMV Infection and Mortality in the Oldest Old: Results from the Belfrail Study. Age Ageing 2015, 44, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Demirci-Çekiç, S.; Özkan, G.; Avan, A.N.; Uzunboy, S.; Çapanoğlu, E.; Apak, R. Biomarkers of Oxidative Stress and Antioxidant Defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Shang, M.; Li, X. Serum MicroRNA-137 Serves as a Novel Biomarker for Cerebral Atherosclerosis Diagnosis and Cerebrovascular Event Prediction. J. Cardiovasc. Pharmacol. 2021, 78, 302–307. [Google Scholar] [CrossRef]
- Kim, C.H.; Jeon, J.P.; Kim, S.-E.; Choi, H.J.; Cho, Y.J. Endovascular Treatment with Intravenous Thrombolysis versus Endovascular Treatment Alone for Acute Anterior Circulation Stroke : A Meta-Analysis of Observational Studies. J. Korean Neurosurg. Soc. 2018, 61, 467–473. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue Plasminogen Activator for Acute Ischemic Stroke. N. Engl. J. Med. 1995, 333, 1581–1588. [Google Scholar] [CrossRef]
- Mustanoja, S.; Meretoja, A.; Putaala, J.; Viitanen, V.; Curtze, S.; Atula, S.; Artto, V.; Häppölä, O.; Kaste, M. Outcome by Stroke Etiology in Patients Receiving Thrombolytic Treatment. Stroke 2011, 42, 102–106. [Google Scholar] [CrossRef]
- Blinc, A.; Planinsic, G.; Keber, D.; Jarh, O.; Lahajnar, G.; Zidansĕk, A.; Demsar, F. Dependence of Blood Clot Lysis on the Mode of Transport of Urokinase into the Clot--a Magnetic Resonance Imaging Study in Vitro. Thromb. Haemost. 1991, 65, 549–552. [Google Scholar] [CrossRef]
- Saqqur, M.; Uchino, K.; Demchuk, A.M.; Molina, C.A.; Garami, Z.; Calleja, S.; Akhtar, N.; Orouk, F.O.; Salam, A.; Shuaib, A.; et al. Site of Arterial Occlusion Identified by Transcranial Doppler Predicts the Response to Intravenous Thrombolysis for Stroke. Stroke 2007, 38, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Ishibashi-Ueda, H.; Iwakiri, T.; Ikeda, Y.; Matsuyama, T.; Hatakeyama, K.; Asada, Y. Thrombus Components in Cardioembolic and Atherothrombotic Strokes. Thromb. Res. 2012, 130, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.A.; Montaner, J.; Arenillas, J.F.; Ribo, M.; Rubiera, M.; Alvarez-Sabín, J. Differential Pattern of Tissue Plasminogen Activator–Induced Proximal Middle Cerebral Artery Recanalization Among Stroke Subtypes. Stroke 2004, 35, 486–490. [Google Scholar] [CrossRef]
- Brunser, A.M.; Lavados, P.M.; Cárcamo, D.A.; Hoppe, A.; Olavarría, V.; Diaz, V.; Rivas, R. Additional Information Given to a Multimodal Imaging Stroke Protocol by Transcranial Doppler Ultrasound in the Emergency Room: A Prospective Observational Study. Cerebrovasc. Dis. 2010, 30, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, B.; Martínez-Sánchez, P.; de Leciñana, M.A.; Egido, J.; Reig-Roselló, G.; Díaz-Otero, F.; Sánchez, V.; Simal, P.; Ximenez-Carrillo, A.; García-Pastor, A.; et al. Efficacy of Intravenous Thrombolysis According to Stroke Subtypes: The Madrid Stroke Network Data. Eur. J. Neurol. 2012, 19, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Liao, X.; Pan, Y.; Shi, Y.; Wang, C.; Wang, Y.; Liu, L.; Zhao, X.; Wang, Y.; et al. Unfavorable Outcome of Thrombolysis in Chinese Patients with Cardioembolic Stroke: A Prospective Cohort Study. CNS Neurosci. Ther. 2015, 21, 657–661. [Google Scholar] [CrossRef]
- Nam, H.S.; Lee, K.-Y.; Kim, Y.D.; Choi, H.-Y.; Cho, H.-J.; Cha, M.-J.; Nam, C.M.; Heo, J.H. Failure of Complete Recanalization Is Associated with Poor Outcome after Cardioembolic Stroke. Eur. J. Neurol. 2011, 18, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Mattle, S.; Galimanis, A.; Kappeler, L.; Fischer, U.; Jung, S.; De Marchis, G.M.; Gralla, J.; Mono, M.-L.; Brekenfeld, C.; et al. Impact of Admission Glucose and Diabetes on Recanalization and Outcome after Intra-Arterial Thrombolysis for Ischaemic Stroke. Int. J. Stroke 2014, 9, 985–991. [Google Scholar] [CrossRef]
- Tramacere, I.; Boncoraglio, G.B.; Banzi, R.; Del Giovane, C.; Kwag, K.H.; Squizzato, A.; Moja, L. Comparison of Statins for Secondary Prevention in Patients with Ischemic Stroke or Transient Ischemic Attack: A Systematic Review and Network Meta-Analysis. BMC Med. 2019, 17, 67. [Google Scholar] [CrossRef]
- Spoiala, E.L.; Cinteza, E.; Vatasescu, R.; Vlaiculescu, M.V.; Moisa, S.M. Statins—Beyond Their Use in Hypercholesterolemia: Focus on the Pediatric Population. Children 2024, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Lonn, E.M.; Dagenais, G.R.; Gao, P.; Lopez-Jaramillo, P.; Zhu, J.; Pais, P.; Avezum, A.; Sliwa, K.; Chazova, I.E.; et al. Antihypertensives and Statin Therapy for Primary Stroke Prevention: A Secondary Analysis of the HOPE-3 Trial. Stroke 2021, 52, 2494–2501. [Google Scholar] [CrossRef] [PubMed]
- Marmolejo-Martínez-Artesero, S.; Casas, C.; Romeo-Guitart, D. Endogenous Mechanisms of Neuroprotection: To Boost or Not to Be. Cells 2021, 10, 370. [Google Scholar] [CrossRef]
- Dukay, B.; Csoboz, B.; Tóth, M.E. Heat-Shock Proteins in Neuroinflammation. Front. Pharmacol. 2019, 10, 920. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Aliyeva, O.G.; Popazova, O.O.; Bukhtiyarova, N.V. Involvement of Heat Shock Proteins HSP70 in the Mechanisms of Endogenous Neuroprotection: The Prospect of Using HSP70 Modulators. Front. Cell. Neurosci. 2023, 17, 1131683. [Google Scholar] [CrossRef] [PubMed]
- England, T.J.; Sprigg, N.; Alasheev, A.M.; Belkin, A.A.; Kumar, A.; Prasad, K.; Bath, P.M. Granulocyte-Colony Stimulating Factor (G-CSF) for Stroke: An Individual Patient Data Meta-Analysis. Sci. Rep. 2016, 6, 36567. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.-K.; Cho, S.-R. Exploring Erythropoietin and G-CSF Combination Therapy in Chronic Stroke Patients. Int. J. Mol. Sci. 2016, 17, 463. [Google Scholar] [CrossRef] [PubMed]
- Roy-O’Reilly, M.A.; Ahnstedt, H.; Spychala, M.S.; Munshi, Y.; Aronowski, J.; Sansing, L.H.; McCullough, L.D. Aging Exacerbates Neutrophil Pathogenicity in Ischemic Stroke. Aging 2020, 12, 436–461. [Google Scholar] [CrossRef]
- Yu, C.Y.; Ng, G.; Liao, P. Therapeutic Antibodies in Stroke. Transl. Stroke Res. 2013, 4, 477–483. [Google Scholar] [CrossRef]
- Mallah, K.; Couch, C.; Borucki, D.M.; Toutonji, A.; Alshareef, M.; Tomlinson, S. Anti-Inflammatory and Neuroprotective Agents in Clinical Trials for CNS Disease and Injury: Where Do We Go From Here? Front. Immunol. 2020, 11, 2021. [Google Scholar] [CrossRef] [PubMed]
- Drieu, A.; Buendia, I.; Levard, D.; Hélie, P.; Brodin, C.; Vivien, D.; Rubio, M. Immune Responses and Anti-Inflammatory Strategies in a Clinically Relevant Model of Thromboembolic Ischemic Stroke with Reperfusion. Transl. Stroke Res. 2020, 11, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Furuya, K.; Takeda, H.; Azhar, S.; McCarron, R.M.; Chen, Y.; Ruetzler, C.A.; Wolcott, K.M.; DeGraba, T.J.; Rothlein, R.; Hugli, T.E.; et al. Examination of Several Potential Mechanisms for the Negative Outcome in a Clinical Stroke Trial of Enlimomab, a Murine Anti-Human Intercellular Adhesion Molecule-1 Antibody: A Bedside-to-Bench Study. Stroke 2001, 32, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tian, T.; Gong, S.-X.; Huang, W.-Q.; Zhou, Q.-Y.; Wang, A.-P.; Tian, Y. Microglia-Associated Neuroinflammation Is a Potential Therapeutic Target for Ischemic Stroke. Neural Regen. Res. 2021, 16, 6. [Google Scholar] [CrossRef]
- Sheng, Z.; Liu, Y.; Li, H.; Zheng, W.; Xia, B.; Zhang, X.; Yong, V.W.; Xue, M. Efficacy of Minocycline in Acute Ischemic Stroke: A Systematic Review and Meta-Analysis of Rodent and Clinical Studies. Front. Neurol. 2018, 9, 1103. [Google Scholar] [CrossRef] [PubMed]
- Bascuñana, P.; Möhle, L.; Brackhan, M.; Pahnke, J. Fingolimod as a Treatment in Neurologic Disorders Beyond Multiple Sclerosis. Drugs R&D 2020, 20, 197–207. [Google Scholar] [CrossRef]
- Emsley, H.C.A. A Randomised Phase II Study of Interleukin-1 Receptor Antagonist in Acute Stroke Patients. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, X.; Chen, L.; Lenahan, C.; Fu, Z.; Fang, Y.; Yu, W. Crosstalk Between the Oxidative Stress and Glia Cells After Stroke: From Mechanism to Therapies. Front. Immunol. 2022, 13, 852416. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The Blood–Brain Barrier: Structure, Regulation, and Drug Delivery. Signal Transduct. Target. Ther. 2023, 8, 217. [Google Scholar] [CrossRef]
- Elendu, C.; Amaechi, D.C.; Elendu, T.C.; Ibhiedu, J.O.; Egbunu, E.O.; Ndam, A.R.; Ogala, F.; Ologunde, T.; Peterson, J.C.; Boluwatife, A.I.; et al. Stroke and Cognitive Impairment: Understanding the Connection and Managing Symptoms. Ann. Med. Surg. 2023, 85, 6057–6066. [Google Scholar] [CrossRef] [PubMed]
- Devereux, N.; Berns, A.M. Evaluation & Treatment of Psychological Effects of Stroke. Del. J. Public Health 2023, 9, 62–69. [Google Scholar] [CrossRef]
- Rochmah, T.N.; Rahmawati, I.T.; Dahlui, M.; Budiarto, W.; Bilqis, N. Economic Burden of Stroke Disease: A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 7552. [Google Scholar] [CrossRef] [PubMed]
- Ya, J.; Pellumbaj, J.; Hashmat, A.; Bayraktutan, U. The Role of Stem Cells as Therapeutics for Ischaemic Stroke. Cells 2024, 13, 112. [Google Scholar] [CrossRef]
- Lin, Q.; Lee, Y.-J.; Yun, Z. Differentiation Arrest by Hypoxia. J. Biol. Chem. 2006, 281, 30678–30683. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, Z.; Yang, W.; Zhang, Q.; Zeng, J. An Update on the Potential of Mesenchymal Stem Cell Therapy for Cutaneous Diseases. Stem Cells Int. 2021, 2021, 8834590. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yang, J.; Fang, J.; Zhou, Y.; Candi, E.; Wang, J.; Hua, D.; Shao, C.; Shi, Y. The Secretion Profile of Mesenchymal Stem Cells and Potential Applications in Treating Human Diseases. Signal Transduct. Target. Ther. 2022, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liu, G.; Halim, A.; Ju, Y.; Luo, Q.; Song, G. Mesenchymal Stem Cell Migration and Tissue Repair. Cells 2019, 8, 784. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Shen, Y.; Chopp, M.; Chen, J. Cell-Based and Pharmacological Neurorestorative Therapies for Ischemic Stroke. Neuropharmacology 2018, 134, 310–322. [Google Scholar] [CrossRef]
- Tsai, M.-J.; Tsai, S.-K.; Hu, B.-R.; Liou, D.-Y.; Huang, S.-L.; Huang, M.-C.; Huang, W.-C.; Cheng, H.; Huang, S.-S. Recovery of Neurological Function of Ischemic Stroke by Application of Conditioned Medium of Bone Marrow Mesenchymal Stem Cells Derived from Normal and Cerebral Ischemia Rats. J. Biomed. Sci. 2014, 21, 5. [Google Scholar] [CrossRef]
- Tao, J.; Cao, X.; Yu, B.; Qu, A. Vascular Stem/Progenitor Cells in Vessel Injury and Repair. Front. Cardiovasc. Med. 2022, 9, 845070. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.; Sobrino, T.; Agulla, J.; Bobo-Jiménez, V.; Ramos-Araque, M.E.; Duarte, J.J.; Gómez-Sánchez, J.C.; Bolaños, J.P.; Castillo, J.; Almeida, Á. Neovascularization and Functional Recovery after Intracerebral Hemorrhage Is Conditioned by the Tp53 Arg72Pro Single-Nucleotide Polymorphism. Cell Death Differ. 2017, 24, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Crapanzano, F.; Schirone, L.; Allegra, A.; Pisano, C.; Ruvolo, G.; Forte, M.; Greco, E.; Cavarretta, E.; Marullo, A.G.M.; et al. Deregulation of Notch1 Pathway and Circulating Endothelial Progenitor Cell (EPC) Number in Patients with Bicuspid Aortic Valve with and without Ascending Aorta Aneurysm. Sci. Rep. 2018, 8, 13834. [Google Scholar] [CrossRef] [PubMed]
- Rakkar, K.; Othman, O.; Sprigg, N.; Bath, P.; Bayraktutan, U. Endothelial Progenitor Cells, Potential Biomarkers for Diagnosis and Prognosis of Ischemic Stroke: Protocol for an Observational Case-Control Study. Neural Regen. Res. 2020, 15, 1300. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, Z.; Wang, J.; Cui, S.; Xu, R.; Wang, Y. Functions and Regulatory Mechanisms of Resting Hematopoietic Stem Cells: A Promising Targeted Therapeutic Strategy. Stem Cell Res. Ther. 2023, 14, 73. [Google Scholar] [CrossRef]
- Courties, G.; Herisson, F.; Sager, H.B.; Heidt, T.; Ye, Y.; Wei, Y.; Sun, Y.; Severe, N.; Dutta, P.; Scharff, J.; et al. Ischemic Stroke Activates Hematopoietic Bone Marrow Stem Cells. Circ. Res. 2015, 116, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Savitz, S.I.; Misra, V.; Kasam, M.; Juneja, H.; Cox, C.S.; Alderman, S.; Aisiku, I.; Kar, S.; Gee, A.; Grotta, J.C. Intravenous Autologous Bone Marrow Mononuclear Cells for Ischemic Stroke. Ann. Neurol. 2011, 70, 59–69. [Google Scholar] [CrossRef]
- Hess, D.C.; Wechsler, L.R.; Clark, W.M.; Savitz, S.I.; Ford, G.A.; Chiu, D.; Yavagal, D.R.; Uchino, K.; Liebeskind, D.S.; Auchus, A.P.; et al. Safety and Efficacy of Multipotent Adult Progenitor Cells in Acute Ischaemic Stroke (MASTERS): A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Neurol. 2017, 16, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Peedicayil, J.; Aaron, S. Epigenetics and Cerebrovascular Diseases. In Neuropsychiatric Disorders and Epigenetics; Elsevier: Amsterdam, The Netherlands, 2024; pp. 287–310. [Google Scholar]
- Peng, J.; Ghosh, D.; Zhang, F.; Yang, L.; Wu, J.; Pang, J.; Zhang, L.; Yin, S.; Jiang, Y. Advancement of Epigenetics in Stroke. Front. Neurosci. 2022, 16, 981726. [Google Scholar] [CrossRef]
- Quaranta, G.; Guarnaccia, A.; Fancello, G.; Agrillo, C.; Iannarelli, F.; Sanguinetti, M.; Masucci, L. Fecal Microbiota Transplantation and Other Gut Microbiota Manipulation Strategies. Microorganisms 2022, 10, 2424. [Google Scholar] [CrossRef]
- Ding, L.-N.; Ding, W.-Y.; Ning, J.; Wang, Y.; Yan, Y.; Wang, Z.-B. Effects of Probiotic Supplementation on Inflammatory Markers and Glucose Homeostasis in Adults With Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2021, 12, 770861. [Google Scholar] [CrossRef] [PubMed]
- Harshfield, E.L.; Sands, C.J.; Tuladhar, A.M.; de Leeuw, F.E.; Lewis, M.R.; Markus, H.S. Metabolomic Profiling in Small Vessel Disease Identifies Multiple Associations with Disease Severity. Brain 2022, 145, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
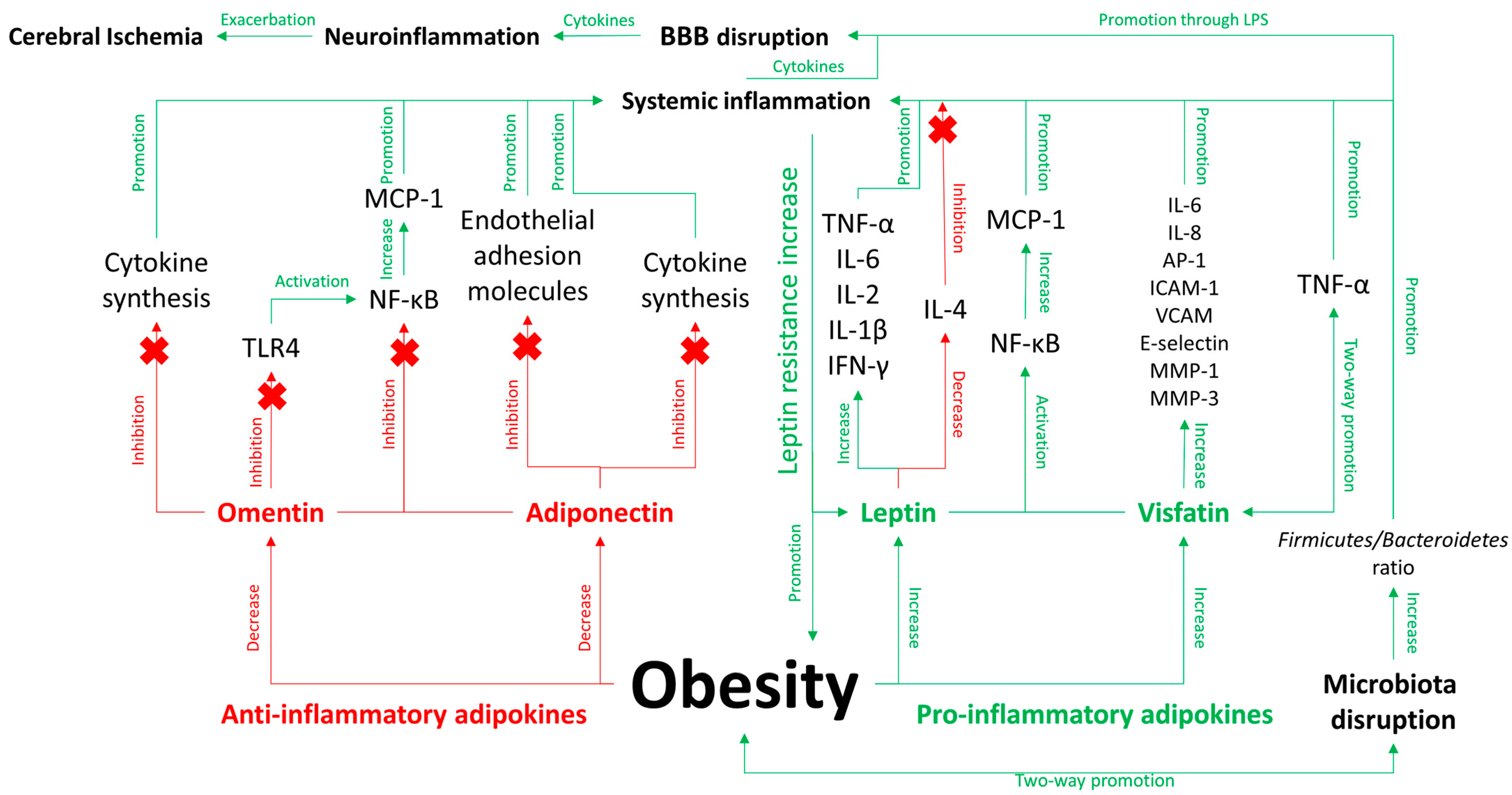
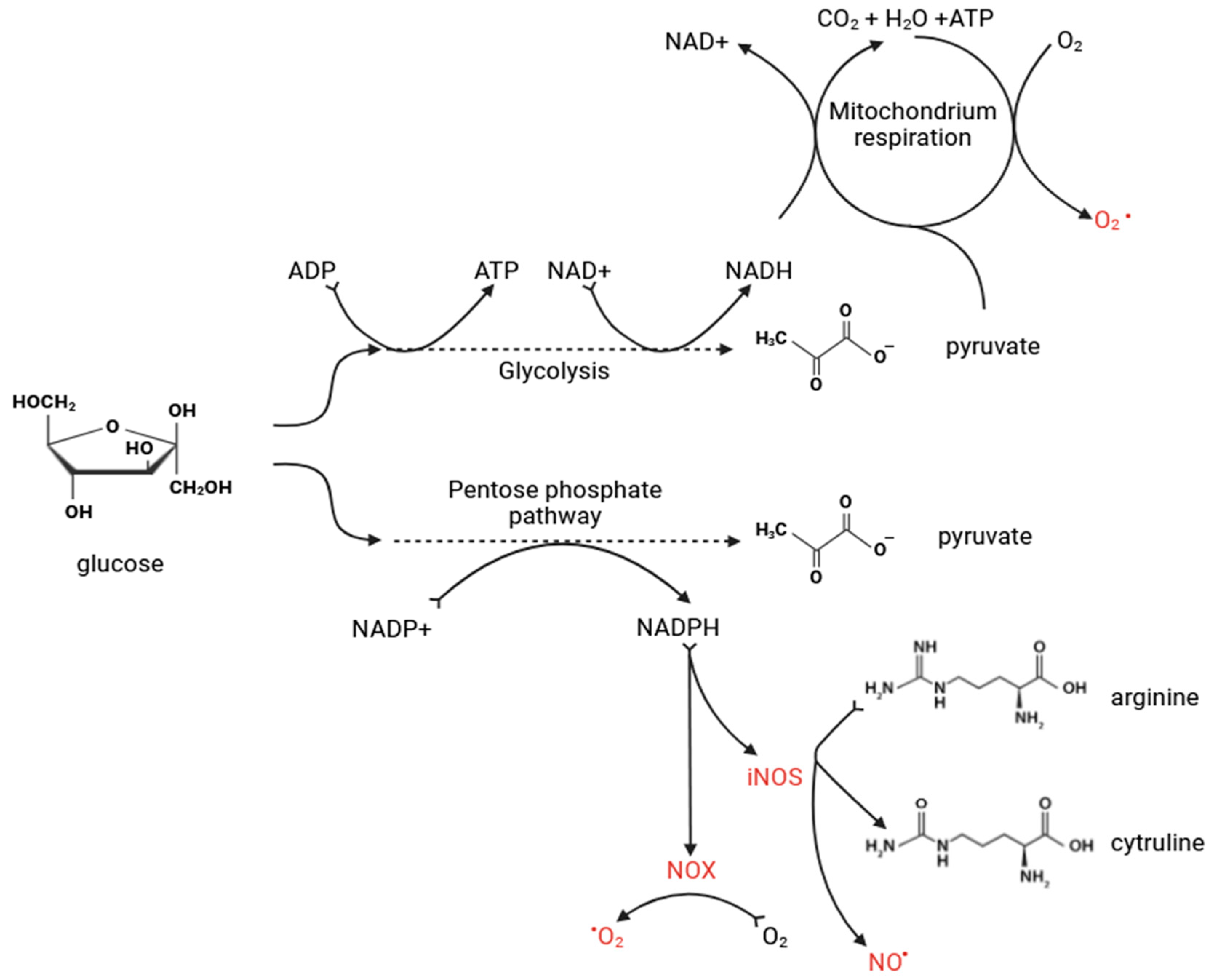
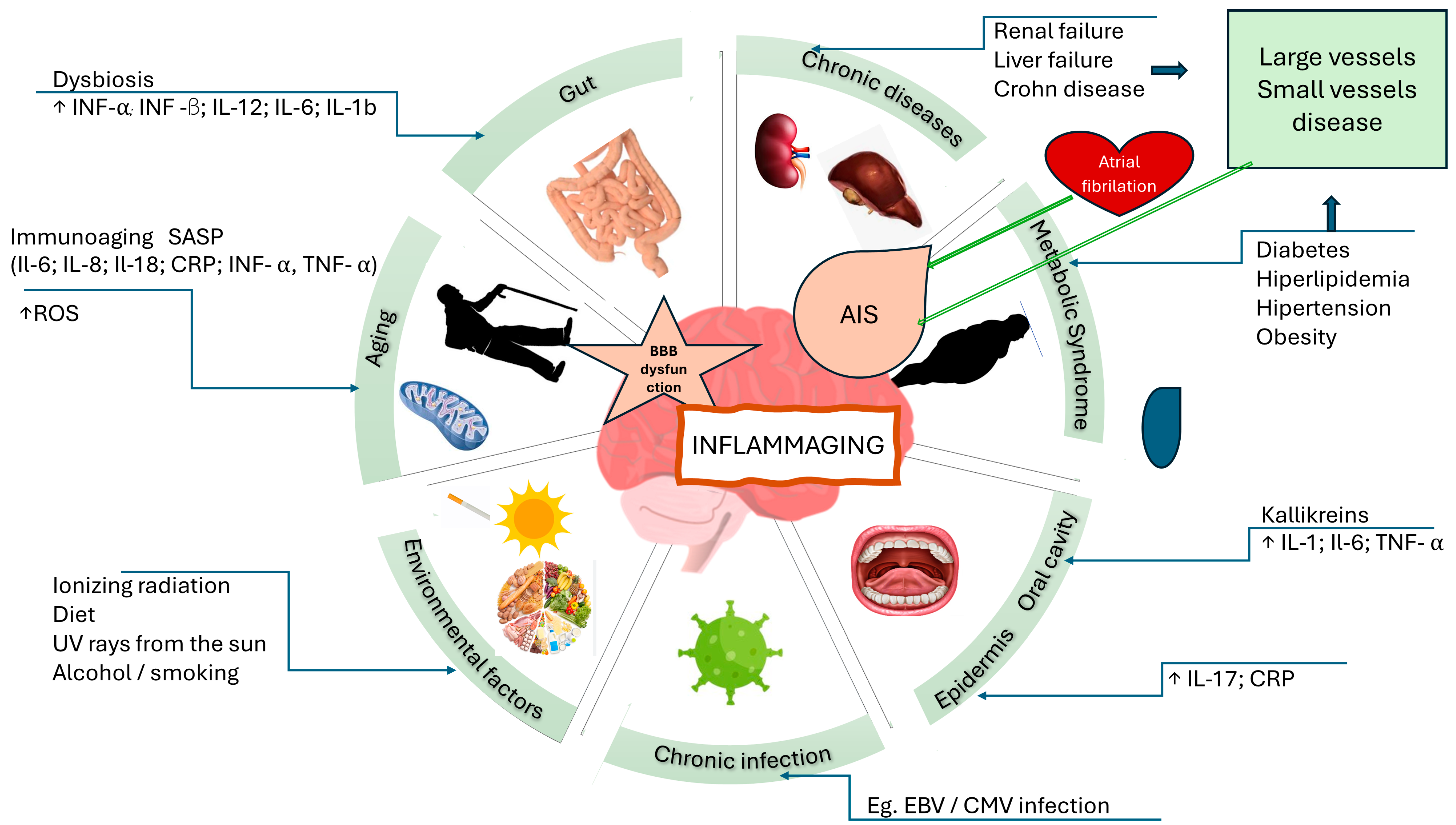

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Agent | Role of Agent in Acute Ischemic Stroke | Role of Agent in Microbiota Dysfunction | Role of Agent in Metabolic Disorders | References |
|---|---|---|---|---|
| Interleukin-1 alpha (IL-1α) | A strong activator of A1 astrocytes, which induce the death of neurons and oligodendrocytes. | - | - | [205,207] |
| Interleukin-1 beta (IL-1β) | Weakens tight junctions of the blood-brain barrier (BBB), causing it to unseal. | Studies showed that the microbiota can promote intestinal inflammation and pathology via NLRP3-mediated IL-1 signaling in mice. | Promotes inflammatory signals in surrounding cells, leading to adipose tissue inflammation. | [90,107,156,286] |
| Interleukin-2 (IL-2) | Secreted by activated microglia and TCD4+ cells in the acute phase of stroke, promoting inflammation. | Gut microbiota could modulate/mediate the efficacy of immunotherapies, which has been observed in cancer immunotherapies. | IL-2 is associated with insulin resistance as well as with several important inflammatory markers. In our obese cohort, two diverse clusters of IL-2 gene expression were identified, and in those with high expression, IL-2 gene expression correlated positively with fasting blood glucose (FBG) and glycated hemoglobin (HbA1c). | [155,287,288,289] |
| Interleukin-4 (IL-4) | Secreted by activated microglia and TCD4+ cells in the acute phase of stroke, promoting oligodendrogenesis. | In mice, extensive antibiotic treatment has caused elevation of IL-4 compared to those treated with probiotics. | Obese mice have lower levels of circulating IL-4; an 8-week IL-4 supplementation improves common phenotypes of obesity. | [155,289,290] |
| Interleukin-6 (IL-6) | Promotes proliferation of microglia and astrocytes, enhancing BBB permeability. | One of the most vital cytokines produced in response to inflammation. | Levels are elevated in obese patients and decrease after reduction in body mass. | [87,90,97,135,149,156,205,275,291,292] |
| Interleukin-8 (IL-8) | Mobilizes and activates neutrophils, causing their infiltration into the ischemic area, aggravating local inflammation, expanding ischemic lesions, and leading to severe morbidity and disability. | - | Diabetes mellitus is associated with higher levels of IL-8 compared to systemic healthy patients. | [87,155,293] |
| Interleukin-10 (IL-10) | A strong anti-inflammatory cytokine that plays a role in resolving inflammation. | IL-10 is emerging as a critical mediator of immunomodulation induced by microbiota, crucial for maintaining host-microbe homeostasis without triggering exaggerated detrimental inflammatory responses. | - | [87,206,294] |
| Interleukin-17 (IL-17) | - | Abrogation of the enteric IL-17RA signaling pathway led to commensal dysbiosis, increased serum GM-CSF concentration, and enhanced predisposition to neuroinflammation in mice. | Inhibits adipogenesis in mice. | [295,296] |
| Interleukin-18 (IL-18) | Highly expressed in atherosclerotic plaques, particularly in unstable plaques. | - | Elevated in subjects with metabolic syndrome and associated with its components, predicting cardiovascular events and mortality. | [156,297] |
| IFN-γ (Interferon gamma) | Secreted in the acute phase of stroke by Th1 lymphocytes. | Microbiota dysbiosis triggers the promotion of T-cell polarization into pro-inflammatory Th1 cells in the small intestine, migrating to the ischemic brain and exacerbating infarct damage. | - | [298,299] |
| TNF-α (Tumor Necrosis Factor alpha) | Secreted by activated microglia, forming neurotoxic astrocytes responsible for killing neural cells in neuroinflammatory settings. | A prominent pro-inflammatory agent that may contribute to septic shock due to extensive stimulation. | Exacerbates insulin resistance and metabolic disorders due to its pro-inflammatory properties. | [87,90,107,135,136,137,139,149,156,177,205,207] |
| MCP-1 (Monocyte Chemoattractant Protein-1) | Plays a critical role in attracting immune cells, especially macrophages, into adipose tissue, promoting the expression of pro-inflammatory proteins. | FMT from patients after bariatric surgery to recipients suffering from metabolic syndrome has been proven to decrease macrophage-attracting factor (MCP-1) in adipose tissue and plasma compared to FMT from donors with metabolic syndrome. | The circulating levels of MCP-1 and IL-8 in the serum were significantly (p < 0.05) higher in obese subjects (BMI > 30 kg/m2) compared with non-obese controls (BMI < 25 kg/m2). | [156,194,251,300] |
| NLRP3 | Plays a pivotal role in intimal inflammation driven by cholesterol accumulation and in atherosclerotic plaques. | Activated by bacterial components, such as LPS. | - | [252,253,301,302,303] |
| Adiponectin | Prevents formation of foam cells. | Reduces production of pro-inflammatory cytokines. | Secreted by adipose tissue, levels negatively correlated with body mass, and has insulin-sensitizing properties. | [151,152,155] |
| Leptin | Elevated levels are found in sclerotic plaques, associated with angiogenesis and inflammation, contributing to atherosclerotic plaque burden and hypertension. | Dysregulated leptin levels influence gut microbiota composition, leading to an imbalance that exacerbates systemic inflammation and metabolic dysfunction | Increased leptin from adipose tissue acts in the hypothalamus to inhibit appetite and increase metabolic rate, reducing adiposity. | [146,147,194] |
| Omentin | - | - | Possesses anti-inflammatory properties similar to adiponectin, involving the activation of endothelial nitric oxide synthesis, regulating blood pressure, and influencing blood vessel relaxation. | [155] |
| Resistin | - | - | Increased levels indicate development of insulin resistance, diabetes, obesity, and cardiovascular disease; may be an early biomarker for metabolic disorders. | [155] |
| CRP (C-reactive Protein) | Chlamydia pneumoniae is involved in forming atherosclerotic plaques. | Porphyromonas gingivalis, a well-known bacterium, causes systemic inflammation, raising CRP levels, which have pro-coagulation and pro-thrombotic effects. | Elevated CRP levels are often found in obese individuals and are associated with an increased risk of developing chronic diseases such as type 2 diabetes, hypertension, and cardiovascular disorders. | [72,73,74,75,76,136] |
| Low-density lipoprotein cholesterol (LDL-c) | Its oxidized form plays a pivotal role in intimal inflammation and atherosclerotic plaque formation. | Lactobacillus acidophilus reduced the levels of malondialdehyde (MDA), oxidized low-density lipoprotein (ox-LDL), and TNF-α in the serum profile of treated mice. | LDL is one of the most prominent biomarkers of metabolic disorders. | [291,292,304] |
| MicroRNAs (miR-137, miR-21, miR-34a, miR-146b-5p, miR-210) | Levels of these miRNAs are elevated in atherosclerotic arteries and could be used as biomarkers in assessing vascular risk. | No circulating miRNAs are validated as biomarkers for routine use in clinical practice; lack of significant comparative studies between miRNAs and common disease biomarkers and high detection costs are the main limitations. | An association between alteration of miRNA expression profiles and progression of GI tract diseases enables the opportunity to explore new biomarkers in stool samples in the future. | [303,305,306,307] |
| Plasma fibrinogen | Its levels can be used as a predictor of carotid atherosclerotic plaque, ischemic stroke, and coronary heart disease. | Dysbiosis in microflora contributes to low-grade inflammation, causing coagulation disturbances. | A significant positive correlation was observed between fibrinogen concentration and HbA1c; negative correlation between fibrinogen concentration and HDL levels. Levels of fibrinogen are higher in obese patients with type 2 diabetes than in obese subjects without type 2 diabetes. Plasma fibrinogen levels correlate with fasting insulin levels and disease state advancement in non-insulin-dependent diabetics. | [291,292,308,309,310] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuszkiewicz, J.; Kukulska-Pawluczuk, B.; Piec, K.; Jarek, D.J.; Motolko, K.; Szewczyk-Golec, K.; Woźniak, A. Intersecting Pathways: The Role of Metabolic Dysregulation, Gastrointestinal Microbiome, and Inflammation in Acute Ischemic Stroke Pathogenesis and Outcomes. J. Clin. Med. 2024, 13, 4258. https://doi.org/10.3390/jcm13144258
Nuszkiewicz J, Kukulska-Pawluczuk B, Piec K, Jarek DJ, Motolko K, Szewczyk-Golec K, Woźniak A. Intersecting Pathways: The Role of Metabolic Dysregulation, Gastrointestinal Microbiome, and Inflammation in Acute Ischemic Stroke Pathogenesis and Outcomes. Journal of Clinical Medicine. 2024; 13(14):4258. https://doi.org/10.3390/jcm13144258
Chicago/Turabian StyleNuszkiewicz, Jarosław, Beata Kukulska-Pawluczuk, Katarzyna Piec, Dorian Julian Jarek, Karina Motolko, Karolina Szewczyk-Golec, and Alina Woźniak. 2024. "Intersecting Pathways: The Role of Metabolic Dysregulation, Gastrointestinal Microbiome, and Inflammation in Acute Ischemic Stroke Pathogenesis and Outcomes" Journal of Clinical Medicine 13, no. 14: 4258. https://doi.org/10.3390/jcm13144258