
Phenotypic Plasticity and Cancer: A System Biology Perspective
 , ,
, ,  and
and 
{kind=link}
{kind=link}
Abstract
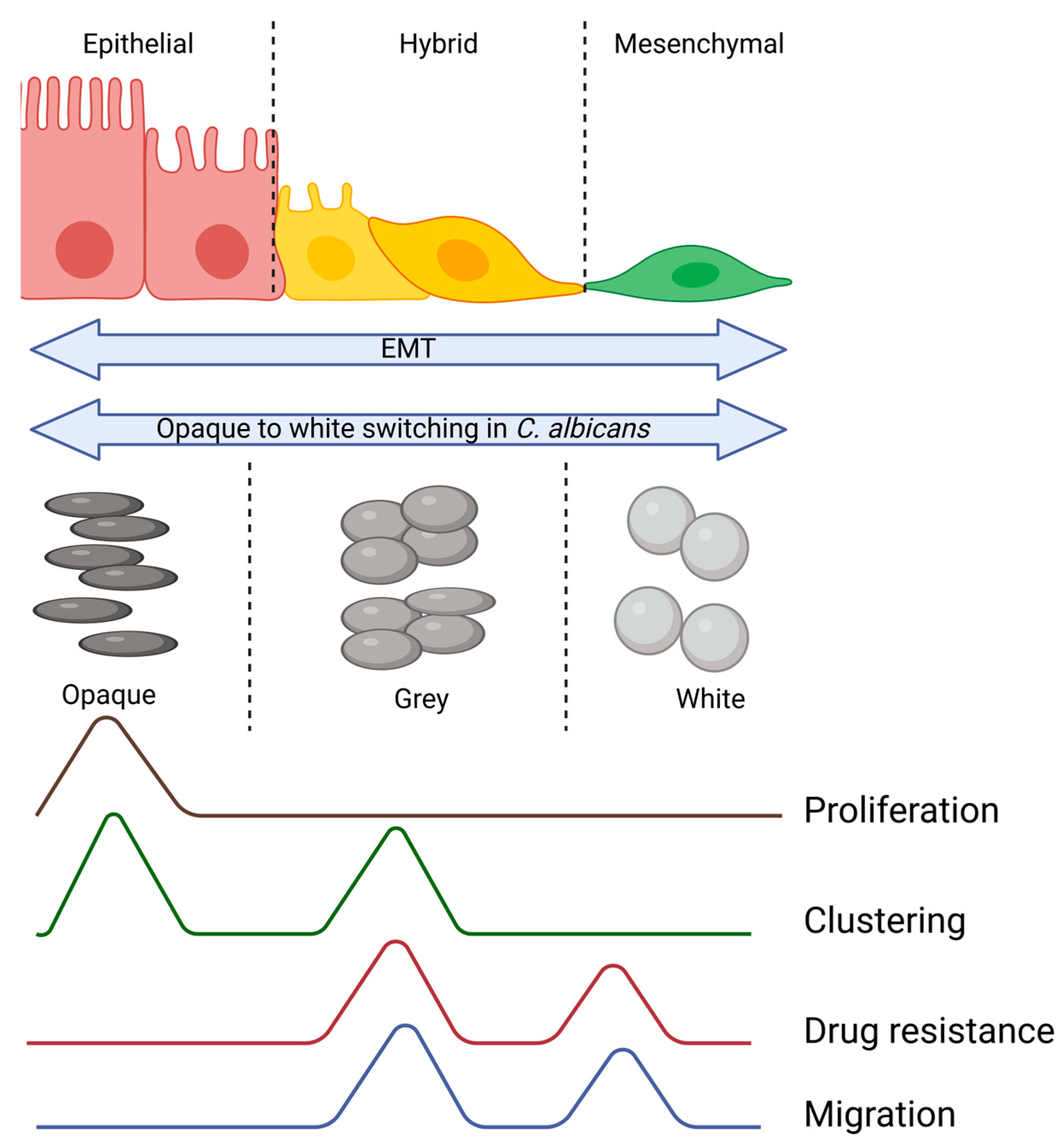
:1. Introduction
2. Hybrid E/M Cells in Cancer Metastasis
2.1. Hybrid E/M Cells in Solid Tumors
2.2. Hybrid E/M Cells in CTCs
2.3. Hybrid E/M Cells and Resistance to Cancer Therapy
3. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Poste, G.; Fidler, I.J. The Pathogenesis of Cancer Metastasis. Nature 1980, 283, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Kang, Y. Distinctive Properties of Metastasis-Initiating Cells. Genes. Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Somarelli, J.A.; Hanash, S.M.; Levine, H. Epithelial/Mesenchymal Plasticity: How Have Quantitative Mathematical Models Helped Improve Our Understanding? Mol. Oncol. 2017, 11, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Front. Oncol. 2018, 8, 325166. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Subbalakshmi, A.R.; Jolly, M.K. The Fundamentals of Phenotypic Plasticity. In Phenotypic Switching; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–21. [Google Scholar]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 1979, 2013, 342. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E.D. Epithelia Suspended in Collagen Gels Can Lose Polarity and Express Characteristics of Migrating Mesenchymal Cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef]
- Font-Clos, F.; Zapperi, S.; Porta, C.A.M. La Topography of Epithelial–Mesenchymal Plasticity. Proc. Natl. Acad. Sci. USA 2018, 115, 5902–5907. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the Tumour Transition States Occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Chakraborty, P.; Levine, H.; Jolly, M.K. A Mechanism for Epithelial-Mesenchymal Heterogeneity in a Population of Cancer Cells. PLoS Comput. Biol. 2020, 16, e1007619. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Meca-Cortés, Ó.; Mateo, F.; De Paz, A.M.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-Mesenchymal Transition Can Suppress Major Attributes of Human Epithelial Tumor-Initiating Cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Donaher, J.L.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-Β4 Identifies Cancer Stem Cell-Enriched Populations of Partially Mesenchymal Carcinoma Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid Epithelial/Mesenchymal Phenotype(s): The ‘Fittest’ for Metastasis? Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef]
- Stylianou, N.; Lehman, M.L.; Wang, C.; Fard, A.T.; Rockstroh, A.; Fazli, L.; Jovanovic, L.; Ward, M.; Sadowski, M.C.; Kashyap, A.S.; et al. A Molecular Portrait of Epithelial–Mesenchymal Plasticity in Prostate Cancer Associated with Clinical Outcome. Oncogene 2019, 38, 913–934. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; Fouquier d’Hérouël, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the Hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Bornes, L.; Belthier, G.; Van Rheenen, J. Clinical Medicine Epithelial-to-Mesenchymal Transition in the Light of Plasticity and Hybrid E/M States. J. Clin. Med. 2021, 10, 2403. [Google Scholar] [CrossRef]
- Sahoo, S.; Nayak, S.P.; Hari, K.; Purkait, P.; Mandal, S.; Kishore, A.; Levine, H.; Jolly, M.K. Immunosuppressive Traits of the Hybrid Epithelial/Mesenchymal Phenotype. Front. Immunol. 2021, 12, 5347. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, A.R.; Ashraf, B.; Jolly, M.K. Biophysical and Biochemical Attributes of Hybrid Epithelial/Mesenchymal Phenotypes. Phys. Biol. 2022, 19, 025001. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the Hybrid Epithelial/Mesenchymal Phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [PubMed]
- Revenu, C.; Gilmour, D. EMT 2.0: Shaping Epithelia through Collective Migration. Curr. Opin. Genet. Dev. 2009, 19, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-Mesenchymal Transition in Cancer: Parallels between Normal Development and Tumor Progression. J. Mammary Gland. Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, M.; Shaikh, A.; Lo, H.C.; Arpino, G.; De Placido, S.; Zhang, X.H.; Cristofanilli, M.; Schiff, R.; Trivedi, M.V. Perspective on Circulating Tumor Cell Clusters: Why It Takes a Village to Metastasize. Cancer Res. 2018, 78, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Joosse, S.A.; Gorges, T.M.; Pantel, K. Biology, Detection, and Clinical Implications of Circulating Tumor Cells. EMBO Mol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Toner, M.; Maheswaran, S.; Haber, D.A. En Route to Metastasis: Circulating Tumor Cell Clusters and Epithelial-to-Mesenchymal Transition. Trends Cancer 2015, 1, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Godin, L.; Balsat, C.; Van Eycke, Y.; Allard, J.; Royer, C.; Remmelink, M.; Pastushenko, I.; Haene, N.D.; Blanpain, C.; Salmon, I.; et al. A Novel Approach for Quantifying Cancer Cells Showing Hybrid Epithelial/Mesenchymal States in Large Series of Tissue Samples: Towards a New Prognostic Marker. Cancers 2020, 12, 906. [Google Scholar] [CrossRef]
- Liao, T.-T.; Yang, M.-H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef]
- Mahmoudabadi, G.; Rajagopalan, K.; Getzenberg, R.H.; Hannenhalli, S.; Rangarajan, G.; Kulkarni, P. Intrinsically Disordered Proteins and Conformational Noise: Implications in Cancer. Cell Cycle 2013, 12, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-Based Regulation of Epithelial–Hybrid–Mesenchymal Fate Determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Abdollahi, B.; Wilkins, O.M.; Lu, H.; Chakraborty, P.; Ognjenovic, N.B.; Muller, K.E.; Jolly, M.K.; Christensen, B.C.; Hassanpour, S.; et al. Phenotypic Heterogeneity Driven by Plasticity of the Intermediate EMT State Governs Disease Progression and Metastasis in Breast Cancer. Sci. Adv. 2022, 8, eabj8002. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. Epithelial-Mesenchymal Transitions: From Cell Plasticity to Concept Elasticity. In Current Topics in Developmental Biology; Academic Press Inc.: Cambridge, MA, USA, 2015; Volume 112, pp. 273–300. [Google Scholar]
- Watanabe, K.; Villarreal-Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary Morphogenesis and Regeneration Require the Inhibition of EMT at Terminal End Buds by Ovol2 Transcriptional Repressor. Dev. Cell 2014, 29, 59–74. [Google Scholar] [CrossRef]
- Subbalakshmi, A.R.; Kundnani, D.; Biswas, K.; Ghosh, A.; Hanash, S.M.; Tripathi, S.C.; Jolly, M.K. NFATc Acts as a Non-Canonical Phenotypic Stability Factor for a Hybrid Epithelial/Mesenchymal Phenotype. Front. Oncol. 2020, 10, 1794. [Google Scholar] [CrossRef]
- Subbalakshmi, A.R.; Sahoo, S.; Biswas, K.; Jolly, M.K. A Computational Systems Biology Approach Identifies SLUG as a Mediator of Partial Epithelial-Mesenchymal Transition (EMT). Cells Tissues Organs 2022, 211, 689–702. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a Hybrid E/M State Is Essential for Tumorigenicity of Basal Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Ren, Z.; Dharmaratne, M.; Liang, H.; Benard, O.; Morales-Gallego, M.; Suyama, K.; Kumar, V.; Fard, A.T.; Kulkarni, A.S.; Prystowsky, M.; et al. Redox Signalling Regulates Breast Cancer Metastasis via Phenotypic and Metabolic Reprogramming Due to P63 Activation by HIF1α. Br. J. Cancer 2024, 130, 908–924. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.; Centonze, G.; Murianni, F.; Orecchia, P.; Andriani, F.; Roato, I.; Gardelli, C.; Balsamo, M.; Moro, M.; Taiè, G.; et al. Hybrid Epithelial-Mesenchymal Status of Lung Cancer Dictates Metastatic Success through Differential Interaction with NK Cells. J. Immunother. Cancer 2024, 12, e007895. [Google Scholar] [CrossRef] [PubMed]
- Topel, H.; Bagirsakci, E.; Comez, D.; Bagci, G.; Cakan-akdogan, G.; Atabey, N. LncRNA HOTAIR Overexpression Induced Downregulation of C-Met Signaling Promotes Hybrid Epithelial/Mesenchymal Phenotype in Hepatocellular Carcinoma Cells. Cell Commun. Signal. 2020, 18, 110. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Fournier-Level, A.; Cooper, H.M.; Murray, M.J. Loss of Neogenin1 in Human Colorectal Carcinoma Cells Causes a Partial EMt and Wound-Healing Response. Sci. Rep. 2019, 9, 4110. [Google Scholar] [CrossRef]
- Saxena, K.; Subbalakshmi, A.R.; Jolly, M.K. Phenotypic Heterogeneity in Circulating Tumor Cells and Its Prognostic Value in Metastasis and Overall Survival. EBioMedicine 2019, 46, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating Tumor Cells from Patients with Advanced Prostate and Breast Cancer Display Both Epithelial and Mesenchymal Markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of Circulating Tumour Cells with a Hybrid (Epithelial/Mesenchymal) Phenotype in Patients with Metastatic Non-Small Cell Lung Cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Quan, Q.; Wang, X.; Lu, C.; Ma, W.; Wang, Y.; Xia, G.; Wang, C.; Yang, G. Cancer Stem-like Cells with Hybrid Epithelial/Mesenchymal Phenotype Leading the Collective Invasion. Cancer Sci. 2020, 111, 467–476. [Google Scholar] [CrossRef]
- Papadaki, M.A.; Stoupis, G.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Circulating Tumor Cells with Stemness and Epithelial-to-Mesenchymal Transition Features Are Chemoresistant and Predictive of Poor Outcome in Metastatic Breast Cancer. Mol. Cancer Ther. 2019, 18, 437–447. [Google Scholar] [CrossRef]
- Huangfu, Y.; Guo, J.; Zhao, Y.; Cao, X.; Han, L. Linking EMT Status of Circulating Tumor Cells to Clinical Outcomes in Lung Cancer. Cancer Manag. Res. 2024, 16, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wu, G.; Cheng, K.S.; Chen, A.; Neoh, K.H.; Chen, S.; Tang, Z.; Lee, P.F.; Dai, M.; Han, R.P.S. CTC Phenotyping for a Preoperative Assessment of Tumor Metastasis and Overall Survival of Pancreatic Ductal Adenocarcinoma Patients. EBioMedicine 2019, 46, 133–149. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, X.; Sun, H.; Fu, Y.; Tian, Y.; Yang, L.; Wang, J.; Xia, F. Association of Preoperative NANOG-Positive Circulating Tumor Cell Levels with Recurrence of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 601668. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Takahashi, H.; Ida, S.; Nagata, Y.; Chikamatsu, K. Epithelial-Mesenchymal Transition Status of Circulating Tumor Cells Is Associated with Tumor Relapse in Head and Neck Squamous Cell Carcinoma. Anticancer Res. 2020, 40, 3559–3564. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Tian, Q.; Teng, Q.; Wurpel, J.N.D.; Zeng, L.; Pan, Y.; Chen, Z. Understanding and Targeting Resistance Mechanisms in Cancer. MedComm 2023, 4, e265. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Furney, S.J.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-Genome Sequencing Reveals Complex Mechanisms of Intrinsic Resistance to BRAF Inhibition. Ann. Oncol. 2014, 25, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, L.; Neitzel, L.R.; Loganathan, S.N.; Tang, N.; Qin, L.; Crispi, E.E.; Guo, Y.; Knapp, S.; Beauchamp, R.D.; et al. The MAPK Pathway Regulates Intrinsic Resistance to BET Inhibitors in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Elkord, E. Acquired Resistance to Cancer Immunotherapy: Role of Tumor-Mediated Immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Mandalà, M.; Massi, D.; Taverna, D.; Tang, H.; Hemmings, B.A.; Xue, G. Acquired Resistance to Clinical Cancer Therapy: A Twist in Physiological Signaling. Physiol. Rev. 2016, 96, 805–829. [Google Scholar] [CrossRef]
- Wilson, C.; Nicholes, K.; Bustos, D.; Lin, E.; Song, Q.; Stephan, J.P.; Kirkpatrick, D.S.; Settleman, J. Overcoming EMT-Associated Resistance to Anti-Cancer Drugs via Src/FAK Pathway Inhibition. Oncotarget 2014, 5, 7328–7341. [Google Scholar] [CrossRef]
- Song, K.A.; Faber, A.C. Epithelial-to-Mesenchymal Transition and Drug Resistance: Transitioning Away from Death. J. Thorac. Dis. 2019, 11, E82–E85. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Corominas-Faja, B.; Vazquez-Martin, S.C.A.; Martin-Castillo, B.; Iglesias, J.M.; López-Bonet, E.; Martin, Á.G.; Menendez, J.A. Epithelial-to-Mesenchymal Transition (EMT) Confers Primary Resistance to Trastuzumab (Herceptin). Cell Cycle 2012, 11, 4020–4032. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.G.; Apoorva, D.; Chandrakala, M.; Snijesh, V.; Anupama, C.; Rajarajan, S.; Sahoo, S.; Mohan, G.; Jayakumar, V.S.; Ramesh, R.S.; et al. Acquisition of Hybrid E/M Phenotype Associated with Increased Migration, Drug Resistance and Stemness Is Mediated by Reduced MiR-18a Levels in ER-Negative Breast Cancer. bioRxiv 2022. preprint. [Google Scholar] [CrossRef]
- Nair, M.G.; Desai, K.; Prabhu, J.S.; Hari, P.S.; Remacle, J.; Sridhar, T.S. Β3 Integrin Promotes Chemoresistance to Epirubicin in MDA-MB-231 through Repression of the pro-Apoptotic Protein, BAD. Exp. Cell Res. 2016, 346, 137–145. [Google Scholar] [CrossRef]
- Zhang, X.; Powell, K.; Li, L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765. [Google Scholar] [CrossRef] [PubMed]
- Bontemps, I.; Lallemand, C.; Biard, D.; Dechamps, N.; Kortulewski, T.; Bourneuf, E.; Siberchicot, C.; Boussin, F.; Chevillard, S.; Campalans, A.; et al. Loss of CD24 Promotes Radiation and Chemo resistance by Inducing Stemness Properties Associated with a Hybrid E/M State in Breast Cancer Cells. Oncol. Rep. 2023, 49, 4. [Google Scholar] [CrossRef] [PubMed]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct Contributions of Partial and Full EMT to Breast Cancer Malignancy. Dev. Cell 2021, 56, 3203–3221.e11. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Steeg, P.S. Tumor Metastasis: Mechanistic Insights and Clinical Challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Pasani, S.; Sahoo, S.; Jolly, M.K. Hybrid E/M Phenotype(s) and Stemness: A Mechanistic Connection Embedded in Network Topology. J. Clin. Med. 2021, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Somarelli, J.A.; Shelter, S.; Jolly, M.K.; Wang, X.; Bartholf Dewitt, S.; Hish, A.J.; Gilja, S.; Eward, W.C.; Ware, K.E.; Levine, H.; et al. Mesenchymal-Epithelial Transition in Sarcomas Is Controlled by the Combinatorial Expression of MiR-200s and GRHL2. Mol. Cell Biol. 2016, 36, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Pickard, A.; Craig, S.G.; Quinn, G.P.; Lambe, S.M.; James, J.A.; McDade, S.S.; McCance, D.J. ΔNp63γ/SRC/Slug Signaling Axis Promotes Epithelial-to-Mesenchymal Transition in Squamous Cancers. Clin. Cancer Res. 2018, 24, 3917–3927. [Google Scholar] [CrossRef]
- Selvaggio, G.; Canato, S.; Pawar, A.; Monteiro, P.T.; Guerreiro, P.S.; Brás, M.M.; Janody, F.; Chaouiya, C. Hybrid Epithelial-Mesenchymal Phenotypes Are Controlled by Microenvironmental Factors. Cancer Res. 2020, 80, 2407–2420. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef]
- Mandal, M.; Ghosh, B.; Anura, A.; Mitra, P.; Pathak, T.; Chatterjee, J. Modeling Continuum of Epithelial Mesenchymal Transition Plasticity. Integr. Biol. 2016, 8, 167–176. [Google Scholar] [CrossRef]
- Tao, L.; Du, H.; Guan, G.; Dai, Y.; Nobile, C.J.; Liang, W.; Cao, C.; Zhang, Q.; Zhong, J.; Huang, G. Discovery of a “White-Gray-Opaque” Tristable Phenotypic Switching System in Candida albicans: Roles of Non-Genetic Diversity in Host Adaptation. PLoS Biol. 2014, 12, e1001830. [Google Scholar] [CrossRef]
- Hnisz, D.; Sehwarzmüller, T.; Kuchler, K. Transcriptional Loops Meet Chromatin: A Dual-Layer Network Controls White-Opaque Switching in Candida albicans. Mol. Microbiol. 2009, 74, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Srikantha, T.; Borneman, A.R.; Daniels, K.J.; Pujol, C.; Wu, W.; Seringhaus, M.R.; Gerstein, M.; Yi, S.; Snyder, M.; Soll, D.R. TOS9 Regulates White-Opaque Switching in Candida albicans. Eukaryot. Cell 2006, 5, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, B.; Staebell, M.; Anderson, J.; Risen, L.; Pfaller, M.; Soll, D.R. “White-Opaque Transition”: A Second High-Frequency Switching System in Candida albicans. J. Bacteriol. 1987, 169, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Hu, J.; Guan, G.; Tao, L.; Du, H.; Li, H.; Huang, G. Discovery of the Gray Phenotype and White-Gray-Opaque Tristable Phenotypic Transitions in Candida Dubliniensis. Virulence 2016, 7, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liao, H.; Pei, L.; Pu, Y. Combatting Persister Cells: The Daunting Task in Post-Antibiotics Era. Cell Insight 2023, 2, 100104. [Google Scholar] [CrossRef]
- Saini, S.; Pearl, J.A.; Rao, C.V. Role of FimW, FimY, and FimZ in Regulating the Expression of Type i Fimbriae in Salmonella Enterica Serovar Typhimurium. J. Bacteriol. 2009, 191, 3003–3010. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Ray, J.C.J.; Narula, J.; Igoshin, O.A. Bistable Responses in Bacterial Genetic Networks: Designs and Dynamical Consequences. Math. Biosci. 2011, 231, 76–89. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subbalakshmi, A.R.; Ramisetty, S.; Mohanty, A.; Pareek, S.; Do, D.; Shrestha, S.; Khan, A.; Talwar, N.; Tan, T.; Vishnubhotla, P.; et al. Phenotypic Plasticity and Cancer: A System Biology Perspective. J. Clin. Med. 2024, 13, 4302. https://doi.org/10.3390/jcm13154302
Subbalakshmi AR, Ramisetty S, Mohanty A, Pareek S, Do D, Shrestha S, Khan A, Talwar N, Tan T, Vishnubhotla P, et al. Phenotypic Plasticity and Cancer: A System Biology Perspective. Journal of Clinical Medicine. 2024; 13(15):4302. https://doi.org/10.3390/jcm13154302
Chicago/Turabian StyleSubbalakshmi, Ayalur Raghu, Sravani Ramisetty, Atish Mohanty, Siddhika Pareek, Dana Do, Sagun Shrestha, Ajaz Khan, Neel Talwar, Tingting Tan, Priya Vishnubhotla, and et al. 2024. "Phenotypic Plasticity and Cancer: A System Biology Perspective" Journal of Clinical Medicine 13, no. 15: 4302. https://doi.org/10.3390/jcm13154302