
Acute Pancreatitis in Individuals with Sickle Cell Disease: A Systematic Review
 , and
, and
Abstract
:1. Introduction
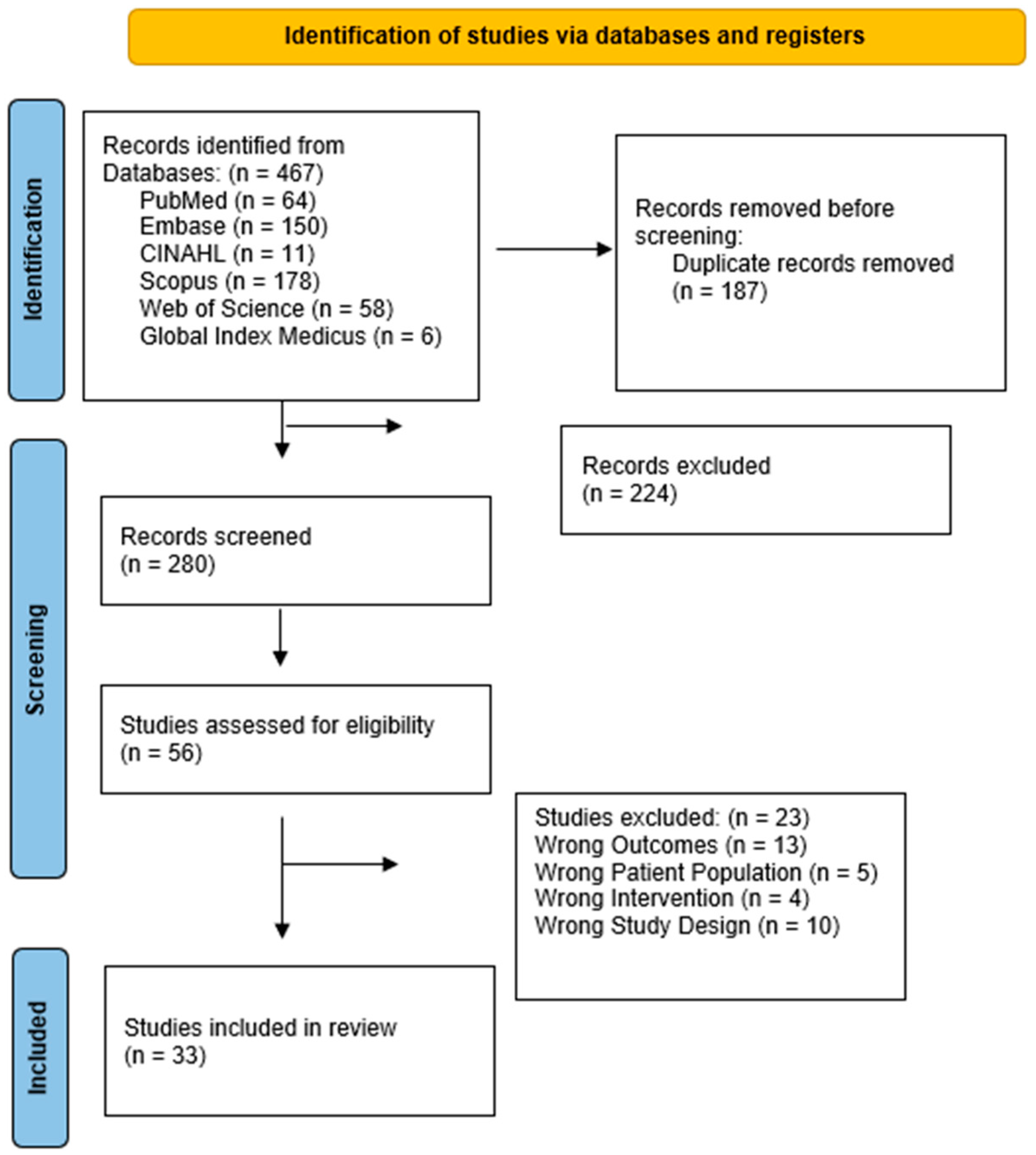
2. Materials and Methods
2.1. Types of Studies
2.2. Population
2.3. Exposure
2.4. Comparison
2.5. Outcomes
- AP prevalence (%) in SCD when compared to the general population.
- Length of hospital stay.
- Need for intensive care admission.
- Mortality.
- Presence of local pancreatic and peripancreatic complications (necrosis, hemorrhage, cysts).
- Need for procedural interventions during index admission.
2.6. Literature Search Including Data Extraction
2.7. Data Synthesis
2.8. Grading of Quality of Evidence
3. Results
3.1. Study Types
3.2. Characteristics of Included Studies
3.3. AP Presentation
3.4. Outcomes
3.4.1. AP Prevalence
3.4.2. AP Complications
3.5. AP Etiology
3.6. Risk of Bias Assessment and Grading of Quality Evidence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomson, A.M.; A McHugh, T.; Oron, A.P.; Teply, C.; Lonberg, N.; Tella, V.V.; Wilner, L.B.; Fuller, K.; Hagins, H.; Aboagye, R.G.; et al. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: A systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023, 10, e585–e599. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, D.; Agodoa, I.; Bhakta, N.; Danese, M.; Pappu, K.; Howard, R.; Gleeson, M.; Halperin, M.; Lanzkron, S. Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared with Those Without Sickle Cell Disease. JAMA Netw. Open 2019, 2, e1915374. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K.; Gupta, K.; Adams-Graves, P. Sickle cell pain: A critical reappraisal. Blood 2012, 120, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- McClish, D.K.; Smith, W.R.; Dahman, B.A.; Levenson, J.L.; Roberts, J.D.; Penberthy, L.T.; Aisiku, I.P.; Roseff, S.D.; Bovbjerg, V.E. Pain site frequency and location in sickle cell disease: The PiSCES project. Pain 2009, 145, 246–251. [Google Scholar] [CrossRef]
- Akingbola, T.S.; Kolude, B.; Aneni, E.C.; Raji, A.; Iwara, K.; Aken’ova, Y.; Soyannwo, O. Abdominal pain in adult sickle cell disease patients: A nigerian experience. Ann. Ib Postgrad. Med. 2011, 9, 100–104. [Google Scholar] [PubMed]
- Morinville, V.D.; Husain, S.Z.; Bai, H.; Barth, B.; Alhosh, R.; Durie, P.R.; Freedman, S.D.; Himes, R.; Lowe, M.E.; Pohl, J.; et al. Definitions of pediatric pancreatitis and survey of present clinical practices. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 261–265. [Google Scholar] [CrossRef]
- Tenner, S.; Baillie, J.; DeWitt, J.; Vege, S.S. American College of Gastroenterology guideline: Management of acute pancreatitis. Am. J. Gastroenterol. 2013, 108, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Krauss, J.S.; Freant, L.J.; Lee, J.R. Gastrointestinal pathology in sickle cell disease. Ann. Clin. Lab Sci. 1998, 28, 19–23. [Google Scholar]
- Walker, T.M.; Hambleton, I.R.; Serjeant, G.R. Gallstones in sickle cell disease: Observations from The Jamaican Cohort study. J. Pediatr. 2000, 136, 80–85. [Google Scholar] [CrossRef]
- Roberts, S.E.; Morrison-Rees, S.; John, A.; Williams, J.G.; Brown, T.H.; Samuel, D.G. The incidence and aetiology of acute pancreatitis across Europe. Pancreatology 2017, 17, 155–165. [Google Scholar] [CrossRef]
- Smolka, V.; Rohanova, M.; Seda, M.; Karaskova, E.; Tkachyk, O.; Zapalka, M.; Volejnikova, J. Etiology and classification of acute pancreatitis in children admitted to ICU using the Pediatric Sequential Organ Failure Assessment (pSOFA) score. Hepatobiliary Pancreat. Dis. Int. 2023, 22, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Gao, C.-F.; Wei, D.; Wang, C.; Ding, S.-Q. Acute pancreatitis: Etiology and common pathogenesis. World J. Gastroenterol. 2009, 15, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Dike, C.R.; Fittro, S.; Oster, R.A.; Morrow, C.D.; Brandow, A.; Demark-Wahnefried, W.; Lebensburger, J. Gastrointestinal symptoms, diagnostic evaluations, and abdominal pathology in children with sickle cell disease. Pediatr. Blood Cancer 2023, 70, e30699. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1. The Cochrane Collaboration. 2008. Available online: http://www.cochrane-handbook.org (accessed on 10 May 2023).
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Basit, H.; Ruan, G.J.; Mukherjee, S. Ranson Criteria. In StatPearls; StatPearls Publishing LLC: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Osvaldt, A.B.; Viero, P.; Borges da Costa, M.S.; Wendt, L.R.; Bersch, V.P.; Rohde, L. Evaluation of Ranson, Glasgow, APACHE-II, and APACHE-O criteria to predict severity in acute biliary pancreatitis. Int. Surg. 2001, 86, 158–161. [Google Scholar] [PubMed]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Abu-El-Haija, M.; Kumar, S.; Szabo, F.; Werlin, S.; Conwell, D.; Banks, P.; Morinville, V.D.; NASPGHAN Pancreas Committee. Classification of Acute Pancreatitis in the Pediatric Population: Clinical Report From the NASPGHAN Pancreas Committee. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Covidence Systematic Review Software VHI, Melbourne, Australia. Available online: www.covidence.org (accessed on 10 May 2023).
- Granholm, A.; Alhazzani, W.; Møller, M.H. Use of the GRADE approach in systematic reviews and guidelines. Br. J. Anaesth. 2019, 123, 554–559. [Google Scholar] [CrossRef]
- Nourallah, H.; Al-Salem, A.H. Diagnostic and Therapeutic ERCP in Children with Sickle Cell Disease. Pediatr. Endosurg. Innov. Tech. 1998, 2, 123–128. [Google Scholar] [CrossRef]
- Ahmed, S.; Siddiqui, A.K.; Siddiqui, R.K.; Kimpo, M.; Russo, L.; Mattana, J. Acute pancreatitis during sickle cell vaso-occlusive painful crisis. Am. J. Hematol. 2003, 73, 190–193. [Google Scholar] [CrossRef]
- Al Hindi, S.; Khalaf, Z.; Nazzal, K.; Nazzal, O.; Ahmed, A.; Alshaibani, L. Acute Pancreatitis in Children: The Clinical Profile at a Tertiary Hospital. Cureus 2021, 13, e14871. [Google Scholar] [CrossRef] [PubMed]
- Al-Salem, A.H.; Nourallah, H. Sequential endoscopic/laparoscopic management of cholelithiasis and choledocholithiasis in children who have sickle cell disease. J. Pediatr. Surg. 1997, 32, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Almudaibigh, A.; Alkasim, F.; Ghareeb, E. Prevalence and outcome of cholelithiasis in children with sickle cell disease at King Saud Medical City, Saudi Arabia. Article. J. Appl. Hematol. 2021, 12, 203. [Google Scholar] [CrossRef]
- Alwabari, A.; Parida, L.; Al-Salem, A.H. Laparoscopic splenectomy and/or cholecystectomy for children with sickle cell disease. Pediatr. Surg. Int. 2009, 25, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Amoako, M.O.; Casella, J.F.; Strouse, J.J. High rates of recurrent biliary tract obstruction in children with sickle cell disease. Pediatr. Blood Cancer 2013, 60, 650–652. [Google Scholar] [CrossRef]
- Badurdeen, D.; Alkhalloufi, K.; Kibreab, A.; Begum, R. Acute Pancreatitis and Sickle Cell Hepatopathy in a Pregnant Patient: 1053. Off. J. Am. Coll. Gastroenterol. 2014, 109, S313. [Google Scholar] [CrossRef]
- Banza, M.I.; Mulefu, J.P.; Lire, L.I.; N’Dwala, Y.T.B.; Tshiamala, I.B.; Cabala, V.P.K. Digestives diseases associated to sickle cell anemia in Lubumbashi: Epidemiological and clinical aspects. Pan Afr. Med J. 2019, 33, 253. [Google Scholar] [CrossRef]
- Barkin, R.L.; Richtsmeier, A.J. Alternative Agents in Pharmacological Management of Sickle Cell Pain Crisis Complicated by Acute Pancreatitis. Am. J. Ther. 1995, 2, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Bogue, C.O.; Murphy, A.J.; Gerstle, J.T.; Moineddin, R.; Daneman, A. Risk factors, complications, and outcomes of gallstones in children: A single-center review. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 303–308. [Google Scholar] [CrossRef]
- Coats, T.; Gardner, K.; Thein, S.L. Gallstones in Sickle Cell Disease: A Single Institution Experience. Blood 2014, 124, 4939. [Google Scholar] [CrossRef]
- Ehab Hanafy, F.A.; Alotaibi, N.; Albalawi, M.; Abuharfil, D.; Altoonisi, M.; Mahmoud, G. Acute pancreatitis in a child with sickle cell disease: A case report. Med. Sci. 2020, 24, 848–854. [Google Scholar]
- Gale, H.I.; Setty, B.N.; Sprinz, P.G.; Doros, G.; Williams, D.D.; Morrison, T.C.; Kalajian, T.A.; Tu, P.; Mundluru, S.N.; Mehta, M.N.; et al. Implications of radiologic-pathologic correlation for gallbladder disease in children and young adults with sickle cell disease. Emerg. Radiol. 2015, 22, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Garikapati, A.; Kumar, S.; Chaturvedi, A.; Bagga, C. Pseudocyst of Pancreas, Rare Crisis in Sickle Cell Disease. J. Evol. Med Dent. Sci. 2020, 9, 2072–2073. [Google Scholar] [CrossRef]
- Hanafy, E.; Altoonisi, M.; Alghuraydh, A.J.; Alatawi, A.; Alsabah, B.; Alzahrani, S.; Attili, I.; Mahmoud, G. Characteristics and Outcomes of Patients with Sickle Cell Disease Admitted to Pediatric Intensive Care: A Retrospective Review. J. Appl. Hematol. 2020, 11, 68. [Google Scholar] [CrossRef]
- Hussain, W.O.N.; Foy, I.; Raslich, M. Acute pancreatitis in sickle cell vaso-occlusive crisis. Consultant 2019, 59, 127–128. [Google Scholar]
- Jasti, S.; Williams, V.; Vamadevan, A.; Gillette, P.; Gress, F. Prevalance of Acute Pancreatitis in Sickle Cell Disease: 236. Off. J. Am. Coll. Gastroenterol. 2008, 103, S91–S92. [Google Scholar] [CrossRef]
- Kumar, A.; Posner, G.; Marsh, F.; Bellvue, R.; Dosik, H. Acute pancreatitis in sickle cell crisis. J. Natl. Med. Assoc. 1989, 81, 91–92. [Google Scholar]
- Kumar, M.C.K.; Thomas, S. Acute pancreatitis in a child with sickle cell anemia. Int. J. Contemp. Pediatr. 2020, 7, 5. [Google Scholar]
- Mehrabani, S.; Tammadoni, A.; Osia, S. Abdominal pain in a patient with sickle cell disease with multiple complications. Turk. Arch. Pediatr. 2019, 54, 267–271. [Google Scholar] [CrossRef]
- Moori, P.L.; Dosis, A.; Ahmad, Z.; Kausar, A.; Triantafyllopoulou, D. Acute Pancreatitis as a Complication of Sickle Cell Anaemia. Reports 2018, 1, 19. [Google Scholar] [CrossRef]
- Pasquier, J.M.; Bertrand, Y.; Tran-Minh, V.A.; Louis, D.; Faure, C.; Bussillet, A.; Clerc, M.; Philippe, N. Iconographic rubric. Acute lithiasic pancreatitis, exceptional cause of abdominal pain crisis in sickle cell anemia patient. Arch. Fr. De Pediatr. 1991, 48, 353–354. [Google Scholar]
- Popat, N.; Kumar, S.; Unadkat, B.S. Acute Cholelithiasis with Acute Pancreatic Calcifications: A Unique Presentation of Sickle Cell Crisis. Cureus 2022, 14, e30272. [Google Scholar] [CrossRef] [PubMed]
- Sack, J.; McCarty, T.R. Consideration of Vaso-occulsive Pancreatitis in a Patient with Sickle Cell Pain Crisis: 1199. Off. J. Am. Coll. Gastroenterol. 2016, 111, S524–S525. [Google Scholar] [CrossRef]
- Sakhalkar, V.M.S.; Rao, S.; Sakhalkar, M.; Rabinowitz, S. Pancreatitis in Children with Sickle Cell Disease. Pediatr. Res. 2004, 55, 307A. [Google Scholar]
- Shah, S.S.S.; Shah, M. Acute Pancreatitis in Sickle Cell Crisis. Am. J. Gastroenterol. 2013, 108, S69. [Google Scholar] [CrossRef]
- Sharma, A.; Khadka, B.; Sharma, A.; Shah, K.B.; Shrestha, A.N. Recurrent acute pancreatitis in an adult female with sickle cell disease: A case report. Ann. Med. Surg. 2023, 85, 37–40. [Google Scholar] [CrossRef]
- Sharma, P.; Hundt, M.; Aguilar, R.; Nader, M.; Yadav, S.; Rawlings, R.; Feuerstadt, P. Annual Hospitalization and Mortality Trends Among Patients With Sickle Cell Disease and Clostridioides difficile Infection: 197. Am. J. Gastroenterol. 2019, 114, S120–S121. [Google Scholar] [CrossRef]
- Sheehan, A.G.; Machida, H.; Butzner, J.D. Acute pancreatitis in a child with sickle cell anemia. J. Natl. Med. Assoc. 1993, 85, 70–72. [Google Scholar]
- Turnbull, T.L.; Houston, P.M.; Batalis, N.I. A Fatal Case of Necrotizing Pancreatitis in Sickle Cell Beta Thalassemia Zero. Am. Surg. 2018, 84, e401–e402. [Google Scholar] [CrossRef]
- Vicari, P.; Gil, M.V.; Cavalheiro Rde, C.; Figueiredo, M.S. Multiple primary choledocholithiasis in sickle cell disease. Intern. Med. 2008, 47, 2169–2170. [Google Scholar] [CrossRef]
- Ziegler, D.W.; Long, J.A.; Philippart, A.I.; Klein, M.D. Pancreatitis in childhood. Experience with 49 patients. Ann. Surg. 1988, 207, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Sakhalkar, V.S.; Miller, S.T.; Rao, S.P.; Soto, M.B.; Paulet, F.; Ramenofsky, M.L.; Gilchrist, B.F.; Bajaj, R.; Rabinowitz, S.S. Pancreatitis in Children with Sickle Cell Disease (SCD). J. Pediatr. Hematol./Oncol. 2004, 22, 371. [Google Scholar] [CrossRef]
- Lankisch, P.G.; Assmus, C.; Pflichthofer, D.; Struckmann, K.; Lehnick, D. Which etiology causes the most severe acute pancreatitis? J. Gastrointest. Cancer 1999, 26, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Frey, C.F.; Zhou, H.; Harvey, D.J.; White, R.H. The incidence and case-fatality rates of acute biliary, alcoholic, and idiopathic pancreatitis in California, 1994–2001. Pancreas 2006, 33, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Agawu, A.; Shults, J.; Smith-Whitley, K.; Feudtner, C. Age- and sex-specific rates of gall bladder disease in children with sickle cell disease. Pediatr. Blood Cancer 2022, 69, e29863. [Google Scholar] [CrossRef]

{kind=link}
| Population: Adult and Pediatric Exposure: Sickle Cell Disease Comparison: No Sickle Cell Disease | |||
|---|---|---|---|
| Outcome | Number of Studies and Effect Size | Certainty of Evidence | Comments |
| Acute Pancreatitis Prevalence | Studies: 2 Reported Prevalence: 2% and 7% | Very Low | Two of the included studies were small and had poor quality based on the Ottawa Scale. No meta-analysis was conducted due to lack of homogenous studies. There was no comparison group. |
| Study | Rating Based on Ottawa Scale | Comments |
|---|---|---|
| Jasti 2008 [39] | Poor | The study population might not have been representative because the patients were recruited from a single institution. There was no comparison group. SCD, however, was defined with electrophoresis and the definition of outcome (AP) was clearly described, but it was not a blinded assessment. |
| Sakhalkar 2004 [47] | Poor | The study population might not have been representative because the patients were recruited from a single institution. There was no comparison group. SCD was defined, and specific genotypes were noted. The definition of outcome (AP) was not clearly described, and it was not a blinded assessment. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dike, C.R.; DadeMatthews, A.; DadeMatthews, O.; Abu-El-Haija, M.; Lebensburger, J.; Smith, A.; Imdad, A. Acute Pancreatitis in Individuals with Sickle Cell Disease: A Systematic Review. J. Clin. Med. 2024, 13, 4712. https://doi.org/10.3390/jcm13164712
Dike CR, DadeMatthews A, DadeMatthews O, Abu-El-Haija M, Lebensburger J, Smith A, Imdad A. Acute Pancreatitis in Individuals with Sickle Cell Disease: A Systematic Review. Journal of Clinical Medicine. 2024; 13(16):4712. https://doi.org/10.3390/jcm13164712
Chicago/Turabian StyleDike, Chinenye R., Adefunke DadeMatthews, Oluwagbemiga DadeMatthews, Maisam Abu-El-Haija, Jeffrey Lebensburger, Abigail Smith, and Aamer Imdad. 2024. "Acute Pancreatitis in Individuals with Sickle Cell Disease: A Systematic Review" Journal of Clinical Medicine 13, no. 16: 4712. https://doi.org/10.3390/jcm13164712