

Clinical History of Patients with Hypertrophic Cardiomyopathy—How to Improve the Initiation Process of the Diagnosis?

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
- type of referral to the hospital:
- -
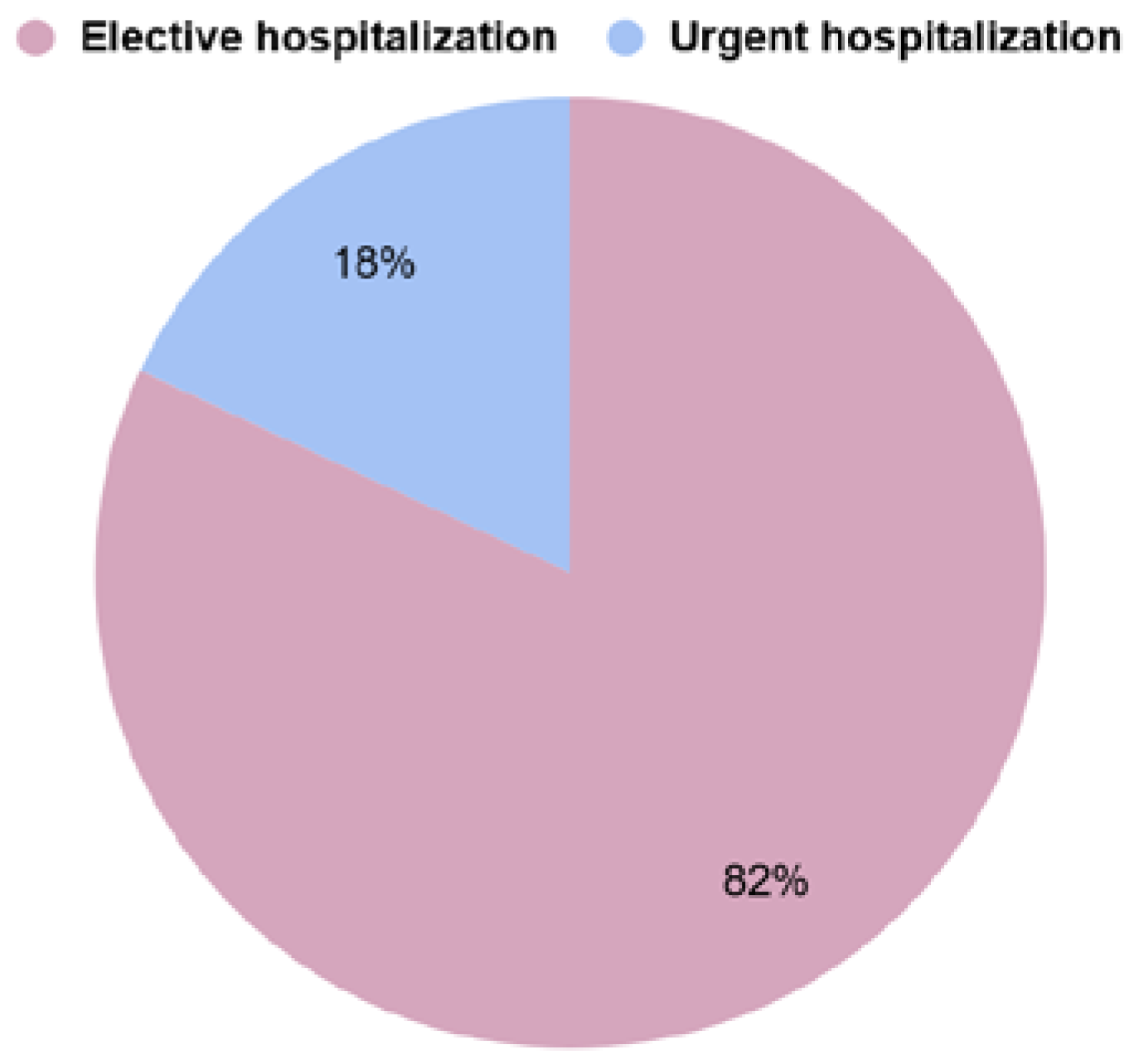
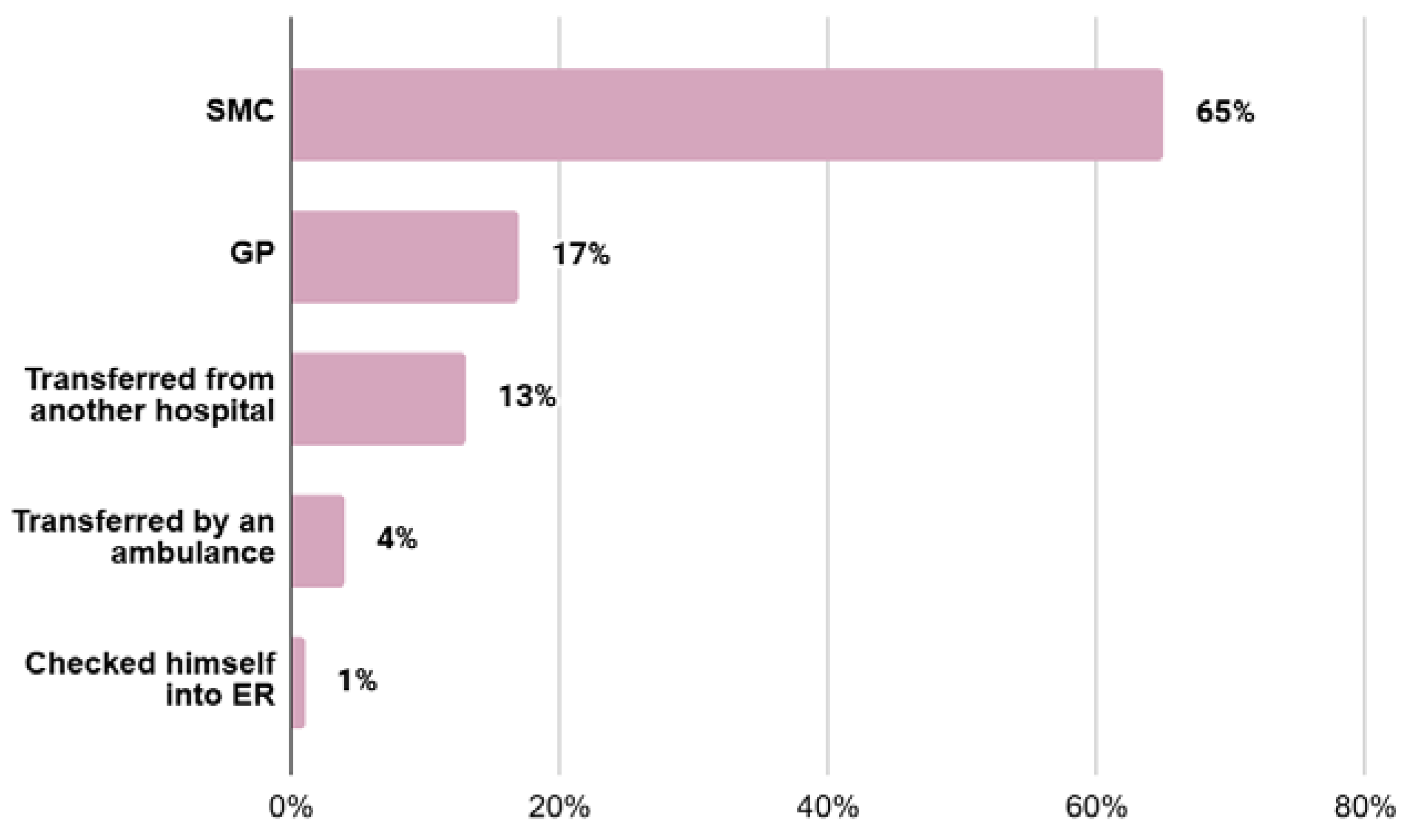
- elective hospitalization (EH)—referrals made by first-line outpatient health care personnel (GP) or referrals from specialised outpatient medical care (SMC) personnel
- -
- urgent hospitalization (UH)—self-referral to the hospital emergency department, transfer from an ambulance to the hospital emergency department, or transfer of the patient from another facility for further diagnosis;
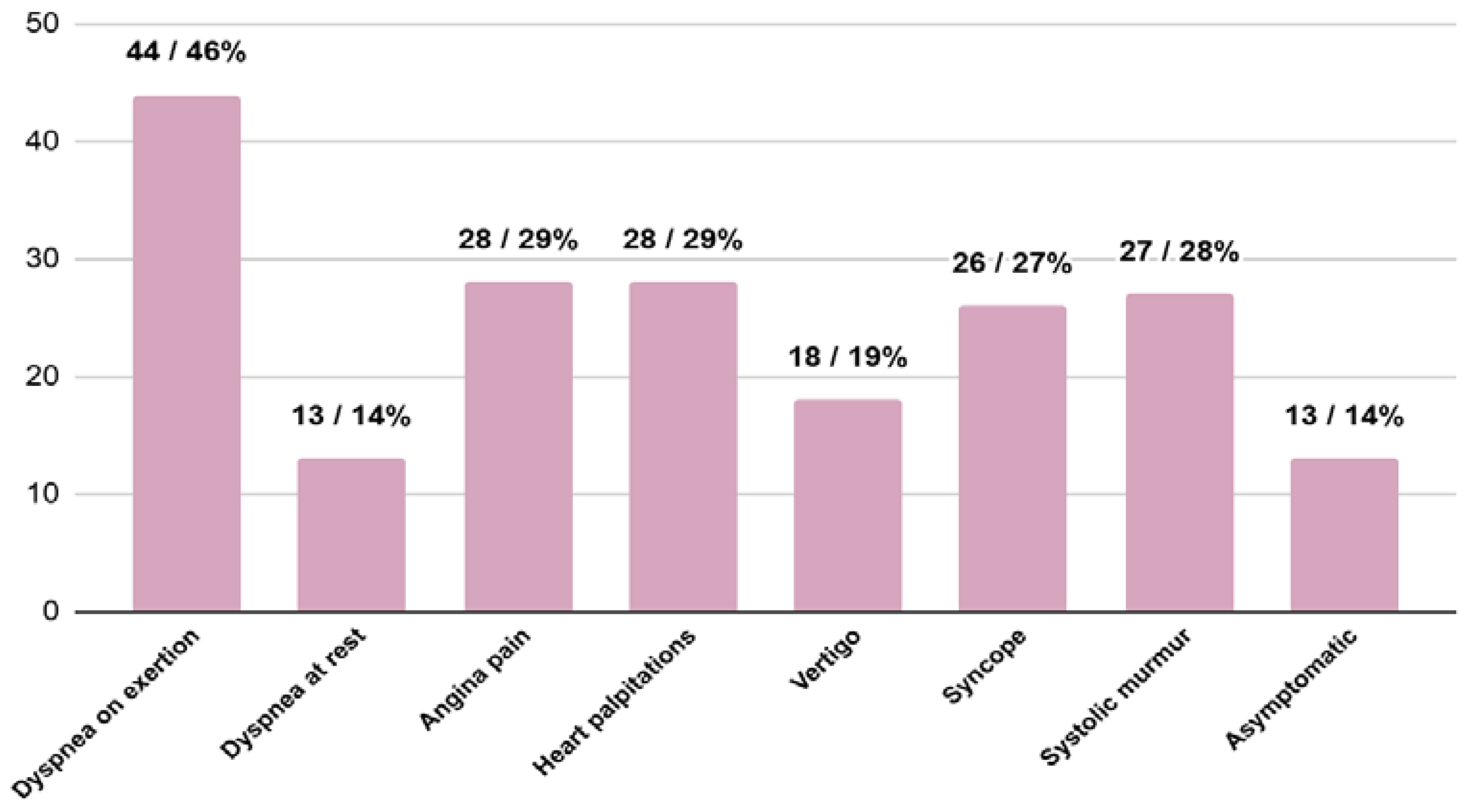
- accompanying signs and symptoms: systolic murmur, dyspnea at rest, dyspnea on exertion, angina pain, palpitations, syncope and vertigo;
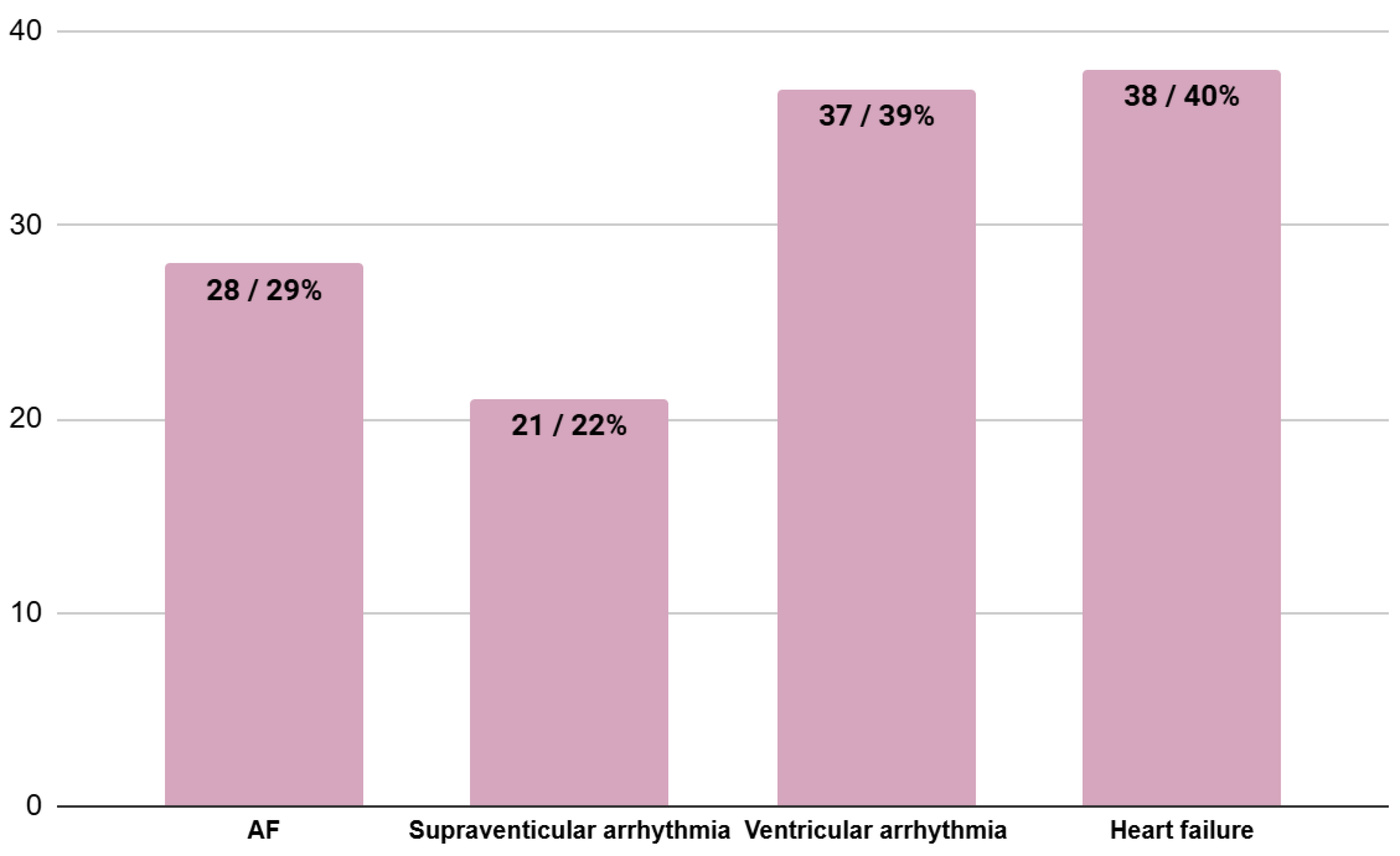
- coexisting heart involvement at the moment of HCM diagnosis (in accordance with patients’ hospital and outpatient records): atrial fibrillation (AF)—any form of AF, supraventricular arrhythmias other than AF, ventricular arrhythmias and heart failure;
- comorbidities (in accordance with patients’ hospital and outpatient records): hypertension, diabetes, prediabetes, lipid metabolism disorders, coronary artery disease, obesity, chronic obstructive pulmonary disease (COPD), chronic kidney disease (CKD) (eGFR < 60 mL/min/m2) and gout;
- family history (in accordance with patients’ hospital and outpatient records): family history of HCM and sudden cardiac death in the family members.
3. Results
3.1. Type of Referral to the Hospital
3.2. Signs and Symptoms at the Moment of HCM Diagnosis
3.3. Coexisting Heart Involvement at the Moment of HCM Diagnosis
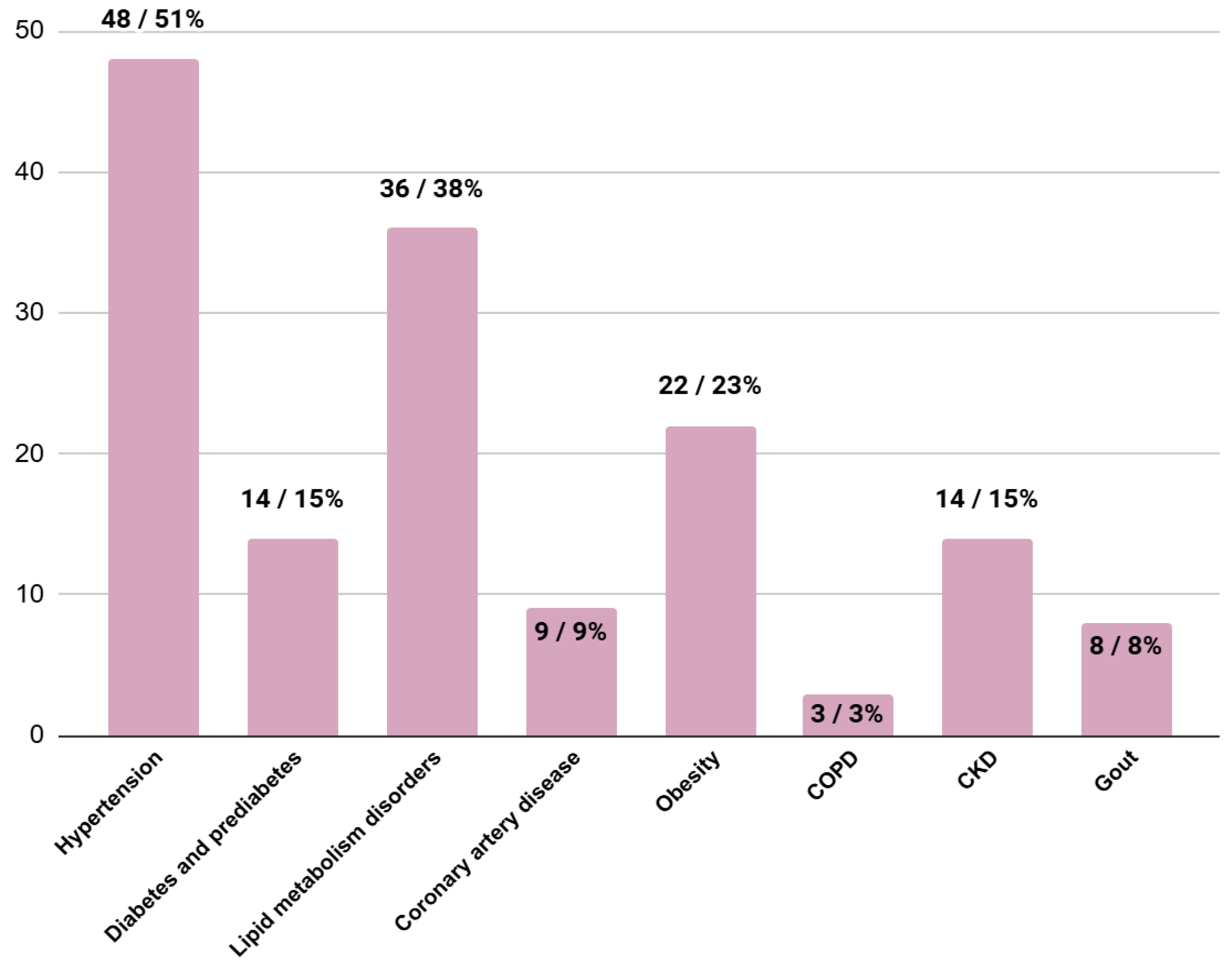
3.4. Comorbidities at the Moment of HCM Diagnosis
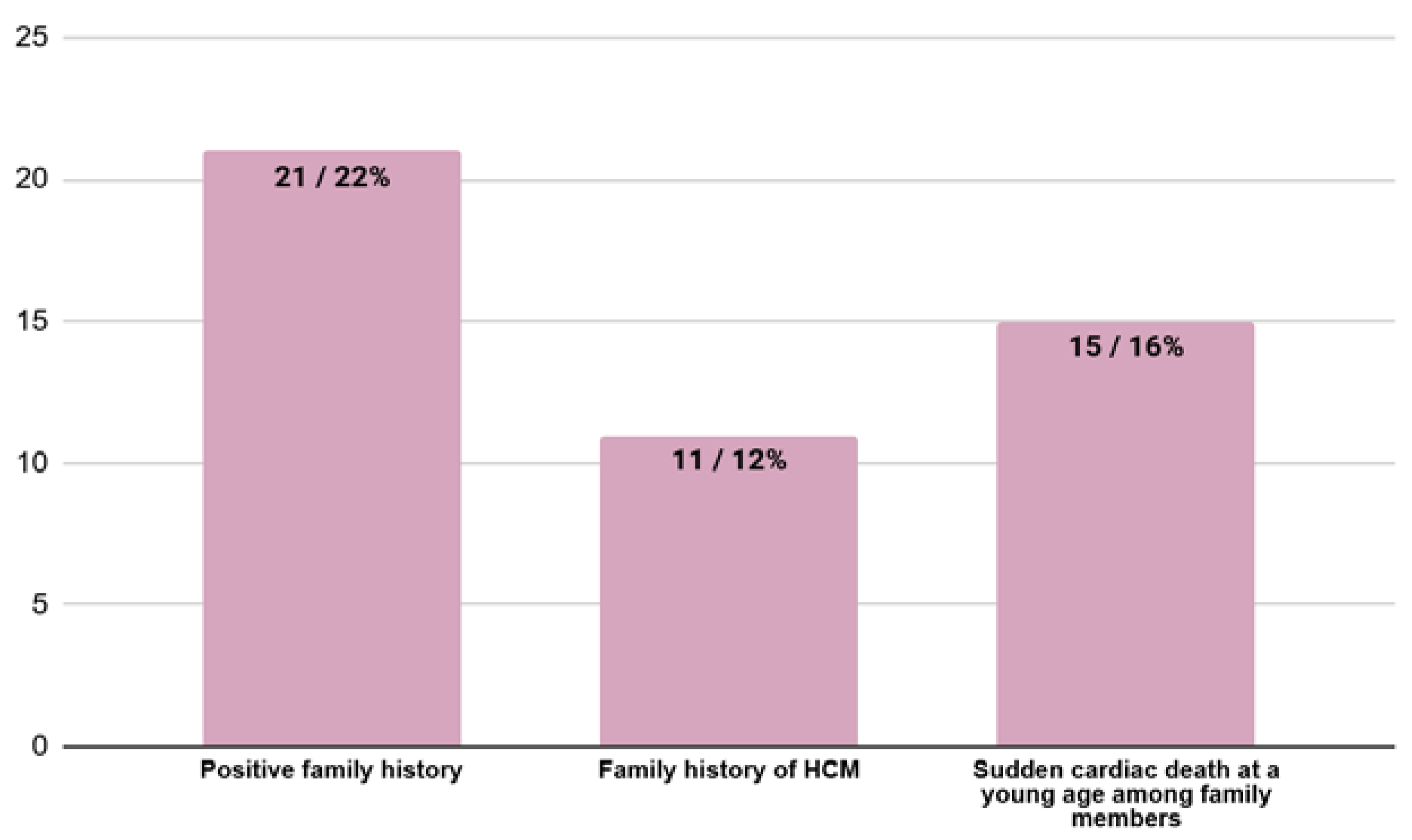
3.5. Family History at the Moment of HCM Diagnosis
3.6. Presence of LVOTO at the Moment of HCM Diagnosis
3.7. Subgroup Characteristics and Analysis—EHs vs. UHs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antunes, M.O.; Scudeler, T.L. Hypertrophic cardiomyopathy. Int. J. Cardiol. Heart Vasc. 2020, 27, 100503. [Google Scholar] [CrossRef] [PubMed]
- Basit, H.; Brito, D.; Sharma, S. Hypertrophic Cardiomyopathy. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar] [PubMed]
- Pellnitz, C.; Geier, C.; Perrot, A.; Dietz, R.; Osterziel, K.J.; Haverkamp, W. Der plötzliche Herztod bei familiärer hypertropher Kardiomyopathie. Identifizierung von Hochrisikopatienten. Dtsch. Med. Wochenschr. 2005, 130, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Geske, J.B.; Ommen, S.R.; Gersh, B.J. Hypertrophic Cardiomyopathy: Clinical Update. JACC Heart Fail. 2018, 6, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, A.D.; Goldman, M.E. Management of hypertrophic cardiomyopathy. Ann. Glob. Health 2014, 80, 35–45. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef]
- Mizia-Stec, K.; Grzybowski, J.; Cegłowska, U.; Wiśniewska, A.; Hałgas, K.; Wybraniec, M.; Pachciński, O.; Stec, M.; Cieśla, D.; Gąsior, M.; et al. Treatment pathways defined as the sequence of visits to the public health system of patients with cardiomyopathies in Poland in the period 2016–2021. Kardiol. Pol. 2024, 82, 500–506. [Google Scholar] [CrossRef]
- Mizia-Stec, K.; Leszek, P.; Cegłowska, U.; Wiśniewska, A.; Hałgas, K.; Wybraniec, M.; Pachciński, O.; Stec, M.; Cieśla, D.; Gąsior, M.; et al. Incidence and prevalence of cardiomyopathies in Poland and outcomes for patients in the years 2016–2020. Kardiol. Pol. 2024, 82, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Canepa, M.; Fumagalli, C.; Tini, G.; Vincent-Tompkins, J.; Day, S.M.; Ashley, E.A.; Mazzarotto, F.; Ware, J.S.; Michels, M.; Jacoby, D.; et al. Temporal Trend of Age at Diagnosis in Hypertrophic Cardiomyopathy: An Analysis of the International Sarcomeric Human Cardiomyopathy Registry. Circ. Heart Fail. 2020, 13, e007230. [Google Scholar] [CrossRef]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef]
- Bercu, B.A.; Diettert, G.A.; Danforth, W.H.; Pund, E.E.; Ahlvin, R.C.; Belliveau, R.R. Pseudoaortic stenosis produced by ventricular hypertrophy. Am. J. Med. 1958, 25, 814–818. [Google Scholar] [CrossRef]
- Melas, M.; Beltsios, E.T.; Adamou, A.; Koumarelas, K.; McBride, K.L. Molecular Diagnosis of Hypertrophic Cardiomyopathy (HCM): In the Heart of Cardiac Disease. J. Clin. Med. 2022, 12, 225. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.K.; Alkhatib, D.; Pour-Ghaz, I.; Isa, S.; Al-Taweel, O.; Ugonabo, I.; Yedlapati, N.; Jefferies, J.L. Hypertrophic Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2023, 10, 106. [Google Scholar] [CrossRef]
- Freundlich, I.M.; McMurray, J.T.; Lehman, J.S. Idiopathic hypertrophic subaortic stenosis. Am. J. Roentgenol. 1967, 100, 284–289. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Makavos, G.; Kairis, C.; Tselegkidi, M.E.; Karamitsos, T.; Rigopoulos, A.G.; Noutsias, M.; Ikonomidis, I. Hypertrophic cardiomyopathy: An updated review on diagnosis, prognosis, and treatment. Heart Fail. Rev. 2019, 24, 439–459. [Google Scholar] [CrossRef]
- Cirino, A.L.; Harris, S.; Lakdawala, N.K.; Michels, M.; Olivotto, I.; Day, S.M.; Abrams, D.J.; Charron, P.; Caleshu, C.; Semsarian, C.; et al. Role of Genetic Testing in Inherited Cardiovascular Disease: A Review. JAMA Cardiol. 2017, 2, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Cannon, R.O.; Rosing, D.R.; Maron, B.J.; Leon, M.B.; Bonow, R.O.; Watson, R.M.; Epstein, S.E. Myocardial ischemia in patients with hypertrophic cardiomyopathy: Contribution of inadequate vasodilator reserve and elevated left ventricular filling pressures. Circulation 1985, 71, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Siontis, K.C.; Geske, J.B.; Ong, K.; Nishimura, R.A.; Ommen, S.R.; Gersh, B.J. Atrial fibrillation in hypertrophic cardiomyopathy: Prevalence, clinical correlations, and mortality in a large high-risk population. J. Am. Heart Assoc. 2014, 3, e001002. [Google Scholar] [CrossRef]
- López-Ponce de Leon, J.D.; Estacio, M.; Giraldo, N.; Escalante, M.; Rodas, Y.; Largo, J.; Lores, J.; Victoria, M.C.; Argote, D.; Florez, N.; et al. Hypertrophic Cardiomyopathy in a Latin American Center: A Single Center Observational Study. J. Clin. Med. 2023, 12, 5682. [Google Scholar] [CrossRef]
- Mascia, G.; Crotti, L.; Groppelli, A.; Canepa, M.; Merlo, A.C.; Benenati, S.; Di Donna, P.; Della Bona, R.; Soranna, D.; Zambon, A.; et al. Syncope in hypertrophic cardiomyopathy (part I): An updated systematic review and meta-analysis. Int. J. Cardiol. 2022, 357, 88–94. [Google Scholar] [CrossRef]
- Bisceglia, I.; Ceci, V.; Giovannini, E.; Santoboni, A.; Sebastiani, F.; Masini, V. Aspetti clinici e criteri diagnostici delle miocardiopatie ipertrofiche simmetriche non ostruttive [Clinical aspects and diagnostic criteria in non-obstructive symmetric hypertrophic myocardiopathy]. G. Ital. Cardiol. 1983, 13, 192–199. [Google Scholar] [PubMed]
- Licordari, R.; Trimarchi, G.; Teresi, L.; Restelli, D.; Lofrumento, F.; Perna, A.; Campisi, M.; de Gregorio, C.; Grimaldi, P.; Calabrò, D.; et al. Cardiac Magnetic Resonance in HCM Phenocopies: From Diagnosis to Risk Stratification and Therapeutic Management. J. Clin. Med. 2023, 12, 3481. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Characteristic | General Group N = 85 | Elective N = 68 | Urgent N = 15 | p |
|---|---|---|---|---|
| Age (years ± SD) | 51 ± 15.2 | 52 ± 14.0 | 46 ± 19.5 | 0.36 |
Sex
| 43/45% 42/44% | 34/41% 34/41% | 8/10% 7/8% | 0.82 |
| Asymptomatic (n/%) | 13/14% | 11/16% | 1/7% | 0.34 |
| Signs and symptoms (n/%) | 71/76% | 57/84% | 14/93% | 0.34 |
| 27/28% | 22/32% | 5/33% | 0.97 |
| 13/14% | 11/24% | 2/13% | 0.78 |
| 44/46% | 38/56% | 6/40% | 0.26 |
| 28/29% | 23/34% | 5/33% | 0.97 |
| 28/29% | 25/37% | 3/20% | 0.21 |
| 26/27% | 17/25% | 9/60% | 0.01 |
| 18/19% | 15/22% | 3/20% | 0.86 |
| Coexisting heart involvement | ||||
| 28/29% | 22/32% | 6/40% | 0.57 |
| 21/22% | 18/27% | 3/20% | 0.60 |
| 37/39% | 28/41% | 8/53% | 0.39 |
| 38/40% | 33/49% | 4/27% | 0.11 |
| Comorbidities | ||||
| 48/51% | 41/60% | 7/47% | 0.33 |
| 14/15% | 9/13% | 5/33% | 0.02 |
| 36/38% | 31/46% | 4/27% | 0.34 |
| 9/9% | 4/6% | 4/27% | 0.01 |
| 22/23% | 18/27% | 3/20% | 0.54 |
| 3/3% | 2/3% | 1/7% | 0.40 |
| 14/15% | 10/15% | 3/20% | 0.43 |
| 8/8% | 6/9% | 1/7% | 0.91 |
| Family history | ||||
| Positive family history (HCM in family, sudden cardiac death in family or both) (n/%) | 21/22% | 17/25% | 4/27% | 0.89 |
| HCM in family (n/%) | 11/12% | 7/10% | 4/27% | 0.09 |
| Sudden cardiac death in family (n/%) | 15/16% | 13/19% | 2/13% | 0.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bieczek, D.; Ściślicka, A.; Adamiec, A.; Cader, A.; Wandasiewicz, M.; Basiaga, B.; Niemiec, M.; Mizia-Stec, K. Clinical History of Patients with Hypertrophic Cardiomyopathy—How to Improve the Initiation Process of the Diagnosis? J. Clin. Med. 2024, 13, 5239. https://doi.org/10.3390/jcm13175239
Bieczek D, Ściślicka A, Adamiec A, Cader A, Wandasiewicz M, Basiaga B, Niemiec M, Mizia-Stec K. Clinical History of Patients with Hypertrophic Cardiomyopathy—How to Improve the Initiation Process of the Diagnosis? Journal of Clinical Medicine. 2024; 13(17):5239. https://doi.org/10.3390/jcm13175239
Chicago/Turabian StyleBieczek, Dominika, Adrianna Ściślicka, Agnieszka Adamiec, Aleksandra Cader, Monika Wandasiewicz, Bartosz Basiaga, Małgorzata Niemiec, and Katarzyna Mizia-Stec. 2024. "Clinical History of Patients with Hypertrophic Cardiomyopathy—How to Improve the Initiation Process of the Diagnosis?" Journal of Clinical Medicine 13, no. 17: 5239. https://doi.org/10.3390/jcm13175239
APA StyleBieczek, D., Ściślicka, A., Adamiec, A., Cader, A., Wandasiewicz, M., Basiaga, B., Niemiec, M., & Mizia-Stec, K. (2024). Clinical History of Patients with Hypertrophic Cardiomyopathy—How to Improve the Initiation Process of the Diagnosis? Journal of Clinical Medicine, 13(17), 5239. https://doi.org/10.3390/jcm13175239