
Advancements and Challenges in Personalized Therapy for BRAF-Mutant Melanoma: A Comprehensive Review

Abstract
:1. Introduction
2. Tumor Heterogeneity and Its Implications for BRAF-Mutant Melanoma
2.1. Introduction to Tumor Heterogeneity and the Tumor Microenvironment
2.2. Genetic and Immune Interactions
2.3. Metastatic Heterogeneity and Immune Evasion in BRAF-Mutant Melanoma
2.4. Mechanisms of Drug Resistance in BRAF V600E-Mutant Melanoma
3. Current Approaches to Personalized Therapy for Patients with B-RAF Mutant Melanoma
3.1. Tumor-Infiltrating Lymphocyte (TIL) Therapy
3.2. CAR T-Cell Therapy
3.3. Biomarker Detection
3.4. Targeted Therapy
3.5. Neoantigens
4. Discussion
4.1. Challenges and Opportunities
4.1.1. Challenges
4.1.2. Opportunities

{kind=link}
{kind=link}
| Treatment Approach | Challenges | Opportunities | References |
|---|---|---|---|
| CAR-T cell therapy | Emergence of treatment-related adverse events, hindrance of CAR-T cell functionality in combination with daBRAFenib and trametinib | Exploring patient responses to other concurrent CAR-T cell and combined targeted therapy regimens | [63,64] |
| TIL therapy | Extensive duration of T-cell harvesting periods, cost of manufacturing, toxicities | Freezing tumor-sample TILs for improved efficiency To mitigate the toxicity associated with IL-2 therapy, alternative strategies have been explored. One such approach involves using mesenchymal stem cells (MSCs) as a vehicle for targeted delivery of IL-2 to reduce systemic toxicity [68] Genetically engineered IL-2 variants have been developed to potentially reduce systemic toxicity while maintaining efficacy in immunotherapy for solid tumors | [31,61,62,72,73,74] |
| 18FDG-PET/CT scanning | Relative limitation of literature regarding artificial intelligence-based techniques in 18FDG-PET/CT scanning for diagnosing melanoma | Multiple PET parameters hold prognostic value for patients with metastatic melanoma undergoing immunotherapy | [70] |
4.2. Dynamic and Adaptations within the Tumor Microenvironment
4.2.1. Impact of Tumor Microenvironment (TME)
4.2.2. Tumor Microenvironment (TME) Adaptation
4.3. Future Directions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current advances in the treatment of BRAF-mutant melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed]
- Castellani, G.; Buccarelli, M.; Arasi, M.B.; Rossi, S.; Pisanu, M.E.; Bellenghi, M.; Lintas, C.; Tabolacci, C. BRAF mutations in melanoma: Biological aspects, therapeutic implications, and circulating biomarkers. Cancers 2023, 15, 4026. [Google Scholar] [CrossRef]
- Ottaviano, M.; Giunta, E.F.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF gene and melanoma: Back to the future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef]
- Kong, B.Y.; Carlino, M.S.; Menzies, A.M. Biology and treatment of BRAF mutant metastatic melanoma. Melanoma Manag. 2016, 3, 33–45. [Google Scholar] [CrossRef]
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U.; ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current state of target treatment in BRAF mutated melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef]
- Giugliano, F.; Crimini, E.; Tarantino, P.; Zagami, P.; Uliano, J.; Corti, C.; Trapani, D.; Curigliano, G.; Ascierto, P.A. First line treatment of BRAF mutated advanced melanoma: Does one size fit all? Cancer Treat. Rev. 2021, 99, 102253. [Google Scholar] [CrossRef]
- Mullard, A. BRAF plus MEK inhibitor combo secures tumour-agnostic FDA approval. Nat. Rev. Drug Discov. 2022, 21, 548. [Google Scholar] [CrossRef]
- Gonzalez-Cao, M.; Rosell, R.; Martin Algarra, S.; Puertolas, T.; Espinosa, E. Sequence of therapies for advanced BRAFV600E/K melanoma. Ann. Transl. Med. 2023, 11, 270. [Google Scholar] [CrossRef]
- Kiyotani, K.; Toyoshima, Y.; Nakamura, Y. Personalized immunotherapy in cancer precision medicine. Cancer Biol. Med. 2021, 18, 955–965. [Google Scholar] [CrossRef]
- Force, J.; Salama, A.K. First-line treatment of metastatic melanoma: Role of nivolumab. Immunotargets Ther. 2017, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor heterogeneity: A great barrier in the age of cancer immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef]
- Ito, T.; Kaku-Ito, Y.; Murata, M.; Ichiki, T.; Kuma, Y.; Tanaka, Y.; Ide, T.; Ohno, F.; Wada-Ohno, M.; Yamada, Y.; et al. Intra- and inter-tumor BRAF heterogeneity in acral melanoma: An immunohistochemical analysis. Int. J. Mol. Sci. 2019, 20, 6191. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Valachis, A.; Ullenhag, G.J. Discrepancy in BRAF status among patients with metastatic malignant melanoma: A meta-analysis. Eur. J. Cancer 2017, 81, 106–115. [Google Scholar] [CrossRef]
- Sakaizawa, K.; Ashida, A.; Kiniwa, Y.; Okuyama, R. BRAF mutation heterogeneity in melanoma lesions. Acta Derm. Venereol. 2020, 100, adv00045. [Google Scholar] [CrossRef]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.M.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 10. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and intertumor heterogeneity in melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Wang, A.; Yuan, Y.; Zhu, B.; Long, H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp. Hematol. Oncol. 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 607. [Google Scholar] [CrossRef] [PubMed]
- Pozniak, J.; Pedri, D.; Landeloos, E.; Van Herck, Y.; Antoranz, A.; Vanwynsberghe, L.; Nowosad, A.; Roda, N.; Makhzami, S.; Bervoets, G.; et al. A TCF4-dependent gene regulatory network confers resistance to immunotherapy in melanoma. Cell 2024, 187, 166–183.e25. [Google Scholar] [CrossRef]
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol. Res. 2017, 123, 95–102. [Google Scholar] [CrossRef]
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Ferretti, G.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor microenvironment: Implications in melanoma resistance to targeted therapy and immunotherapy. Cancers 2020, 12, 2870. [Google Scholar] [CrossRef]
- Ding, L.; Kim, M.; Kanchi, K.L.; Dees, N.D.; Lu, C.; Griffith, M.; Fenstermacher, D.; Sung, H.; Miller, C.A.; Goetz, B.; et al. Clonal architectures and driver mutations in metastatic melanomas. PLoS ONE 2014, 9, e111153. [Google Scholar] [CrossRef]
- Birkeland, E.; Zhang, S.; Poduval, D.; Geisler, J.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; Hovig, E.; Myklebost, O.; Knappskog, S.; et al. Patterns of genomic evolution in advanced melanoma. Nat. Commun. 2018, 9, 2665. [Google Scholar] [CrossRef]
- Takata, M.; Morita, R.; Takehara, K. Clonal heterogeneity in sporadic melanomas as revealed by loss-of-heterozygosity analysis. Int. J. Cancer 2000, 85, 492–497. [Google Scholar] [CrossRef]
- Lin, Z.; Meng, X.; Wen, J.; Corral, J.M.; Andreev, D.; Kachler, K.; Schett, G.; Chen, X.; Bozec, A. Intratumor heterogeneity correlates with reduced immune activity and worse survival in melanoma patients. Front. Oncol. 2020, 10, 596493. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J. Clin. Oncol. 2021, 39, 2656–2666, Correction in J. Clin. Oncol. 2021, 39, 2972. https://doi.org/10.1200/JCO.21.01866. [Google Scholar] [CrossRef] [PubMed]
- Betof Warner, A.; Corrie, P.G.; Hamid, O. Tumor-infiltrating lymphocyte therapy in melanoma: Facts to the future. Clin. Cancer Res. 2023, 29, 1835–1854. [Google Scholar] [CrossRef] [PubMed]
- Parums, D. Editorial: First regulatory approval for adoptive cell therapy with autologous tumor-infiltrating lymphocytes (tils)—Lifileucel (amtagvi). Med. Sci. Mon. 2024, 30, e944927-1. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Lewis, K.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (til) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the c-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Qi, R. Dynamics of circulating cytokines and chemokines during and after tumor-infiltrating lymphocyte cell therapy with lifileucel in advanced melanoma patients. J. Clin. Oncol. 2024, 42 (Suppl. S16), 9594. [Google Scholar] [CrossRef]
- Byron, S.A.; Loch, D.S.; Wellens, C.L.; Wortmann, A.; Wu, J.; Wang, J.; Pollock, P.M. Sensitivity to the mek inhibitor e6201 in melanoma cells is associated with mutant braf and wildtype pten status. Mol. Cancer 2012, 11, 75. [Google Scholar] [CrossRef]
- Sarnaik, A.; Khushalani, N.I.; Chesney, J.A.; Lewis, K.D.; Medina, T.M.; Kluger, H.M.; Thomas, S.S.; Musibay, E.D.; Pavlick, A.C.; Whitman, E.D.; et al. Long-term follow up of lifileucel (LN-144) cryopreserved autologous tumor infiltrating lymphocyte therapy in patients with advanced melanoma progressed on multiple prior therapies. J. Clin. Oncol. 2020, 38, 10006. [Google Scholar] [CrossRef]
- Sharnaik, A.; Thomas, S.; Davar, D.; Kirkwood, J.; Kluger, H.; Lutzky, J.; Wilson, M.; Pavlick, A.C.; Curti, B.D.; Whitman, E.D.; et al. Safety and efficacy of cryopreserved autologous tumor infiltrating lymphocyte therapy (ln-144, lifileucel) in advanced metastatic melanoma patients previously treated with at least one prior systemic therapy. J. Clin. Oncol. 2019, 37 (Suppl. S8), 136. [Google Scholar] [CrossRef]
- Warner, A.; Postow, M.; Panageas, K.; Smithy, J.; Schoenfeld, A.; Wolchok, J.; Shoushtari, A. A pilot trial of autologous tumor infiltrating lymphocytes (lifileucel, ln-144) for patients with asymptomatic melanoma brain metastases. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS9606. [Google Scholar] [CrossRef]
- Overwijk, W.; Tagliaferri, M.; Zalevsky, J. Engineering il-2 to give new life to T cell immunotherapy. Annu. Rev. Med. 2021, 72, 281–311. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Bennett, A.D.; Melenhorst, J.J.; et al. Enhancing CAR T-cell persistence through ICOS and 4–1BB co-stimulation. J. Clin. Investig. 2018, 128, 2123–2138. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR-T cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Spiliopoulou, P.; Holanda Lopes, C.D.; Spreafico, A. Promising and minimally invasive biomarkers: Targeting melanoma. Cells 2023, 13, 19. [Google Scholar] [CrossRef]
- Peng, K.; Zhang, Y.; Liu, D.; Chen, J. MMP2 is a immunotherapy related biomarker and correlated with cancer-associated fibroblasts infiltrate in melanoma. Cancer Cell Int. 2023, 23, 26. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.; Mignion, L.; Jordan, B.F. Metabolic profiling to assess response to targeted and immune therapy in melanoma. Int. J. Mol. Sci. 2024, 25, 1725. [Google Scholar] [CrossRef]
- Carlino, M.S.; Saunders, C.A.B.; Haydu, L.E.; Menzies, A.M.; Martin Curtis, C., Jr.; Lebowitz, P.F.; Kefford, R.F.; Long, G.V. (18)F-labelled fluorodeoxyglucose-positron emission tomography (FDG-PET) heterogeneity of response is prognostic in daBRAFenib treated BRAF mutant metastatic melanoma. Eur. J. Cancer 2013, 49, 395–402. [Google Scholar] [CrossRef]
- Pallocca, M.; Molineris, I.; Berrino, E.; Marcozzi, B.; Betti, M.; Levati, L.; D’Atri, S.; Menin, C.; Madonna, G.; Ghiorzo, P.; et al. Comprehensive genomic profiling on metastatic melanoma: Results from a network screening from 7 Italian cancer centres. J. Transl. Med. 2024, 22, 29. [Google Scholar] [CrossRef]
- Sun, C.; España, S.; Richarz, N.; Solé-Blanch, C.; Boada, A.; Martinez-Cardús, A.; Chu, A.; Liu, Z.; Manzano, J.L. Targeted therapy or immunotherapy in BRAF-mutated metastatic melanoma: A Spanish center’s decade of experience. Front. Oncol. 2024, 14, 1322116. [Google Scholar] [CrossRef]
- Stopfer, L.E.; Rettko, N.J.; Leddy, O.; Mesfin, J.M.; Brown, E.; Winski, S.; Bryson, B.; Wells, J.A.; White, F.M. MEK inhibition enhances presentation of targetable MHC-I tumor antigens in mutant melanomas. Proc. Natl. Acad. Sci. USA 2022, 119, e2208900119. [Google Scholar] [CrossRef]
- Stadler, J.C.; Keller, L.; Mess, C.; Bauer, A.T.; Koett, J.; Geidel, G.; Heidrich, I.; Vidal-Y-Sy, S.; Andreas, A.; Stramaglia, C.; et al. Prognostic value of von Willebrand factor levels in patients with metastatic melanoma treated by immune checkpoint inhibitors. J. Immunother. Cancer 2023, 11, e006456. [Google Scholar] [CrossRef]
- Kang, K.; Xie, F.; Mao, J.; Bai, Y.; Wang, X. Significance of tumor mutation burden in immune infiltration and prognosis in cutaneous melanoma. Front. Oncol. 2020, 10, 573141. [Google Scholar] [CrossRef]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Törngren, T.; Kvist, A.; et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1738, Correction in Nat. Commun. 2020, 11, 1714. https://doi.org/10.1038/s41467-020-15531-2. [Google Scholar] [CrossRef]
- Lin, S.H.; Yc, L.; Yao, L.; Kalhor, N.; Carter, B.W.; Altan, M.; Tsao, A.S. Phase II trial of concurrent atezolizumab with chemoradiation for unresectable nsclc. J. Thorac. Oncol. 2020, 15, 248–257. [Google Scholar] [CrossRef]
- Soltantoyeh, T.; Akbari, B.; Karimi, A.; Mahmoodi Chalbatani, G.; Ghahri-Saremi, N.; Hadjati, J.; Hamblin, M.R.; Mirzaei, H.R. Chimeric antigen receptor (CAR) T cell therapy for metastatic melanoma: Challenges and road ahead. Cells 2021, 10, 1450. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Truong, N.T.H.; Gardam, B.; Yu, W.; Ebert, L.M.; Johnson, A.; Yeo, E.C.F.; Wittwer, N.L.; Tapia Rico, G.; Logan, J.; et al. Safety and biological outcomes following a phase 1 trial of GD2-specific CAR-T cells in patients with GD2-positive metastatic melanoma and other solid cancers. J. Immunother. Cancer 2024, 12, e008659. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular markers and targets in melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Gibney, G.T.; Zaemes, J.; Shand, S.; Shah, N.; Swoboda, D.M.; Gardner, K.; Radfar, A.; Petronic-Rosic, V.; Reilly, M.J.; Al-Refaie, W.B.; et al. Pet/ct scan and biopsy-driven approach for safe anti-pd-1 therapy discontinuation in patients with advanced melanoma. J. Immunother. Cancer 2021, 9, e002955. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844, Erratum in Lancet 2020, 396, 466. https://doi.org/10.1016/S0140-6736(20)31721-9. [Google Scholar] [CrossRef] [PubMed]
- Inman, B.A.; Longo, T.A.; Ramalingam, S.; Harrison, M.R. Atezolizumab: A pd-l1–blocking antibody for bladder cancer. Clin. Cancer Res. 2017, 23, 1886–1890. [Google Scholar] [CrossRef]
- Müller, P.C.; Ertural, C.; Hempelmann, J.; Dronskowski, R. Crystal orbital bond index: Covalent bond orders in solids. J. Phy. Chem. 2021, 125, 7959–7970. [Google Scholar] [CrossRef]
- Lepist, E.; Phan, T.K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.P.; Ray, A.S. Cobicistat boosts the intestinal absorption of transport substrates, including hiv protease inhibitors and gs-7340, in vitro. Antimicrob. Agents Chemother. 2012, 56, 5409–5413. [Google Scholar] [CrossRef]
- Filippi, L.; Bianconi, F.; Schillaci, O.; Spanu, A.; Palumbo, B. The role and potential of 18F-FDG PET/CT in malignant melanoma: Prognostication, monitoring response to targeted and immunotherapy, and radiomics. Diagnostics 2022, 12, 929. [Google Scholar] [CrossRef]
- Kazemi, M.H.; Sadri, M.; Najafi, A.; Rahimi, A.; Baghernejadan, Z.; Khorramdelazad, H.; Falak, R. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front. Immunol. 2022, 13, 1018962. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Dörrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK inhibitors influence the function of reprogrammed T cells: Consequences for adoptive T-cell therapy. Int. J. Mol. Sci. 2018, 19, 289. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Solovyeva, V.V.; James, V.; Arkhipova, S.S.; Gomzikova, M.O.; Garanina, E.E.; Akhmetzyanova, E.R.; Tazetdinova, L.G.; Khaiboullina, S.F.; Rizvanov, A.A. Human mesenchymal stem cells overexpressing interleukin 2 can suppress proliferation of neuroblastoma cells in co-culture and activate mononuclear cells in vitro. Bioengineering 2020, 7, 59. [Google Scholar] [CrossRef]
- Hartimath, S.V.; Manuelli, V.; Zijlma, R.; Signore, A.; Nayak, T.K.; Freimoser-Grundschober, A.; Klein, C.; Dierckx, R.A.; de Vries, E.F. Pharmacokinetic properties of radiolabeled mutant interleukin-2v: A pet imaging study. Oncotarget 2018, 9, 7162–7174. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Reardon, D.A. Neoantigen vaccine generates intratumoral t cell responses in phase ib glioblastoma trial. Nature 2018, 565, 234–239. [Google Scholar] [CrossRef]
- Wu, R.; Forget, M.A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L.G. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef]
- Hilke, F.J.; Sinnberg, T.; Gschwind, A.; Niessner, H.; Demidov, G.; Amaral, T.; Ossowski, S.; Bonzheim, I.; Röcken, M.; Riess, O.; et al. Distinct mutation patterns reveal melanoma subtypes and influence immunotherapy response in advanced melanoma patients. Cancers 2020, 12, 2359. [Google Scholar] [CrossRef]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 72. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Y.; Ali, N.M.; Zhang, B.; Cui, X. The role of innate immune cells in the tumor microenvironment and research progress in anti-tumor therapy. Front. Immunol. 2022, 13, 1039260. [Google Scholar] [CrossRef]
- Zhao, Y.; Shen, M.; Wu, L.; Yang, H.; Yao, Y.; Yang, Q.; Du, J.; Liu, L.; Li, Y.; Bai, Y. Stromal cells in the tumor microenvironment: Accomplices of tumor progression? Cell Death Dis. 2023, 14, 587. [Google Scholar] [CrossRef]
- Yan, C.; Chen, S.C.; Ayers, G.D.; Nebhan, C.A.; Roland, J.T.; Weiss, V.L.; Johnson, D.B.; Richmond, A. Proximity of immune and tumor cells underlies response to BRAF/MEK-targeted therapies in metastatic melanoma patients. NPJ Precis. Oncol. 2022, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zadeh, S.; Pizzolla, A.; Thia, K.; Gyorki, D.E.; McArthur, G.A.; Scolyer, R.A.; Long, G.; Wilmott, J.S.; Andrews, M.C.; et al. Characterization of the treatment-naive immune microenvironment in melanoma with BRAF mutation. J. Immunother. Cancer 2022, 10, e004095. [Google Scholar] [CrossRef] [PubMed]
- Dabrosin, N.; Sloth Juul, K.; Bæhr Georgsen, J.; Andrup, S.; Schmidt, H.; Steiniche, T.; Heide Øllegaard, T.; Bønnelykke Behrndtz, L. Innate immune cell infiltration in melanoma metastases affects survival and is associated with BRAFV600E mutation status. Melanoma Res. 2019, 29, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Schöllhorn, A.; Gouttefangeas, C.; Schuhmacher, J. CD4+ T cells: Multitasking cells in the duty of cancer immunotherapy. Cancers 2021, 13, 596. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Min, K.W.; Choe, J.Y.; Kwon, M.J.; Lee, H.K.; Kang, H.S.; Nam, E.S.; Cho, S.J.; Park, H.R.; Min, S.K.; Seo, J.; et al. BRAF and NRAS mutations and antitumor immunity in Korean malignant melanomas and their prognostic relevance: Gene set enrichment analysis and CIBERSORT analysis. Pathol. Res. Pract. 2019, 215, 152671. [Google Scholar] [CrossRef]
- Patton, E.E.; Mueller, K.L.; Adams, D.J.; Anandasabapathy, N.; Aplin, A.E.; Bertolotto, C.; Bosenberg, M.; Ceol, C.J.; Burd, C.E.; Chi, P.; et al. Melanoma models for the next generation of therapies. Cancer Cell 2021, 39, 610–631. [Google Scholar] [CrossRef]
- Helgadóttir, H.; Drakensjö, I.; Girnita, A. Personalized medicine in malignant melanoma: Towards patient tailored treatment. Front. Oncol. 2018, 8, 202. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.; Hwu, P. Loss of pten promotes resistance to t cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Xu, J.; Mu, S.; Wang, Y.; Yu, S.; Wang, Z. Recent advances in immunotherapy and its combination therapies for advanced melanoma: A review. Front. Oncol. 2024, 14, 1400193. [Google Scholar] [CrossRef]
- Tang, J.; Gong, Y.; Ma, X. Bispecific antibodies progression in malignant melanoma. Front. Pharmacol. 2022, 13, 837889. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Ribas, A.; Robert, C.; Schachter, J.; Nyakas, M.; Daud, A.; Hamid, O. Association of braf v600e/k mutation status and prior braf/mek inhibition with pembrolizumab outcomes in advanced melanoma. JAMA Oncol. 2020, 6, 1256. [Google Scholar] [CrossRef] [PubMed]
- Sung, E.; Ko, M.; Won, J.-Y.; Jo, Y.; Park, E.; Kim, H.; Choi, E.; Jung, U.-J.; Jeon, J.; Kim, Y.; et al. Lag-3xpd-l1 bispecific antibody potentiates antitumor responses of t cells through dendritic cell activation. Mol. Ther. 2022, 30, 2800–2816. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, V.G.; Waldhauer, I.; Freimoser-Grundschober, A.; Richard, M.; Fahrni, L.; Bommer, E.; Claus, C.; Sam, J.; Colombetti, S.; Sutmuller, R.; et al. Abstract lb-389: Combination of tyrp1-tcb, a novel t cell bispecific antibody for the treatment of melanoma, with immunomodulatory agents. Cancer Res. 2020, 80, LB-389. [Google Scholar] [CrossRef]
- Payandeh, Z.; Yarahmadi, M.; Nariman-Saleh-Fam, Z.; Tarhriz, V.; Islami, M.; Aghdam, A.M.; Eyvazi, S. Immune therapy of melanoma: Overview of therapeutic vaccines. J. Cell. Physiol. 2019, 234, 14612–14621. [Google Scholar] [CrossRef]
- Guerrisi, A.; Falcone, I.; Valenti, F.; Rao, M.; Gallo, E.; Ungania, S.; Maccallini, M.T.; Fanciulli, M.; Frascione, P.; Morrone, A.; et al. Artificial intelligence and advanced melanoma: Treatment management implications. Cells 2022, 11, 3965. [Google Scholar] [CrossRef]
- Higgins, H. Recent advances in the field of artificial intelligence for precision medicine in patients with a diagnosis of metastatic cutaneous melanoma. Diagnostics 2023, 13, 3483. [Google Scholar] [CrossRef]

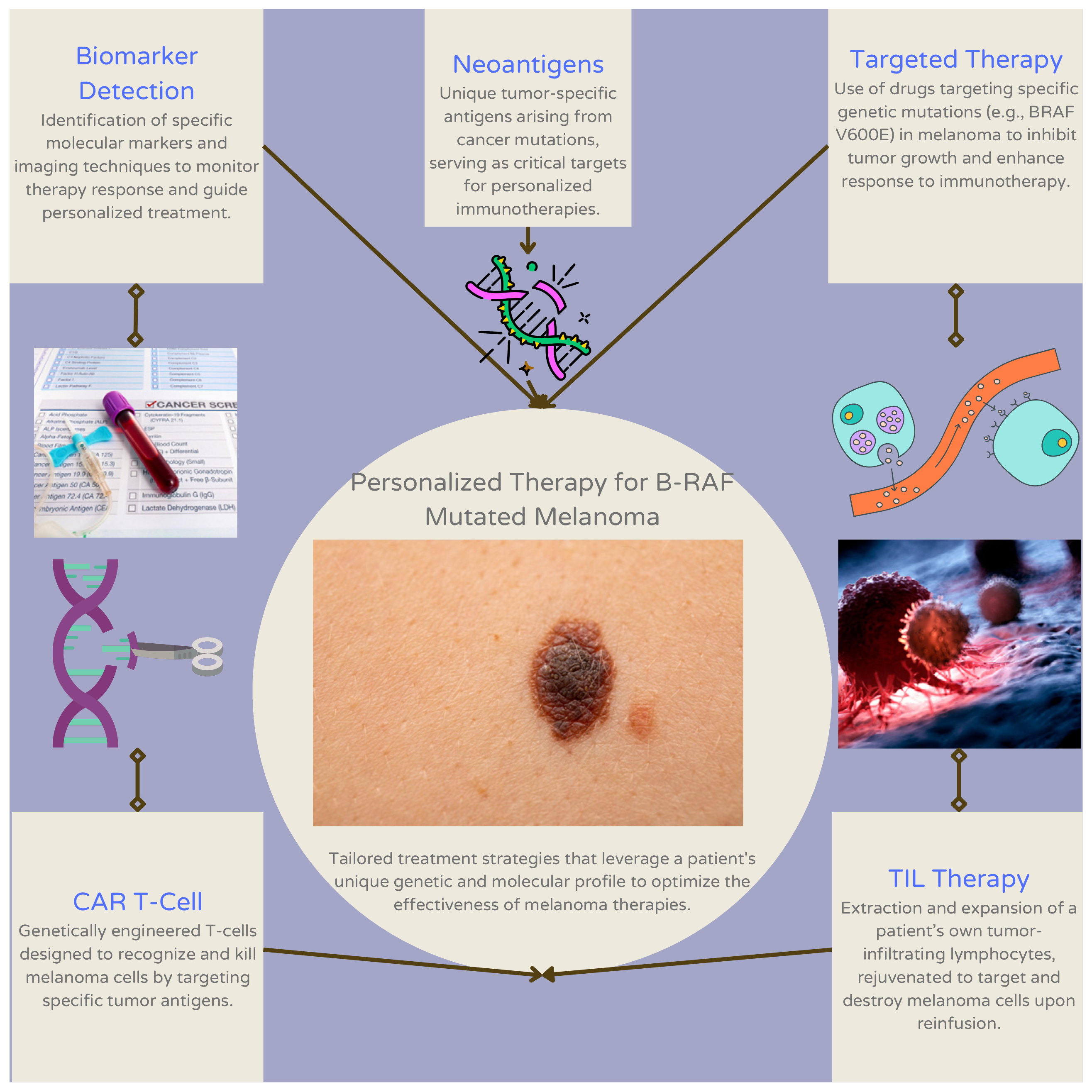
| Type of Approach | Examples | Description of Approach | References |
|---|---|---|---|
| Tumor-infiltrating lymphocyte (TIL) therapy | Adoptive cell therapy, Lifileucel | Isolation of tumor sample via ex vivo expansion and consequent removal of TILs from the tumor microenvironment | [31,32] |
| T-cell therapy | Chimeric antigen receptor (CAR)-T cell therapy | Genetic modification and tumoral targeting of T-lymphocytes expressing CARs | [58] |
| Biomarker detection | Circulating tumor DNA, extracellular vesicle-melanoma membrane-bound proteins (LNGFR, MCAM, MCSP, and ERBB3) | Measurement of biomarker levels to predict patient response to immunotherapy or targeted treatment | [47] |
| Targeted therapy | BRAFi, MEKi | Enhancement of personalized immunotherapy using supplementary targeted therapy | [55] |
| Neoantigens | KRAS G12D | Vaccination with synthesized neoantigen to stimulate a targeted immune response | [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shebrain, A.; Idris, O.A.; Jawad, A.; Zhang, T.; Xing, Y. Advancements and Challenges in Personalized Therapy for BRAF-Mutant Melanoma: A Comprehensive Review. J. Clin. Med. 2024, 13, 5409. https://doi.org/10.3390/jcm13185409
Shebrain A, Idris OA, Jawad A, Zhang T, Xing Y. Advancements and Challenges in Personalized Therapy for BRAF-Mutant Melanoma: A Comprehensive Review. Journal of Clinical Medicine. 2024; 13(18):5409. https://doi.org/10.3390/jcm13185409
Chicago/Turabian StyleShebrain, Abdulaziz, Omer A. Idris, Ali Jawad, Tiantian Zhang, and Yan Xing. 2024. "Advancements and Challenges in Personalized Therapy for BRAF-Mutant Melanoma: A Comprehensive Review" Journal of Clinical Medicine 13, no. 18: 5409. https://doi.org/10.3390/jcm13185409