
Pathogenesis and Surgical Treatment of Congenitally Corrected Transposition of the Great Arteries (ccTGA): Part III

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Definition and Pathogenesis of ccTGA
3. Anatomical Variants of ccTGA
4. Anomalies Associated with ccTGA
5. Tricuspid Valve Regurgitation (TR)
6. Systemic Right Ventricle (sRV) Dysfunction
7. Conduction System and Conduction Disorders in ccTGA
8. Symptomatology of ccTGA
9. Prenatal Diagnosis of ccTGA
10. Diagnosis of ccTGA
10.1. Heart Auscultation
10.2. Electrocardiogram (ECG) in ccTGA
10.3. Chest Radiography (Chest X-ray)
10.4. Echocardiography (Cardiac Ultrasound)
10.5. Cardiac Magnetic Resonance (CMR)
10.6. Computed Tomography in ccTGA
10.7. Cardiac Catheterization
10.8. Cardiovascular Three-Dimensional (3D) Printing
10.9. Ergospirometry Test
11. Cardiac Biomarkers. Clinical Significance of BNP and NT-proBNP Concentrations in Patients with ccTGA
12. Pregnancy
13. Surgical Treatment Strategies for ccTGA
13.1. Indications for Surgical Intervention
13.2. Physiological Correction Strategy for ccTGA
13.3. Anatomical Correction Strategy for ccTGA
14. Fontan Operation for ccTGA
15. Postoperative Period and Treatment Results
16. Cardiac Devices Supporting Circulation
16.1. Defibrillators and Stimulators
16.2. Cardiac Resynchronization Therapy (CRT)
16.3. Ventricular Assist Device (VAD)
17. Cardiac Transplantation
18. Summary
19. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASO | arterial switch operation |
| AtrSR | atrial switch repair |
| AV | atrioventricular |
| BNP | B-type natriuretic peptide |
| cc-TGA | congenitally corrected transposition of the great arteries |
| CD | color Doppler |
| CHD | congenital heart defect |
| CM | cardiomyocytes |
| CMR | cardiac magnetic resonance |
| CRT | cardiac resynchronization therapy |
| CS | coronary sinus |
| CT | computed tomography |
| DA | ductus arteriosus |
| D-TGA | dextro transposition of the great arteries |
| ECG | electrocardiogram |
| ECHO | echocardiography |
| FEV1 | forced expiratory volume in one second |
| GLS | global longitudinal deformation |
| LA | left atrium |
| LBBAP | left bundle branch area pacing |
| L-TGA | L-looped transposition of the great arteries |
| LV | left ventricle |
| LVOT | left ventricle outflow tract |
| LVOTO | left ventricle outflow tract obstruction |
| NT-proBNP | N-terminal pro-B-type natriuretic peptide |
| PA | pulmonary artery |
| PAB | pulmonary artery banding |
| PS | pulmonary stenosis |
| RA | right atrium |
| RV | right ventricle |
| SS | situs solitus |
| SI | situs inversus |
| sRV | systemic right ventricle |
| STE | speckle tracking echocardiography |
| TR | tricuspid valve regurgitation |
| TV | tricuspid valve |
| VAD | ventricular assist devices |
| VSD | ventricular septal defect |
| X-ray | radiological examination |
References
- Samánek, M.; Vorísková, M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: A prospective Bohemia survival study. Pediatr. Cardiol. 1999, 20, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Wallis, G.A.; Debich-Spicer, D.; Anderson, R.H. Congenitally corrected transposition. Orph. J. Rare Dis. 2011, 6, 22–44. [Google Scholar] [CrossRef] [PubMed]
- Filippov, A.A.; Del Nido, P.J.; Vasilyev, N.V. Management of systemic right ventricular failure in patients with congenitally corrected transposition of the great arteries. Circulation 2016, 134, 1293–1302. [Google Scholar] [CrossRef]
- Nikaidoh, H. Aortic translocation and biventricular outflow tract reconstruction. A new surgical repair for transposition of the great arteries associated with ventricular septal defect and pulmonary stenosis. J. Thorac. Cardiovasc. Surg. 1984, 88, 365–372. [Google Scholar] [CrossRef]
- Yamagishi, M.; Shuntoh, K.; Matsushita, T.; Fujiwara, K.; Shinkawa, T.; Miyazaki, T.; Kitamura, N. Half-turned truncal switch operation for complete transposition of the great arteries with ventricular septal defect and pulmonary stenosis. J. Thorac. Cardiovasc. Surg. 2003, 125, 966–968. [Google Scholar] [CrossRef]
- Rastelli, G.C. A new approach to “anatomic” repair of transposition of the great arteries. Mayo Clinic. Proc. 1969, 44, 1–12. [Google Scholar]
- Von Rokitansky, K.F. Die Defekte der Scheidewande des Herzens; W. Braumuller: Vienna, Austria, 1875; pp. 83–86. [Google Scholar]
- Warnes, C.A. Transposition of the Great Arteries. Circulation 2006, 114, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Goldmuntz, E.; Bamford, R.; Karkera, J.D.; dela Cruz, J.; Roessler, E.; Muenke, M. CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. Am. J. Hum. Genet. 2002, 70, 776–780. [Google Scholar] [CrossRef]
- Van Praagh, R. Segmental approach to diagnosis. In Nadasʹ Pediatric Cardiology; Fyler, D.C., Nadas, A., Eds.; Hanley and Belfus: Philadelphia, PA, USA, 1992. [Google Scholar]
- Freedom, R.M.; Culham, G.; Rowe, R.D. The criss-cross heart and superoinferior ventricular heart, an angiographic study. Am. J. Cardiol. 1978, 42, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Hornung, T.S.; Calder, L. Congenitally corrected transposition of the great arteries. Heart 2010, 96, 1154–1161. [Google Scholar] [CrossRef]
- Van Praagh, R.; Papagiannis, J.; Grünenfelder, J.; Bartram, U.; Martanovic, P. Pathologic anatomy of corrected transposition of the great arteries: Medical and surgical implications. Am. Heart J. 1998, 135, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Allwork, S.P.; Bentall, H.H.; Becker, A.E. Congenitally corrected transposition of the great arteries: Morphological study of 32 cases. Am. J. Cardiol. 1976, 38, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.; Valente, A.M.; Manso, P.H.; Gali, L.G.; Braggion-Santos, M.F.; Rocha, J.M.; de Andrade Vicente, W.V.; Schmidt, A. Congenitally Corrected Transposition of the Great Arteries in the Adult. Braz. J. Cardiovasc. Surg. 2022, 37, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, B.N.; Arabi, T.Z.; Shafqat, A.; Rab, S.A.; Razak, A.; Albert-Brotons, D.C. Heart failure in systemic right ventricle: Mechanisms and therapeutic options. Front. Cardiovasc. Med. 2023, 9, 1064196. [Google Scholar] [CrossRef]
- Van Praagh, R.; Van Praagh, S. Anatomically corrected transposition of the great arteries. Br. Heart J. 1967, 29, 112–119. [Google Scholar] [CrossRef]
- Anderson, R.H.; Becker, A.E.; Arnold, R.; Wilkinson, J.L. The conducting tissues in congenitally corrected transposition. Circulation 1974, 50, 911–923. [Google Scholar] [CrossRef]
- Baruteau, A.; Abrams, D.J.; Ho, S.Y.; Thambo, J.; McLeod, C.J.; Shah, M.J. Cardiac conduction system in congenitally corrected transposition of the great arteries and its clinical relevance. J. Am. Heart Assoc. 2017, 6, e007759. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Madrid, A.; Paul, T.; Abrams, D.; Aziz, P.F.; Blom, N.A.; Chen, J.; Chessa, M.; Combes, N.; Dagres, N.; Diller, G.; et al. Arrhythmias in congenital heart disease: A position paper of the European Heart Rhythm Association (EHRA), Association for European Paediatric and Congenital Cardiology (AEPC), and the European Society of Cardiology (ESC) Working Group on Grown-up Congenital Heart Disease, Endorsed by HRS, PACES, APHRS, and SOLAECE. Europace 2018, 20, 1719–1753. [Google Scholar]
- Huhta, J.C.; Maloney, J.D.; Ritter, D.G.; Ilstrup, D.M.; Feldt, R.H. Complete atrioventricular block in patients with atrioventricular discordance. Circulation 1983, 67, 1374–1377. [Google Scholar] [CrossRef]
- Freedom, R.M. Congenitally corrected transposition of the great arteries: Definitions and pathologic anatomy. Prog. Pediatr. Cardiol. 1999, 10, 3–16. [Google Scholar] [CrossRef]
- Prieto, L.R.; Hordof, A.J.; Secic, M.; Rosenbaum, M.S.; Gersony, W.M. Progressive tricuspid valve disease in patients with congenitally corrected transposition of the great arteries. Circulation 1998, 98, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, P.; Klisiewicz, A.; Lubiszewska, B.; Lipczynska, M.; Konka, M.; Kusmierczyk, M.; Hoffman, P. Functional anatomy of tricuspid regurgitation in patients with systemic right ventricles. J. Am. Soc. Echocardiogr. 2010, 23, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.R.; Danielson, G.K.; McGoon, D.C.; Lie, J.T. Ebstein’s anomaly of the left-sided tricuspid valve: Pathological anatomy of the valvular malformation. Circulation 1978, 58, 187–191. [Google Scholar]
- Van Dissel, A.C.; Opotowsky, A.R.; Burchill, L.J.; Aboulhosn, J.; Grewal, J.; Lubert, A.M.; Antonova, P.; Shah, S.; Cotts, T.; John, A.S. End-stage heart failure in congenitally corrected transposition of the great arteries: A multicentre study. Eur. Heart J. 2023, 44, 3278–3291. [Google Scholar] [CrossRef] [PubMed]
- Fusco, F.; Scognamiglio, G.; Sorice, D.; Abbate, M.; Altobelli, I.; Sarubbi, B. Biventricular performance in adults with a systemic right ventricle: New insights from myocardial work analysis. Int. J. Cardiovasc. Imaging 2024, 40, 1067–1079. [Google Scholar] [CrossRef]
- Yoshida, H.; Shinkawa, T.; Yamagata, A.; Furuta, A.; Niinami, H. Long-term Outcomes of Surgical Repair for Corrected Transposition of the Great Arteries. Ann. Thorac. Surg. 2024, 25, S0003-4975(24)00517-4. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.K.S. Congenitally corrected transposition of the great arteries. J. Thorac. Dis. 2020, 12, 1213–1218. [Google Scholar] [CrossRef]
- Silvetti, M.S.; Favoccia, C.; Saputo, F.A.; Tamburri, I.; Mizzon, C.; Campisi, M.; Gimigliano, F.; Rinelli, G.; Rava, L.; Drago, F. Three-dimensional-mapping-guided permanent conduction system pacing in paediatric patients with congenitally corrected transposition of the great arteries. Europace 2023, 25, 1482–1490. [Google Scholar] [CrossRef]
- Hosseinpour, A.R.; McCarthy, K.P.; Griselli, M.; Sethia, B.; Ho, S.Y. Congenitally corrected transposition: Size of the pulmonary trunk and septal malalignment. Ann. Thorac. Surg. 2004, 77, 2163–2166. [Google Scholar] [CrossRef]
- Thiene, G.; Nava, A.; Rossi, L. The conduction system in corrected transposition with situs inversus. Eur. J. Cardiol. 1977, 6, 57–70. [Google Scholar]
- Lundstrom, U.; Bull, C.; Wyse, R.K.; Somerville, J. The natural and “unnatural” history of congenitally corrected transposition. Am. J. Cardiol. 1990, 65, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, M.K.; Silverman, N.H.; Moon-Grady, A.J.; Tong, E.; Nourse, J.; Sorenson, B.; Lee, J.; Hornberger, L.K. Prenatal detection of congenital heart disease. J. Pediatr. 2009, 155, 26–31.e1. [Google Scholar] [CrossRef]
- Graham, T.P.; Bernard, Y.D.; Mellen, B.G.; Celermajer, D.; Baumgartnera, H.; Cetta, F.; Connolly, H.M.; Davidson, W.R.; Dellborg, M.; Fostera, E.; et al. Long-term outcome in congenitally corrected transposition of the great arteries: A multi-institutional study. J. Am. Coll. Cardiol. 2000, 36, 255–261. [Google Scholar] [CrossRef]
- Kowalik, E.; Jakubowska, E.; Hoffman, P. Congenitally corrected transposition of the great arteries in a 72 year old man-a case report. Cardiol. Pol. 2004, 61, 56–58. [Google Scholar]
- Piran, S.; Veldtman, G.; Siu, S.; Webb, G.D.; Liu, P.P. Heart failure and ventricular dysfunction in patients with single or systemic right ventricles. Circulation 2002, 105, 1189–1194. [Google Scholar] [CrossRef]
- Sharland, G.; Tingay, R.; Jones, A.; Simpson, J. Atrioventricular and ventriculoarterial discordance (congenitally corrected transposition of the great arteries): Echocardiographic features, associations, and outcome in 34 fetuses. Heart 2005, 91, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Van Praagh, R. What determines whether the great arteries are normally or abnormally related ? Am. J. Cardiol. 2016, 118, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Biliciler-Denktas, G.; Feldt, R.H.; Connolly, H.M.; Weaver, A.L.; Puga, F.J.; Danielson, G.K. Early and late results of operations for defects associated with corrected transposition and other anomalies with atrioventricular discordance in a pediatric population. J. Thorac. Cardiovasc. Surg. 2001, 122, 234–241. [Google Scholar] [CrossRef]
- Mah, K.; Friedberg, M.K. Congenitally corrected transposition of the great arteries: Situs solitus or inversus. Circ. Cardiovasc. Imaging 2014, 7, 849–851. [Google Scholar] [CrossRef]
- Agarwal, A.; Samad, F.; Kalvin, L.; Bush, M.; Tajik, A.J. A great imitator in adult cardiology practice: Congenitally corrected transposition of the great arteries. Congenit. Heart Dis. 2017, 12, 143–152. [Google Scholar] [CrossRef]
- Perloff, J.K. Congenitally corrected transposition of the great arteries. In The Clinical Recognition of Congenital Heart Disease; W.B Saunders Company: Philadelphia, PA, USA, 1970; pp. 44–60. [Google Scholar]
- Rudski, L.G.; Lai, W.W.; Afilalo, J.; Hua, L.; Handschumacher, M.D.; Chandrasekaran, K.; Solomon, S.D.; Louie, E.K.; Schiller, N.B. Guidelines for the echocardiographic assessment of the right heart in adults: A report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J. Am. Soc. Echocardiogr. 2010, 23, 685–713. [Google Scholar] [PubMed]
- Lipczyńska, M.; Szymański, P.; Kumor, M.; Klisiewicz, A.; Mazurkiewicz, Ł.; Hoffman, P. Global longitudinal strain may identify preserved systolic function of the systemic right ventricle. Can. J. Cardiol. 2015, 31, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, E.; Mazurkiewicz, L.; Kowalski, M.; Klisiewicz, A.; Marczak, M.; Hoffman, P. Echocardiography vs magnetic resonance imaging in assessing ventricular function and systemic atrioventricular valve status in adults with congenitally corrected transposition of the great arteries. Echocardiography 2016, 33, 1697–1702. [Google Scholar] [CrossRef] [PubMed]
- Timóteo, A.T.; Branco, L.M.; Rosa, S.A.; Galrinho, A.; Sousa, L.; Oliveira, J.A.; Pinto, M.F.; Agapito, A.F.; Ferreira, R.C. Longitudinal strain by two-dimensional speckle tracking to assess ventricular function in adults with transposition of the great arteries: Can serial assessment be simplified? Rev. Port. Cardiol. 2018, 37, 739–745. [Google Scholar] [CrossRef]
- Schneider, M.; Beichl, M.; Nietsche, C.; Beitzke, D.; Porenta, G.; Beran, G.; Vonbank, K.; Hauser, J.; Hengstenberg, C.; Goliasch, G.; et al. Systematic evaluation of systemic right ventricular function. J. Clin Med. 2020, 9, 107. [Google Scholar] [CrossRef]
- Diller, G.P.; Dimopoulos, K.; Okonko, D.; Li, W.; Babu-Narayan, S.V.; Broberg, C.S.; Johansson, B.; Bouzas, B.; Mullen, M.J.; Poole-Wilson, P.A.; et al. Exercise intolerance in adult congenital heart disease: Comparative severity, correlates, and prognostic implication. Circulation 2005, 112, 828–835. [Google Scholar] [CrossRef]
- Kalogeropoulos, A.P.; Deka, A.; Border, W.; Pernetz, M.A.; Georgiopoulou, V.V.; Kiani, J.; McConnell, M.; Lerakis, S.; Butler, J.; Martin, R.P.; et al. Right ventricular function with standard and speckle-tracking echocardiography and clinical events in adults with D-transposition of the great arteries post atrial switch. J. Am. Soc. Echokardiogr. 2012, 25, 304–312. [Google Scholar] [CrossRef]
- Saremi, F.; Gera, A.; Ho, S.Y.; Hijazi, Z.M.; Sánchez-Quintana, D. CT and MR imaging of the pulmonary valve. Radiographics 2014, 34, 51–71. [Google Scholar] [CrossRef]
- Jonas, R.A. Comprehensive Surgical Management of Congenital Heart Disease, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Batteux, C.; Haidar, M.A.; Bonnet, D. 3D-Printed Models for Surgical Planning in Complex Congenital Heart Diseases: A Systematic Review. Front. Pediatr. 2019, 7, 23. [Google Scholar] [CrossRef]
- Sahayaraj, R.A.; Ramanan, S.; Subramanyan, R.; Cherian, K.M. 3D printing to model surgical repair of complex congenitally corrected transposition of the great arteries. World J. Pediatr. Congenit. Heart Surg. 2019, 10, 373–375. [Google Scholar] [CrossRef]
- Celi, S.; Gasparotti, E.; Capellini, K.; Vignali, E.; Fanni, B.M.; Ali, L.; Cantinotti, M.; Murzi, M.; Bertiego, B.; Santoro, G.; et al. 3D printing in modern cardiology. Curr. Pharm. Des. 2021, 27, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Fredriksen, P.; Chen, A.; Veldtman, G.; Hechter, S.; Therrien, J.; Webb, G. Exercise capacity in adult patients with congenitally corrected transposition of the great arteries. Heart 2001, 85, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Shafer, K.M.; Mann, N.; Hehn, R.; Tikkanen, A.U.; Valente, A.M.; Geva, T.; Gauthier, N.; Rhodes, J. Relationship between exercise parameters and noninvasive indices of right ventricular function in patients with biventricular circulation and systemic right ventricle. Congenit. Heart Dis. 2015, 10, 457–465. [Google Scholar] [CrossRef]
- Rog, B.; Salapa, K.; Okolska, M.; Dluzniewska, N.; Werynski, P.; Podolec, P.; Tomkiewicz-Pajak, L. Clinical evaluation of exercise capacity in adults with systemic right ventricle. Tex. Heart Inst. J. 2019, 46, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.P.; Sharma, R.; Li, W.; Leenarts, M.; Kalra, P.R.; Kemp, M.; Coats, A.J.S.; Anker, S.D.; Gatzouliset, M.A. Neurohormonal activation and the chronic heart failure syndrome in adults with congenital heart disease. Circulation 2002, 106, 92–99. [Google Scholar] [CrossRef]
- Westhoff-Bleck, M.; Podewski, E.; Tutarel, O.; Wenzel, D.; Cappello, C.; Bertram, H.; Bauersachs, J.; Widder, J. Prognostic value of NT-proBNP in patients with systemic morphological right ventricles: A single-centre experience. Int. J. Cardiol. 2013, 169, 433–438. [Google Scholar] [CrossRef]
- Kowalik, E.; Klisiewicz, A.; Rybicka, J.; Biernacka, E.K.; Hoffman, P. High sensitivity cardiac troponin T and systemic right ventricular function in adults with congenitally corrected transposition of the great arteries. Int. J. Cardiol. 2017, 241, 168–172. [Google Scholar] [CrossRef]
- Kowalik, E.; Klisiewicz, A.; Kowalski, M.; Rybicka, J.; Baranowski, R.; Biernacka, E.K.; Hoffman, P. High-Sensitive Cardiac Troponin T and Systemic Right Ventricular Area Predict Outcomes in Adults with Congenitally Corrected Transposition. Can. J. Cardiol. 2018, 34, 1129–1136. [Google Scholar] [CrossRef]
- Geenen, L.W.; Baggen, V.J.M.; van den Bosch, A.E.; Eindhoven, J.A.; Cuypers, J.A.A.E.; Witsenburg, M.; Boersmy, E.; Roos-Hesselink, J.W. Prognostic value of soluble ST2 in adults with congenital heart disease. Heart 2019, 105, 999–1006. [Google Scholar] [CrossRef]
- De Lemos, J.A.; McGuire, D.K.; Drazner, M.H. B-type natriuretic peptide in cardiovascular disease. Lancet 2003, 362, 316–322. [Google Scholar] [CrossRef]
- Tang, W.H. B-type natriuretic peptide: A critical review. Congest. Heart Fail. 2007, 13, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Palazzuoli, A.; Gallotta, M.; Quatrini, I.; Nuti, R. Natriuretic peptides (BNP and NT- proBNP): Measurement and relevance in heart failure. Vasc. Health Risk. Manag. 2010, 6, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Zandstra, T.E.; Nederend, M.; Jongbloed, M.R.M.; Kiès, P.; Vliegen, H.W.; Bouma, B.J.; Tops, L.F.; Schalij, M.J.; Egorova, A.D. Sacubitril/valsartan in the treatment of systemic right ventricular failure. Heart 2021, 107, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Raman, S.V.; Hoffman, T.M.; Hayes, J.; Daniels, C.J. Serum markers of systemic right ventricular function and exercise performance. Pediatr. Cardiol. 2008, 29, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.A.; Meurling, C.J.; Holmqvist, F.; Waktare, J.E.P.; Thilén, U.J. The diagnostic and prognostic value of brain natriuretic peptides in adults with a systemic morphologically right ventricle or Fontan-type circulation. Int. J. Cardiol. 2007, 114, 345–351. [Google Scholar] [CrossRef]
- Chow, P.C.; Cheung, E.W.Y.; Chong, C.Y.; Lun, K.S.; Yung, T.C.; Wong, K.T.; Chau, A.K.T.; Cheung, Y.F. Brain natriuretic peptide as a biomarker of systemic right ventricular function in patients with transposition of great arteries after atrial switch operation. Int. J. Cardiol. 2008, 127, 192–197. [Google Scholar] [CrossRef]
- Popelova, J.R.; Tomkova, M.; Tomek, J. NT-proBNP predicts mortality in adults with transposition of the great arteries late after Mustard or Senning correction. Congenit. Heart Dis. 2017, 12, 448–457. [Google Scholar] [CrossRef]
- Kowalik, E.; Kwiatek-Wrzosek, A.; Klisiewicz, A.; Lutyńska, A.; Biernacka, E.K.; Kowalski, M.; Hoffman, P. Systemic right ventricle in elderly patients with congenitally corrected transposition of the great arteries: Clinical profile, cardiac biomarkers, and echocardiographic parameters. Anatol. J. Cardiol. 2020, 24, 92–96. [Google Scholar] [CrossRef]
- Kowalik, E.; Klisiewicz, A.; Biernacka, E.K.; Hoffman, P. Pregnancy and long-term cardiovascular outcomes in women with congenitally corrected transposition of the great arteries. Int. J. Gynaecol. Obstet. 2014, 125, 154–157. [Google Scholar] [CrossRef]
- Therrien, J.; Barnes, I.; Somerville, J. Outcome of pregnancy in patients with congenitally corrected transposition of the great arteries. Am. J. Cardiol. 1999, 84, 820–824. [Google Scholar] [CrossRef]
- Wissocque, L.; Mondesert, B.; Dubart, A.E. Late diagnosis of isolated congenitally corrected transposition of the great arteries in a 92-year old woman. Eur. J. Cardiothorac. Surg. 2016, 49, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Helsen, F.; De Meester, P.; Van Keer, J.; Gabriels, C.; Van De Bruaene, A.; Herijgers, P.; Rega, F.; Meyns, B.; Gewillig, M.; Troost, E.; et al. Pulmonary outflow obstruction protects against heart failure in adults with congenitally corrected transposition of the great arteries. Int. J. Cardiol. 2015, 196, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Paladini, D.; Volpe, P.; Marasini, M.; Russo, M.G.; Vassallo, M.; Gentile, M.; Calabrò, R. Diagnosis, characterization and outcome of congenitally corrected transposition of the great arteries in the fetus: A multicenter series of 30 cases. Ultrasound Obstet. Gynecol. 2006, 27, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Spigel, Z.; Binsalamah, Z.M.; Caldarone, C. Congenitally Corrected Transposition of the Great Arteries: Anatomic, Physiologic Repair, and Palliation. Semin. Thorac. Cardiovasc. Surg. Pediatr. Card. Surg. Annu. 2019, 22, 32–42. [Google Scholar] [CrossRef]
- Beauchesne, L.M.; Warnes, C.A.; Connolly, H.M.; Ammash, N.M.; Tajik, A.J. Danielson GK. Outcome of the unoperated adult who presents with congenitally corrected transposition of the great arteries. J. Am. Coll. Cardiol. 2002, 40, 285–290. [Google Scholar] [CrossRef]
- De Leval, M.R.; Bastos, P.; Stark, J. Surgical technique to reduce the risks of heart block following closure of ventricular septal defect in atrioventricular discordance. J. Thorac. Cardiovasc. Surg. 1979, 78, 515–526. [Google Scholar] [CrossRef]
- Karl, T.R. The role of the Fontan operation in the treatment of congenitally corrected transposition of the great arteries. Ann. Pediatr. Cardiol. 2011, 4, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Dibardino, D.J.; Kleeman, K.; Bove, E.L. A method of transcutaneously adjustable pulmonary artery banding for staged left ventricular retraining. J. Thorac. Cardiovasc. Surg. 2012, 144, 553–556. [Google Scholar] [CrossRef]
- Winlaw, D.S.; McGuirk, S.P.; Balmer, C.; Langley, S.M.; Griselli, M.; Stümper, O.; De Giovanni, J.V.; Wright, J.G.; Thorne, S.; Barron, D.J.; et al. Intention-totreat analysis of pulmonary artery banding in conditions with a morphological right ventricle in the systemic circulation with a view to anatomic biventricular repair. Circulation 2005, 111, 405–411. [Google Scholar] [CrossRef]
- Bove, E.L. Congenitally corrected transposition of the great arteries: Ventricle to pulmonary artery connection strategies. Semin. Thorac. Cardiovasc. Surg. 1995, 7, 139–144. [Google Scholar]
- Karl, T.R.; Cochrane, A.D. Congenitally corrected transposition of the great arteries. In Pediatric Cardiac Surgery, 3rd ed.; Mavroudis, C., Backer, C., Eds.; Mosby: St. Louis, MO, USA, 2003; pp. 476–495. [Google Scholar]
- Ilbawi, M.N.; DeLeon, S.Y.; Backer, C.L.; Duffy, C.E.; Muster, A.J.; Zales, V.R.; Paul, M.H.; Idriss, F.S. An alternative approach to the surgical management of physiologically corrected transposition with ventricular septal defect and pulmonary stenosis or atresia. J. Thorac. Cardiovasc. Surg. 1990, 100, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Zubrzycki, M.; Schramm, R.; Costard-Jäckle, A.; Morshuis, M.; Gummert, J.F.; Zubrzycka, M. Pathogenesis and Surgical Treatment of Dextro-Transposition of the Great Arteries (D-TGA): Part II. J. Clin. Med. 2024, 13, 4823. [Google Scholar] [CrossRef]
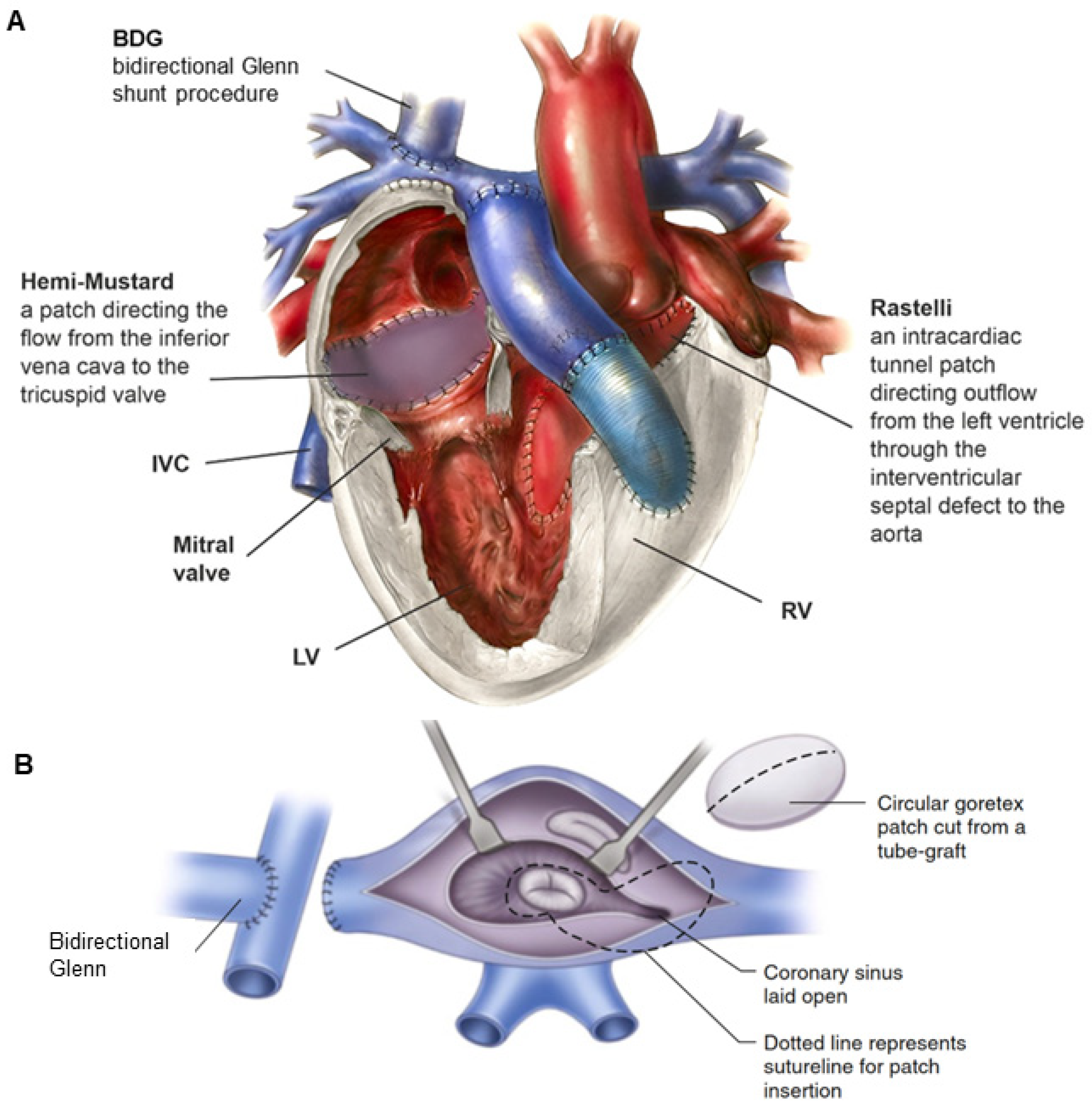
- Malhotra, S.P.; Reddy, V.M.; Qiu, M.; Pirolli, T.J.; Barboza, L.; Reinhartz, O.; Hanley, F.L. The hemi-Mustard/bidirectional Glenn atrial switch procedure in the double-switch operation for congenitally corrected transposition of the great arteries: Rationale and midterm results. J. Thorac. Cardiovasc. Surg. 2011, 141, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Weixler, W.H.M.; Kramer, P.; Murin, P.; Romanchenko, O.; Cho, M.-Y.; Ovroutski, S.; Hübler, M.; Berger, F.; Photiadis, J. Anatomic Repair of Congenitally Corrected Transposition: Reappraisal of Eligibility Criteria. Pediatr. Cardiol. 2022, 43, 1214–1222. [Google Scholar] [CrossRef]
- Ibrahimiye, A.N.; Mainwaring, R.D.; Patrick, W.L.; Downey, L.; Yarlagadda, V.; Hanley, F.L. Left ventricular retraining and double switch in patients with congenitally corrected transposition of the great arteries. World J. Pediatr. Congenit. Heart. Surg. 2017, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, T.; Matsumura, G.; Konuma, T.; Yamazaki, K.; Kurosawa, H.; Imai, Y. Long-term prognosis of double-switch operation for congenitally corrected transposition of the great arteries. Eur. J. Cardiothorac. Surg. 2012, 42, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Myers, P.O.; del Nido, P.J.; Geva, T.; Bautista-Hernandez, V.; Chen, P.; Mayer, J.E., Jr.; Emani, S.M. Impact of age and duration of banding on left ventricular preparation before anatomic repair for congenitally corrected transposition of the great arteries. Ann. Thorac. Surg. 2013, 96, 603–610. [Google Scholar] [CrossRef]
- Zartner, P.A.; Schneider, M.B.; Asfour, B.; Hraška, V. Enhanced left ventricular training in corrected transposition of the great arteries by increasing the preload. Eur. J. Cardiothorac. Surg. 2016, 49, 1571–1576. [Google Scholar] [CrossRef]
- Hraska, V.; Duncan, B.W.; Mayer, J.E., Jr.; Freed, M.; del Nido, P.J.; Jonas, R.A. Long-term outcome of surgically treated patients with corrected transposition of the great arteries. J. Thorac. Cardiovasc. Surg. 2005, 129, 182–191. [Google Scholar] [CrossRef]
- Quinn, D.W.; McGuirk, S.P.; Metha, C.; Nightingale, P.; de Giovanni, J.V.; Dhillon, R.; Miller, P.; Stumper, O.; Wright, J.G.; Barron, D.J.; et al. The morphologic left ventricle that requires training by means of pulmonary artery banding before the double-switch procedure for congenitally corrected transposition of the great arteries is at risk of late dysfunction. J. Thorac. Cardiovasc. Surg. 2008, 135, 1137–1144. [Google Scholar] [CrossRef]
- Duncan, B.W.; Mee, R.B.B.; Mesia, C.I.; Qureshi, A.; Rosenthal, G.L.; Seshadri, S.G.; Lane, G.K.; Latson, L.A. Results of the double switch operation for congenitally corrected transposition of the great arteries. Eur. J. Cardiothorac. Surg. 2003, 24, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.C.; Yu, J.H.; Brizard, C.P.; Mee, R.B.B. Long-term results of left ventricular dysfunction after atrial switch procedures. J. Thorac. Cardiovasc. Surg. 2004, 127, 975–981. [Google Scholar] [CrossRef]
- Fontan, F.; Baudet, E. Surgical repair of tricuspid atresia. Thorax 1971, 26, 240–248. [Google Scholar] [CrossRef]
- Liu, R.; Pang, K.; Rui, L.; Zhang, G.; Wang, C.; Li, S. Congenitally corrected transposition with left ventricular outflow obstruction and cardiac malposition: One-and-a-half ventricular repair vs. Fontan pathway? Front. Cardiovasc. Med. 2022, 9, 938118. [Google Scholar] [CrossRef] [PubMed]
- Rychik, J.; Atz, A.M.; Celermajer, D.S.; Deal, B.J.; Gatzoulis, M.A.; Gewillig, M.H.; Hsia, T.Y.; Hsu, D.T.; Kovacs, A.H.; McCrindle, B.W.; et al. Evaluation and Management of the Child and Adult with Fontan Circulation: A scientific statement from the American Heart Association. Circulation 2019, 140, E234–E284. [Google Scholar] [CrossRef]
- Van den Eynde, J.; Bartelse, S.; Rijnberg, F.M.; Kutty, S.; Jongbloed, M.R.; de Bruin, C.; Hazekamp, M.G.; Le Cessie, S.; Roest, A.W. Somatic growth in single ventricle patients: A systematic review and meta-analysis. Acta Pediatr. 2023, 112, 186–199. [Google Scholar] [CrossRef]
- Hörer, J.; Schreiber, C.; Krane, S.; Prodan, Z.; Cleuziou, J.; Vogt, M.; Holper, K.; Lange, R. Outcome after surgical repair/palliation of congenitally corrected transposition of the great arteries. Thorac. Cardiovasc. Surg. 2008, 56, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Stamm, C.; Friehs, I.; Mayer, J.E., Jr.; Zurakowski, D.; Triedman, J.K.; Moran, A.M.; Walsh, E.P.; Lock, J.E.; Jonas, R.A.; Del Nido, P.J. Long-term results of the lateral tunnel Fontan operation. J. Thorac. Cardiovasc. Surg. 2001, 121, 28–41. [Google Scholar] [CrossRef]
- Gewillig, M.; Goldberg, D.J. Failure of the Fontan circulation. Heart Fail. Clin. 2014, 10, 105–116. [Google Scholar] [CrossRef]
- Surrey, L.F.; Glatz, A.C.; Dodds, K.; O’Byrne, M.L.; Lin, H.C. Hepatic fibrosis is universal following fontan operation, and severity is associated with time from surgery: A liver biopsy and hemodynamic study. J. Am. Heart Assoc. 2017, 6, e004809. [Google Scholar]
- Shin’oka, T.; Kurosawa, H.; Imai, Y.; Aoki, M.; Ishiyama, M.; Sakamoto, T.; Miyamoto, S.; Hobo, K.; Ichihara, Y. Outcomes of definitive surgical repair for congenitally corrected transposition of the great arteries or double outlet right ventricle with discordant atrioventricular connections: Risk analyses in 189 patients. J. Thorac. Cardiovasc. Surg. 2007, 133, 1318–1328.e4. [Google Scholar] [CrossRef] [PubMed]
- Ilbawi, M.N.; Ocampo, C.B.; Allen, B.S.; Barth, M.J.; Roberson, D.A.; Chiemmongkoltip, P.; Arcilla, R.A. Intermediate results of the anatomic repair for congenitally corrected transposition. Ann. Thorac. Surg. 2002, 73, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Langley, S.M.; Winlaw, D.S.; Stumper, O.; Dhillon, R.; De Giovanni, J.V.; Wright, J.G.; Miller, P.; Sethia, B.; Barron, D.J.; Brawn, W.J. Midterm results after restoration of the morphologically left ventricle to the systemic circulation in patients with congenitally corrected transposition of the great arteries. J. Thorac. Cardiovasc. Surg. 2003, 125, 1229–1241. [Google Scholar] [CrossRef]
- Hofferberth, S.C.; Alexander, M.E.; Mah, D.Y.; Bautista-Hernandez, V.; del Nido, P.J.; Fynn-Thompson, F. Impact of pacing on systemic ventricular function in L-transposition of the great arteries. J. Thorac. Cardiovasc. Surg. 2016, 151, 131–138. [Google Scholar] [CrossRef]
- Kapa, S.; Vaidya, V.; Hodge, D.O.; McLeod, C.J.; Connolly, H.M.; Warnes, C.A.; Asirvatham, S.J. Right ventricular dysfunction in congenitally corrected transposition of the great arteries and risk of ventricular tachyarrhythmia and sudden death. Int. J. Cardiol. 2018, 258, 83–89. [Google Scholar] [CrossRef]
- Bottega, N.A.; Kapa, S.; Edwards, W.D.; Connolly, H.M.; Munger, T.M.; Warnes, C.A.; Asirvatham, S.J. The cardiac veins in congenitally corrected transposition of the great arteries: Delivery options for cardiac devices. Heart Rhythm. 2009, 6, 1450–1456. [Google Scholar] [CrossRef]
- De Becker, B.; O’Neill, L.; Pierard, S.; Le Polain De Waroux, J.B. Cardiac resynchronization therapy using conduction system pacing after double-switch surgery for congenitally corrected transposition of the great arteries: A case report. Eur. Heart J. Case Rep. 2023, 7, ytad382. [Google Scholar] [CrossRef]
- Jauvert, G.; Rousseau-Paziaud, J.; Villain, E.; Iserin, L.; Hidden-Lucet, F.; Ladouceur, M.; Sidi, D. Effects of cardiac resynchronization therapy on echocardiographic indices, functional capacity, and clinical outcomes of patients with a systemic right ventricle. Europace 2009, 11, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, R.K.; Moore, J.P.; Taverne, Y.; Bramer, W.M.; Bogers, A.; de Groot, N.M.S. Cardiac resynchronization therapy for the failing systemic right ventricle: A systematic review. Int. J. Cardiol. 2020, 318, 74–81. [Google Scholar] [CrossRef]
- Yeo, W.T.; Jarman, J.W.E.; Li, W.; Gatzoulis, M.A.; Wong, T. Adverse impact of chronic subpulmonary left ventricular pacing on systemic right ventricular function in patients with congenitally corrected transposition of the great arteries. Int. J. Cardiol. 2014, 171, 184–191. [Google Scholar] [CrossRef]
- Vijayaraman, P.; Ponnusamy, S.S.; Cano, Ó.; Sharma, P.S.; Naperkowski, A.; Subsposh, F.A.; Moscal, P.; Bednarek, A.; Dal Forno, A.R.; Young, W.; et al. Left bundle branch area pacing for cardiac resynchronization therapy: Results from the International LBBAP Collaborative Study Group. JACC Clin. Electrophysiol. 2021, 7, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, X.; Su, L.; Wu, S.; Xia, X.; Vijayaraman, P. A beginner’s guide to permanent left bundle branch pacing. Heart Rhythm. 2019, 16, 1791–1796. [Google Scholar] [CrossRef]
- Huang, J.; Slaughter, M.S. HeartWare ventricular assist device placement in a patient with congenitally corrected transposition of the great arteries. J. Thorac. Cardiovasc. Surg. 2013, 145, e23–e25. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Rega, F.; Burkhoff, D.; Meyns, B. The use of a CircuLite micro-pump for congenitally corrected transposition of the great arteries. Eur. J. Cardiothorac. Surg. 2012, 42, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Lam, W.W.; Rodriguez, F.H.; Ermis, P.R.; Simpson, L.; Frazier, O.H.; Franklin, W.J.; Parekh, D.R. Clinical outcomes after ventricular assist device implantation in adults with complex congenital heart disease. J. Heart Lung Transplant. 2013, 32, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Schramm, R.; Gummert, J.F. Heart transplantation: Current situation. Chirurgie 2024, 95, 101–107. [Google Scholar] [CrossRef]
- Muñoz-Guijosa, C.; Ginel, A.; Montiel, J.; Brosa, V.; Mirabet, S.; Bayés-Genis, A.; Padró, J.M. Orthotopic Heart Transplantation in Patients with Transposition of the Great Arteries. Rev. Esp. Cardiol. 2009, 62, 216–219. [Google Scholar] [CrossRef]
- Duncan, B.W.; Mee, R.B. Management of the failing systemic right ventricle. Semin. Thorac. Cardiovasc. Surg. 2005, 17, 160–169. [Google Scholar] [CrossRef]
- Mongeon, F.P.; Connolly, H.M.; Dearani, J.A.; Li, Z.; Warnes, C.A. Congenitally corrected transposition of the great arteries ventricular function at the time of systemic atrioventricular valve replacement predicts long-term ventricular function. J. Am. Coll. Cardiol. 2011, 57, 2008–2017. [Google Scholar] [CrossRef]
- Van Son, J.A.; Danielson, G.K.; Huhta, J.C.; Warnes, C.A.; Edwards, W.D.; Schaff, V.Y.; Puga, F.J.; Ilstrup, D.M. Late results of systemic atrioventricular valve replacement in corrected transposition. J. Thorac. Cardiovasc. Surg. 1995, 109, 642–652. [Google Scholar] [CrossRef]
- Sue, S.H.; Wei, J.; Chuang, Y.C.; Chang, C.Y.; Lee, W.C.; Lee, S.L. Cardiac transplantation for congenitally corrected transposition of the great arteries: A case report. Transplant. Proc. 2008, 40, 2844–2845. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubrzycki, M.; Schramm, R.; Costard-Jäckle, A.; Morshuis, M.; Grohmann, J.; Gummert, J.F.; Zubrzycka, M. Pathogenesis and Surgical Treatment of Congenitally Corrected Transposition of the Great Arteries (ccTGA): Part III. J. Clin. Med. 2024, 13, 5461. https://doi.org/10.3390/jcm13185461
Zubrzycki M, Schramm R, Costard-Jäckle A, Morshuis M, Grohmann J, Gummert JF, Zubrzycka M. Pathogenesis and Surgical Treatment of Congenitally Corrected Transposition of the Great Arteries (ccTGA): Part III. Journal of Clinical Medicine. 2024; 13(18):5461. https://doi.org/10.3390/jcm13185461
Chicago/Turabian StyleZubrzycki, Marek, Rene Schramm, Angelika Costard-Jäckle, Michiel Morshuis, Jochen Grohmann, Jan F. Gummert, and Maria Zubrzycka. 2024. "Pathogenesis and Surgical Treatment of Congenitally Corrected Transposition of the Great Arteries (ccTGA): Part III" Journal of Clinical Medicine 13, no. 18: 5461. https://doi.org/10.3390/jcm13185461
APA StyleZubrzycki, M., Schramm, R., Costard-Jäckle, A., Morshuis, M., Grohmann, J., Gummert, J. F., & Zubrzycka, M. (2024). Pathogenesis and Surgical Treatment of Congenitally Corrected Transposition of the Great Arteries (ccTGA): Part III. Journal of Clinical Medicine, 13(18), 5461. https://doi.org/10.3390/jcm13185461