
Diagnosis, Management and Outcome of Truncus Arteriosus Communis Diagnosed during Fetal Life—Cohort Study and Systematic Literature Review

 , , , and
, , , and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Data Collection of Local Cases
2.2. Literature Review and Analysis
2.3. Methodological Evaluation of Included Case Series
2.4. Analysis of Included Studies and Data Synthesis
2.5. Classification of Morphological TAC Subtypes
2.5.1. Collett and Edwards (1949)
2.5.2. Original Van Praagh Classification (1964)
2.5.3. Modified Van Praagh Classification (2000)
2.5.4. Russell et al. (2011)
2.5.5. Statistical Analysis
3. Results
3.1. Analysis of Local TAC Cases
3.2. Review and Comparison with Published Case Series
Selection and Evaluation of Methodological Quality of Included Case Series in the Literature
3.3. Study Characteristics of Reviewed Literature
3.4. Diagnostic Accuracy and Pregnancy Outcome of Fetal Echocardiography
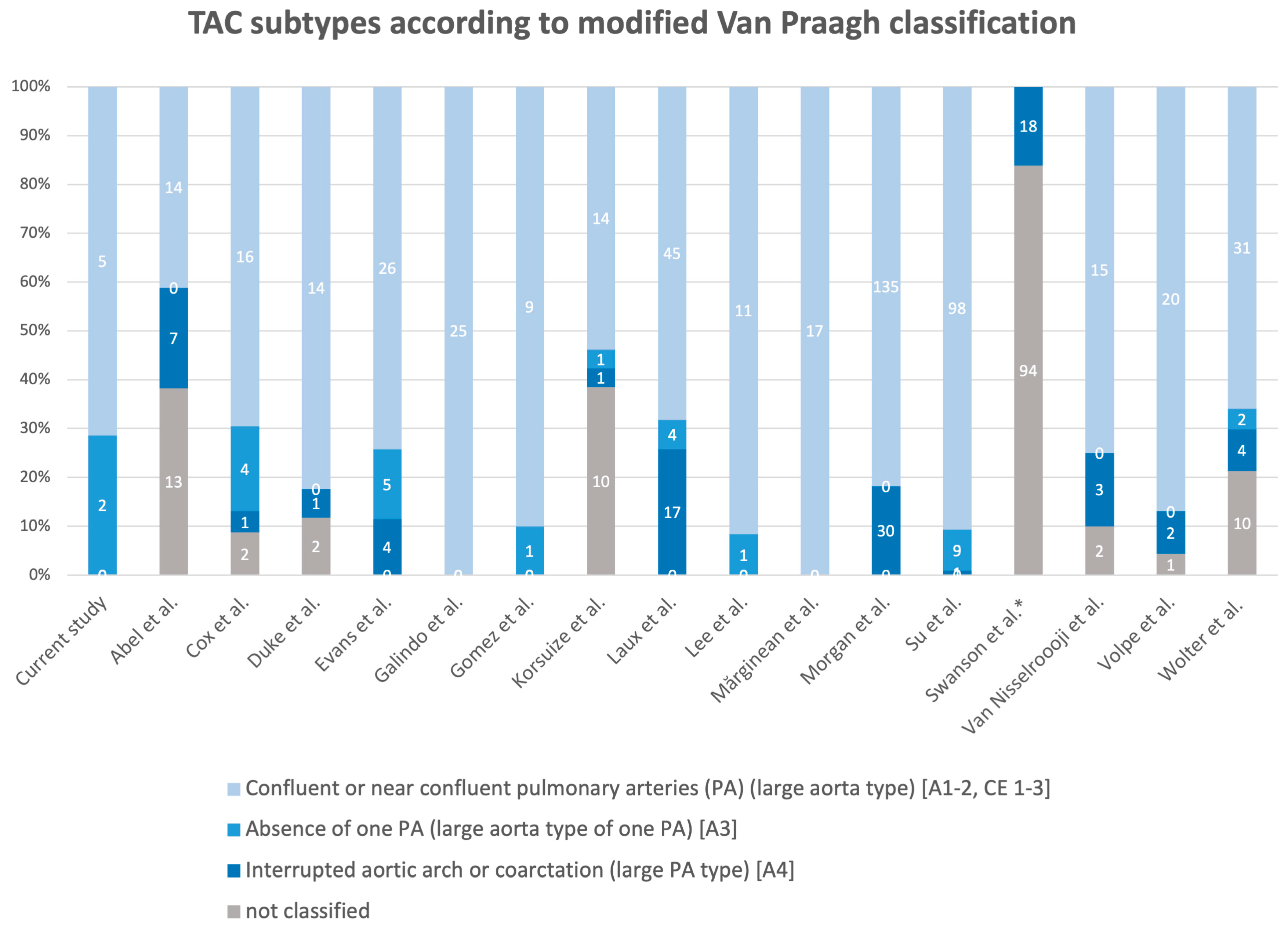
3.5. Classification Systems
3.6. Associated Truncal Valve Regurgitation or Stenosis
3.7. Associated Extracardiac Anomalies
3.8. Genetic Analysis
3.9. Termination of Pregnancy
3.10. Outcome and Survival
4. Discussion
4.1. Risk Stratification in Utero
4.2. Integration of Genetic Counseling
4.3. Impact of Prenatal Diagnosis on Postnatal Outcomes
4.4. Limitations
5. Conclusions
What Does This Study Add to the Clinical Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Collett, R.W.; Edwards, J.E. Persistent truncus arteriosus: A classification according to anatomic types. Surg. Clin. N. Am. 1949, 29, 1245–1270. [Google Scholar] [CrossRef] [PubMed]
- Van Praagh, R.; Van Praagh, S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. Am. J. Cardiol. 1965, 16, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Calder, L.; Van Praagh, R.; Van Praagh, S.; Sears, W.P.; Corwin, R.; Levy, A.; Keith, J.D.; Paul, M.H. Clinical, angiocardiographic, and pathologic findings in 100 patients. Am. Heart J. 1976, 92, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.L. Congenital Heart Surgery Nomenclature and Database Project: Truncus arteriosus. Ann. Thorac. Surg. 2000, 69, 50–55. [Google Scholar] [CrossRef]
- Russell, H.M.; Jacobs, M.L.; Anderson, R.H.; Mavroudis, C.; Spicer, D.; Corcrain, E.; Backer, C.L. A simplified categorization for common arterial trunk. J. Thorac. Cardiovasc. Surg. 2011, 141, 645–653. [Google Scholar] [CrossRef]
- Hazekamp, M.G.; Barron, D.J.; Dangel, J.; Homfray, T.; Jongbloed, M.R.; Voges, I.; ESC Scientific Document Group. Consensus document on optimal management of patients with common arterial trunk. Cardiol. Young 2021, 31, 915–939. [Google Scholar] [CrossRef]
- Patel, A.; Costello, J.M.; Backer, C.L.; Pasquali, S.K.; Hill, K.D.; Wallace, A.S.; Jacobs, J.P.; Jacobs, M.L. Prevalence of Noncardiac and Genetic Abnormalities in Neonates Undergoing Cardiac Operations: Analysis of The Society of Thoracic Surgeons Congenital Heart Surgery Database. Ann. Thorac. Surg. 2016, 102, 1607–1614. [Google Scholar] [CrossRef]
- Hardin, J.; Carmichael, S.L.; Selvin, S.; Lammer, E.J.; Shaw, G.M. Increased prevalence of cardiovascular defects among 56,709 California twin pairs. Am. J. Med. Genet. Pt. A 2009, 149, 877–886. [Google Scholar] [CrossRef]
- Swanson, T.M.; Selamet Tierney, E.S.; Tworetzky, W.; Pigula, F.; McElhinney, D.B. Truncus Arteriosus: Diagnostic Accuracy, Outcomes, and Impact of Prenatal Diagnosis. Pediatr. Cardiol. 2009, 30, 256–261. [Google Scholar] [CrossRef]
- Ferdman, B.; Singh, G. Persistent truncus arteriosus. Curr. Treat Options Cardiovasc. Med. 2003, 5, 429–438. [Google Scholar] [CrossRef]
- Derridj, N.; Villemain, O.; Khoshnood, B.; Belhadjer, Z.; Gaudin, R.; Raisky, O.; Bonnet, D. Outcomes after common arterial trunk repair: Impact of the surgical technique. J. Thorac. Cardiovasc. Surg. 2021, 162, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, M.L.; Mercer-Rosa, L.; Zhao, H.; Zhang, X.; Yang, W.; Cassedy, A.; Fogel, M.A.; Rychik, J.; Tanel, R.E.; Marino, B.S.; et al. Morbidity in children and adolescents after surgical correction of truncus arteriosus communis. Am. Heart J. 2013, 166, 512–518. [Google Scholar] [CrossRef]
- Abel, J.S.; Berg, C.; Geipel, A.; Gembruch, U.; Herberg, U.; Breuer, J.; Brockmeier, K.; Gottschalk, I. Prenatal diagnosis, associated findings and postnatal outcome of fetuses with truncus arteriosus communis (TAC). Arch. Gynecol. Obs. 2021, 304, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Murad, M.H.; Sultan, S.; Haffar, S.; Bazerbachi, F. Methodological quality and synthesis of case series and case reports. BMJ Evid. Based Med. 2018, 23, 60–63. [Google Scholar] [CrossRef]
- Lo, C.K.-L.; Mertz, D.; Loeb, M. Newcastle-Ottawa Scale: Comparing reviewers’ to authors’ assessments. BMC Med. Res. Methodol. 2014, 14, 45. [Google Scholar] [CrossRef]
- Bourdial, H.; Jamal-Bey, K.; Edmar, A.; Caillet, D.; Wuillai, F.; Bernede-Bauduin, C.; Boumahni, B.; Robillard, P.Y.; Kauffmann, E.; Laffitte, A.; et al. Congenital heart defects in La Réunion Island: A 6-year survey within a EUROCAT-affiliated congenital anomalies registry. Cardiol. Young 2012, 22, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.; Husain, N.; Jhaveri, S.; Geiger, M.; Berhane, H.; Patel, S. Fetal Echocardiographic Variables Associated with Pre-Surgical Mortality in Truncus Arteriosus: A Pilot Study. Pediatr. Cardiol. 2023, 44, 1397–1405. [Google Scholar] [CrossRef]
- Duke, C.; Sharland, G.K.; Jones, A.M.R.; Simpson, J.M. Echocardiographic features and outcome of truncus arteriosus diagnosed during fetal life. Am. J. Cardiol. 2001, 88, 1379–1384. [Google Scholar] [CrossRef]
- Evans, W.N.; Acherman, R.J.; Ciccolo, M.L.; Lehoux, J.; Galindo, A.; Rothman, A.; Mayman, G.A.; Restrepo, H. Common arterial trunk in the era of high prenatal detection rates: Results of neonatal palliation and primary repair. J. Card. Surg. 2021, 36, 4090–4094. [Google Scholar] [CrossRef]
- Galindo, A.; Mendoza, A.; Arbues, J.; Graneras, A.; Escribano, D.; Nieto, O. Conotruncal anomalies in fetal life: Accuracy of diagnosis, associated defects and outcome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 146, 55–60. [Google Scholar] [CrossRef]
- Gómez, O.; Soveral, I.; Bennasar, M.; Crispi, F.; Masoller, N.; Marimon, E.; Bartrons, J.; Gratacós, E.; Martinez, J.M. Accuracy of Fetal Echocardiography in the Differential Diagnosis between Truncus Arteriosus and Pulmonary Atresia with Ventricular Septal Defect. Fetal Diagn. Ther. 2016, 39, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Korsuize, N.A.; Bakhuis, W.; van Wijk, B.; Grotenhuis, H.B.; Ter Heide, H.; de Lara, M.C.; Fejzic, Z.; Schoof, P.H.; Haas, F.; Steenhuis, T.J. Truncus arteriosus from prenatal diagnosis to clinical outcome: A single-centre experience. Cardiol. Young 2024, 34, 1–7. [Google Scholar] [CrossRef]
- Laux, D.; Derridj, N.; Stirnemann, J.; Lucron, H.; Stos, B.; Levy, M.; Houyel, L.; Bonnet, D. Accuracy and impact of prenatal diagnosis of common arterial trunk. Ultrasound Obstet. Gynecol. 2022, 60, 223–233. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Won, H.-S.; Lee, B.S.; Kim, E.A.R.; Kim, Y.H.; Park, J.J.; Yun, T.J. Prenatal Diagnosis of Common Arterial Trunk: A Single-Center’s Experience. Fetal Diagn. Ther. 2013, 34, 152–157. [Google Scholar] [CrossRef]
- Marginean, C.; Gozar, L.; Mărginean, C.O.; Suciu, H.; Togănel, R.; Muntean, I.; Mureșan, M.C. Prenatal diagnosis of the fetal common arterial trunk. A case series. Med. Ultrason. 2018, 1, 100. [Google Scholar] [CrossRef]
- Morgan, C.T.; Tang, A.; Fan, C.-P.; Golding, F.; Manlhiot, C.; van Arsdell, G.; Honjo, O.; Jaeggi, E. Contemporary Outcomes and Factors Associated with Mortality After a Fetal or Postnatal Diagnosis of Common Arterial Trunk. Can. J. Cardiol. 2019, 35, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, S.; Glickstein, J.; Printz, B.; Allan, L.D.; Altmann, K.; Solowiejczyk, D.E.; Simpson, L.; Perez-Delboy, A.; Kleinman, C.S. Prenatal Diagnosis of Conotruncal Malformations: Diagnostic Accuracy, Outcome, Chromosomal Abnormalities, and Extracardiac Anomalies. Amer. J. Perinatol. 2006, 23, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Li, T.; Ma, B.; Wang, A.L.; Wei, C.M.; Tie, H.X.; Guo, W. Clinical value of ultrasound in diagnosis and classification of common arterial trunk. J. Clin. Ultrasound 2023, 51, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Van Nisselrooij, A.E.L.; Herling, L.; Clur, S.; Linskens, I.H.; Pajkrt, E.; Rammeloo, L.A.; Ten Harkel, A.D.; Hazekamp, M.G.; Blom, N.A.; Haak, M.C. The prognosis of common arterial trunk from a fetal perspective: A prenatal cohort study and systematic literature review. Prenat. Diagn. 2021, 41, 754–765. [Google Scholar] [CrossRef]
- Volpe, P. Common arterial trunk in the fetus: Characteristics, associations, and outcome in a multicentre series of 23 cases. Heart 2003, 89, 1437–1441. [Google Scholar] [CrossRef]
- Wolter, A.; Haessig, A.; Kurkevych, A.; Weichert, J.; Bosselmann, S.; Mielke, G.; Bedei, I.A.; Schenk, J.; Widriani, E.; Axt-Fliedner, R. Prenatal Diagnosis, Course and Outcome of Patients with Truncus Arteriosus Communis. J. Clin. Med. 2024, 13, 4465. [Google Scholar] [CrossRef] [PubMed]
- Russell, H.M.; Pasquali, S.K.; Jacobs, J.P.; Jacobs, M.L.; O’Brien, S.M.; Mavroudis, C.; Backer, C.L. Outcomes of Repair of Common Arterial Trunk with Truncal Valve Surgery: A Review of The Society of Thoracic Surgeons Congenital Heart Surgery Database. Ann. Thorac. Surg. 2012, 93, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D.; McElhinney, D.B.; Reddy, M.; Petrossian, E.; Silverman, N.H.; Hanley, F.L. Neonatal repair of truncus arteriosus: Continuing improvement in outcomes. Ann. Thorac. Surg. 2001, 72, 391–395. [Google Scholar] [CrossRef]
- Brown, J.W.; Ruzmetov, M.; Okada, Y.; Vijay, P.; Turrentine, M.W. Truncus arteriosus repair: Outcomes, risk factors, reoperation and management. Eur. J. Cardiothorac. Surg. 2001, 20, 221–227. [Google Scholar] [CrossRef]
- Konstantinov, I.E.; Karamlou, T.; Blackstone, E.H.; Mosca, R.S.; Lofland, G.K.; Caldarone, C.A.; Williams, W.G.; Mackie, A.S.; McCrindle, B.W. Truncus Arteriosus Associated with Interrupted Aortic Arch in 50 Neonates: A Congenital Heart Surgeons Society Study. Ann. Thorac. Surg. 2006, 8, 214–222. [Google Scholar] [CrossRef]
- Jones, B.A.; Conaway, M.R.; Spaeder, M.C.; Dean, P.N. Hospital Survival After Surgical Repair of Truncus Arteriosus with Interrupted Aortic Arch: Results from a Multi-institutional Database. Pediatr. Cardiol. 2021, 42, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Buckley, J.R.; Costello, J.M.; Smerling, A.J.; Sassalos, P.; Amula, V.; Cashen, K.; Riley, C.M.; Bakar, A.M.; Iliopoulos, I.; Jennings, A.; et al. Contemporary Multicenter Outcomes for Truncus Arteriosus with Interrupted Aortic Arch. Ann. Thorac. Surg. 2023, 115, 144–150. [Google Scholar] [CrossRef]
- Mitta, A.; Vogel, A.D.; Korte, J.E.; Brennan, E.; Bradley, S.M.; Kavarana, M.N.; Konrad Rajab, T.; Kwon, J.H. Outcomes in Primary Repair of Truncus Arteriosus with Significant Truncal Valve Insufficiency: A Systematic Review and Meta-analysis. Pediatr. Cardiol. 2023, 44, 1649–1657. [Google Scholar] [CrossRef]
- Mastropietro, C.W.; Amula, V.; Sassalos, P.; Buckley, J.R.; Smerling, A.J.; Iliopoulos, I.; Riley, C.M.; Jennings, A.; Cashen, K.; Narasimhulu, S.S.; et al. Characteristics and operative outcomes for children undergoing repair of truncus arteriosus: A contemporary multicenter analysis. J. Thorac. Cardiovasc. Surg. 2019, 157, 2386–2398. [Google Scholar] [CrossRef]
- Mille, F.K.; Shankar, V.R. Truncus arteriosus survival outcomes: Does 22q 11.2 deletion matter? J. Card. Surg. 2020, 35, 3263–3265. [Google Scholar] [CrossRef]
- Buckley, J.R.; Amula, V.; Sassalos, P.; Costello, J.M.; Smerling, A.J.; Iliopoulos, I.; Jennings, A.; Riley, C.M.; Cashen, K.; Narasimhulu, S.S.; et al. Multicenter Analysis of Early Childhood Outcomes After Repair of Truncus Arteriosus. Ann. Thorac. Surg. 2019, 107, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Guariento, A.; Doulamis, I.P.; Staffa, S.J.; Gellis, L.; Oh, N.A.; Kido, T.; Mayer, J.E.; Baird, C.W.; Emani, S.M.; Zurakowski, D.; et al. Long-term outcomes of truncus arteriosus repair: A modulated renewal competing risks analysis. J. Thorac. Cardiovasc. Surg. 2022, 163, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Hook, J.E.; Delany, D.R.; Buckley, J.R.; Chowdhury, S.M.; Kavarana, M.N.; Costello, J.M. Factors Associated with Mortality and Adverse Outcomes After Truncus Arteriosus Repair. Ann. Thorac. Surg. 2023, 116, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Naimo, P.S.; Konstantinov, I.E. Surgery for Truncus Arteriosus: Contemporary Practice. Ann. Thorac. Surg. 2021, 1, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Ramirez, C.; Ibarra, C.; Binsalamah, Z.M.; Adachi, I.; Heinle, J.S.; McKenzie, E.D.; Caldarone, C.A.; Imamura, M. Right Ventricle to Pulmonary Artery Conduit Size Is Associated with Conduit and Pulmonary Artery Reinterventions After Truncus Arteriosus Repair. Semin. Thorac. Cardiovasc. Surg. 2022, 34, 1003–1009. [Google Scholar] [CrossRef]
- Bonilla-Ramirez, C.; Ibarra, C.; Binsalamah, Z.M.; Adachi, I.; Heinle, J.S.; McKenzie, E.D.; Caldarone, C.A.; Imamura, M. Coronary Artery Anomalies Are Associated with Increased Mortality After Truncus Arteriosus Repair. Ann. Thorac. Surg. 2021, 112, 2005–2011. [Google Scholar] [CrossRef]
- Baschat, A.A.; Muench, M.V.; Gembruch, U. Coronary artery blood flow velocities in various fetal conditions. Ultrasound Obstet. Gynecol. 2003, 21, 426–429. [Google Scholar] [CrossRef]
- Hörer, J. Current spectrum, challenges and new developments in the surgical care of adults with congenital heart disease. Cardiovasc. Diagn. Ther. 2018, 8, 754–764. [Google Scholar] [CrossRef]
- Tay, H.; Naimo, P.S.; Huang, L.; Fricke, T.A.; Brink, J.; d’Udekem, Y.; Brizard, C.P.; Konstantinov, I.E. Long-term quality of life in adults following truncus arteriosus repair. Interact. CardioVasc. Thorac. Surg. 2019, 29, 950–954. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Evaluation Questions |
|---|---|
| Selection * | Do the patients represent the whole experience of the center? Is the selection method clear and does it ensure that all cases of suspected TAC are reported? |
| Ascertainment ** | Was the presence of a suspected or confirmed TAC adequately ascertained? Was the outcome of TAC-suspected cases adequately ascertained? |
| Causality * | Was follow-up long enough for the evaluation of the outcome of TAC-suspected pregnancies? |
| Reporting * | Was the study’s approach with regard to inclusion and analysis criteria sufficiently described to allow the case series to be replicated by other researchers or for practitioners to draw relevant conclusions for their own practice? |
| Prenatal Diagnosis | GA at Referral | Pregnancy Outcome | Postnatal Diagnosis | Surgical Outcome | Microdeletion 22q11 |
Associated Cardiac |
Anomalies Extracardiac/Pregnancy-Related |
Survival at Last Follow-Up | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | TAC type 2 | 22 + 2 | Elective CD | TAC type 1 | Primary repair | - | RAA, ASD 2, PDA, PLSVC | IUGR anal atresia with vestibular fistula | alive (23 m) |
| 2 | TAC type 1 | 22 + 0 | Emergency CD | TAC type 1 | Post-OP-NND after pulmonary artery banding | negative | Truncal valve insufficiency, PFO, myocardial hypertrophy, single coronary ostium | Polyhydramnios | NND (8 d) |
| 3 | TAC type 1 | 16 + 2 | TOP | - | - | - | Truncal valve insufficiency and stenosis, mitral and tricuspid insufficiency, | Hypoplastic thymus | TOP |
| 4 | TAC type 1 | 23 + 3 | TOP | - | - | negative | Hypoplastic left ventricle | missing left tibia, bilateral clubfeet, thickened left thumb, short nasal bone, prefrontal skin edema, thickened neck | TOP |
| 5 | TAC type 1 | 21 + 5 | Elective CD | Pulmonary atresia with VSD | - | - | - | - | - |
| 6 | TAC type 1 | 19 + 0 | SVD | TAC type 1 | Primary repair | - | - | Polyhydramnios | alive (21 m) |
| 7 | TAC type 1 | 17 + 0 | TOP | - | - | - | Hypoplastic left ventricle, mitral atresia with VSD | Cerebral ventriculomegaly, micrognathia | TOP |
| 8 | TAC type 1 | 24 + 6 | Elective re-CD | TAC type 1 | Primary repair | negative | - | - | alive (1 m) |
| 9 | TAC type 1 | 20 + 5 | VD after IoL * | DORV | - | - | - | - | - |
| 10 | TAC type 1 | 20 + 3 | VAVD * | TAC type 1 | Primary repair | - | RAA | Penile hypospadias | alive (44 m) |
| 11 | TAC type 1 | 19 + 4 | TOP | - | - | negative [Trisomy 13] | Overriding aorta, VSD | Alobular holoprosencephaly, microcephaly, enlarged cisterna magna, bilateral microphthalmy, proboscis, cleft lip and palate, bilateral nephromegaly, bilateral oligodactyly of the feet, bilateral polydactyly of the hands | TOP |
| 12 | TAC type 3 | 33 + 6 | Elective CD | Pulmonary atresia with VSD (with one great right sided MAPCA arising from DAO and perfusion of the LPA via DA from the aortic arch) | - | - | RAA | - | - |
| 13 | TAC type 2 | 23 + 4 | SVD | TAC type 2 | Primary repair | negative | RAA | - | alive (33 m) |
| 14 | TAC type 2 | 28 + 3 | VD after IoL * | TAC type 2 | Primary repair | - | Peripheral pulmonary stenosis | - | alive (46 m) |
| Study | Current Study | Abel et al. [13] | Bourdial et al. [16] | Cox et al. [17] | Duke et al. [18] | Evans et al. [19] | Galindo et al. [20] | Gómez et al. [21] | Korsuize et al. [22] | Laux et al. [23] | Lee et al. [24] | Mărgi-nean et al. [25] | Morgan et al. [26] | Sivanan-dam et al. [27] | Su et al. [28] | Swanson et al. [9] | v. Nissel-rooji et al. [29] | Volpe et al. [30] | Wolter et al. [31] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Methodological quality | |||||||||||||||||||
| Selection * | * | * | * | - | * | * | * | * | * | * | * | * | * | (*) | * | * | * | * | |
| Ascertainment ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | * | ** | ** | ** | |
| Causality * | * | * | * | * | * | * | * | * | * | * | * | * | * | (*) | - | * | * | * | |
| Reporting * | * | (*) | * | * | (*) | * | * | * | * | * | - | * | * | - | - | * | * | * | |
| Study characteristics | |||||||||||||||||||
| Country | GER | GER | RE | USA | UK | USA | ES | ES | NL | FR | KR | RO | CA | USA | CN | CA | NL | IT | GER, UA |
| Collection period | 2019–2024 | 2010–2018 | 2002–2007 | 2010–2020 | 1990–1999 | 2006–2021 | 2005–2009 | 2006–2013 | 2005–2020 | 2011–2019 | 2003–2012 | 2009–2017 | 1990–2014 | 1994–2003 | 2017–2022 | 1992–2007 | 2002–2016 | 1993–2002 | 2008–2021 |
| Initial diagnosis | pre | pre | pre + post | pre | pre | pre + post | pre + post | pre | pre + post | pre + post | pre | pre | pre + post | pre | pre | pre + post | pre | pre | pre |
| Included cases | |||||||||||||||||||
| Prenatal | 11 | 34 | 16 | 23 | 17 | 32 | 11 | 10 | 19 | 66 | 12 | 17 | 49 | 11 | 108 | 43 | 38 | 23 | 47 |
| Postnatal | - | - | 6 | - | - | 7 | 2 | - | 7 | 16 | - | - | 116 | - | - | 93 | - | - | - |
| Total cases | 11 | 34 | 22 | 23 | 17 | 39 | 13 | 10 | 26 | 82 | 12 | 17 | 165 | 11 | 108 | 136 | 38 | 23 | 47 |
| Diagnostic accuracy | |||||||||||||||||||
| TAC diagnosis | 79% | 88% | no data | no data | 87% | 33%/93% # | 79% | 80% | 68% | 84% | 71% | no data | 6%/45% * | 100% | no data | 88% | 82% | 96% | no data |
| TAC subtype | 86% | 91% | no data | no data | no data | no data | no data | 100% | 92% | 80% | no data | 59% | no data | 100% | no data | no data | no data | 100% | 100% |
| Pregnancy outcome of prenatal diagnoses | |||||||||||||||||||
| GA at referral ([weeks]) | 22 (16–33) | 22 (13–28) | - | 24 ± 6.3 | 22 (18–32) | - | 24 (12–40) | 20 (15–37) | - | 26 | 24 (21–34) | 24 (15–36) | 22 (16–35) | 27 (16–35) | 23 (13–37) | - | 20 (16–34) | 25 (17–36) | 22 (12–37) |
| TOP | 4 (36%) | 14 (41%) | 14 (87.5%) | 4 (17%) | 4 (24%) | 2 (6%) | 5 (36%) | 9 (75%) | 2 (11%) | 9 (12%) | 4 (33%) | 8 (47%) | 11 (22%) | 1 (9%) | 66 (61%) | 17 (40%) | 18 (48%) | 8 (35%) | 10 (21%) |
| IUFD | 0 | 1 (3%) | 0 | 2 (11%) | 0 | 2 (7%) | 2 (22%) | 0 (0) | 1 (6%) | 0 (0) | 0 | - | 1 (23%) | - | 9 (8%) | 2 (5%) | 2 (10) | 2 (9%) | 2 (4%) |
| Live births | 7 (64%) | 19 (56%) | 2 (12.5%) | 18 (74%) | 13 (76%) | 28 (88%) | 6 (43%) | 3 (25%) | 10 (53%) | 62 (79%) | 8 (67%) | 4 (24%) | 37 (75%) | 9 (82%) | 33 (31%) | 24 (56%) | 18 (42%) | 13 (57%) | 35 (75%) |
| Study | Case No° | TAC Subtype | Prenatal Dysplasia | Postnatal Dysplasia | TV Regurgitation | TV Stenosis | Surgery | Outcome | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Prenatal | Postnatal | Prenatal | Postnatal | |||||||
| Duke et al. [18] | 4 | I | + | + | - | + | + | + | yes | postOP-NND |
| 7 | I | + | + | + | + | + | + | no | preOP-NND | |
| 8 | I | + | - | + | + | - | + | yes | alive | |
| Gomez et al. [21] | 3 | I | + | / | + | / | - | - | / | TOP |
| 5 | I | + | / | + | / | - | - | / | TOP | |
| Lee et al. [24] | 1 | I | + | + | + | + | + | + | yes | alive |
| 4 | I | + | + | + | + | + | + | yes | alive | |
| 5 | I | + | + | + | + | + | + | yes | alive | |
| 8 | I | + | + | + | + | + | + | yes | alive | |
| 12 | I | + | / | + | / | + | / | / | TOP | |
| Van Nisselrooji et al. [29] | 14 | / | + | / | + | / | + | / | / | TOP |
| 16 | / | + | / | + | ? | - | / | / | TOP | |
| 19 | / | + | / | + | / | - | / | / | IUFD | |
| 20 | / | + | / | + | / | - | / | / | IUFD | |
| 36 | / | + | ? | - | ? | + | ? | yes | alive | |
| Volpe et al. [30] | 6 | I | + | + | + | + | + | + | no | preOP-NND |
| 9 | I | + | + | - | - | + | + | yes | alive | |
| 15 | I | + | + | + | + | + | + | yes | postOP-NND | |
| Study | Current study | Abel et al. | Bourdial et al. | Cox et al. | Duke et al. | Evans et al. | Galindo et al. | Gómez et al. | Kor-suize et al. | Laux et al. | Lee et al. | Mărgi-nean et al. | Morgan et al. | Sivanan-dam et al. | Su et al. | Swanson et al. | v. Nissel-rooji et al. | Volpe et al. | Wolter et al. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolated TAC | 5 (45%) | 9 (26%) | - | - | 6 (35%) | - | 6 (46%) | 4 (50%) | - | 67 (82%) | 9 (75%) | 7 (41%) | - | - | 22 (20%) | - | 15 (39%) | 11 (48%) | 6 (13%) |
| Cardiac anomalies | |||||||||||||||||||
| Truncal valve defects (%) | 2 (18%) | 8 (23%) | - | ? | 8 (47%) | 1 † (3%) | - | 2 (25%) | 8 (42%) | 35 (61%) | 8 (67%) | 3 (18%) | 64 (39%) | - | - | 32 (29%) | 9 (24%) | 9 (39%) | - |
| 1 (9%) | - | - | 9 (47%) | 5 (29%) | - | - | 0 | 1 (5%) | 12 (21%) | - | 0 | 28 (17%) | - | - | 13 (12%) | 3 (8%) | 5 (22%) | 8 (17%) |
| 2 (18%) | - | - | 9 (47%) | 3 (18%) | 1 † (3%) | - | 2 (25%) | 7 (37%) | 23 (41%) | - | 3 (18%) | 36 (22%) | - | - | 19 (17%) | 6 (16%) | 4 (17%) | 14 (30%) |
| Right aortic arch | 3 (27%) | 6 (18%) | - | ? | - | - | - | 1 (13%) | 4 (21%) | 11 (13%) | 2 (17%) | - | 44 (27%) | - | 9 (8%) | 34 (30%) | 8 (21%) | 3 (13%) | 4 (9%) |
| Extracardiac anomalies | 6 (55%) | 26 (76%) | - | 10 (43%) | 4 (24%) | - | 7 (54%) | 4 (50%) | - | 15 (18%) | 2 (17%) | 5 (29%) | - | 3 (38%) | 86 (80%) | 20 (18%) | 17 (45%) | 10 (43%) | 17 (36%) |
| Genetic testing | |||||||||||||||||||
| Tested cases | 6 (43%) | 34 (100%) | - | 21 (91%) | 10 (59%) | 35 (90%) | 13 (100%) | 10 (100%) | 14 (54%) | 78 (95%) | 12 (100%) | 4 (24%) | 129 (78%) | - | - | 68 (50%) | 38 (100%) | 22 (96%) | 37 (78%) |
| Number of genetic anomalies in total (% *) | 1 (17%) | 13 (38%) | - | 12 (57%) | - | 16 (45%) | 4(30%) | 4 (40%) | 3 (21%) | 39 (50%) | 2 (17%) | 0 | 51 (40%) | - | - | - | 14 (37%) | 8 (36%) | 15 (41%) |
| Microdeletion 22q11 (% *) | 0 | 6 (18%) | 4 (25%) | 4 (19%) | 3 (30%) | 11 (31%) | 2 (15%) | 1 (10%) | 1 (7%) | 18 (23%) | 0 | 0 | 39 (30%) | - | - | 32 (47%) | 7 (18%) | 6 (27%) | 9 (24%) |
| Others (% *) | 1 (17%) | 7 (21%) | - | 8 (38%) | - | 5 (14%) | 2 (15%) | 3 (30%) | 2 (14%) | 21 (27%) | 2 (17%) | 0 | 12 (9%) | - | - | - | 7 (18%) | 2 (9%) | 6 (16%) |
| Study | Current Study | Abel et al. | Bourdial et al. | Cox et al. | Duke et al. | Evans et al. | Galindo et al. | Gómez et al. | Korsuize et al. | Laux et al. | Lee et al. | Mărgi-nean et al. | Morgan et al. | Sivanan-dam et al. | Su et al. | Swanson et al. | v. Nissel-rooji et al. | Volpe et al. | Wolter et al. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Live births | 7 | 19 | 8 | 17 | 13 | 35 | 8 | 3 | 16 | 78 | 8 | 4 | 165 | 8 | 16 | 117 | 18 | 13 | 35 |
| pre-OP NND | 1 (14%) | 0 (0) | - | 2 (11%) | 5 (38%) | 1 (3%) | 2 (25%) | 1 (33%) | 1 (6%) | 11 (14%) | 1 (13%) | 0 | 16 (10%) | 0 | - | 4 (3%) | 3 (17%) | 3 (23%) | 3 (9%) |
| Surgeries in total | 7 (100%) | 14 (74%) | - | 15 (88%) | 8 (62%) | 34 (97%) | 6 (75%) | 1 (100%) | 16 (100%) | 67 (86%) | 7 (88%) | 3 (75%) | 137 (83.03) | - | - | 113 (96,5%) | 15 (83%) | 10 (77%) | 32 (91%) |
| 6 (86%) | 13 (93%) | - | 15 (100%) | 7 (88%) | 26 (76%) | - | 1 (100%) | 16 (100%) | 64 (96%) | 7 (100%) | 3 (100%) | 137 (100%) | - | - | 113 (100%) | 15 (100%) | 6 (60%) | 32 (100%) |
| 1 (14%) | 0 | - | 1 (6%) | 1 (12%) | 8 (24%) | - | 0 | 0 | 3 (4%) | 0 | 1 (25%) | 0 | - | - | 0 | 0 | 2 (20%) | 0 |
| 0 | 1 (7%) | - | 0 | 0 | 0 | - | 0 | 0 | - | 0 | 0 | 0 | - | - | 0 | 0 | 2 (20%) | 0 |
| Peri- and post-OP-NND | 1 (14%) | 5 (38%) | - | 1 (7%) | 3 (38%) | 2 (6%) | 1 (17%) | 0 | 0 | - | 1 (14%) | 2 (66%) | 25 (18%) | 1 (12%) | - | 11 (10%) | 1 (7%) | 2 (25%) | 5 (16%) |
| CHD | 0 | 0 | - | - | 1 (8%) | 1 (3%) | 2 (25%) | 0 | 0 | - | 0 | 0 | 12 (7%) | - | - | - | 2 (11%) | - | - |
| Intention-to-treat-survival | 6/7 (86%) | 14/20 (70%) | - | 14/17 (82%) | 5/12 (42%) | 24/26 (92%) | 3/? | 1/1 (100%) | 16/16 (100%) | 16/64 (25%) | 6/8 (75%) | 1/3 (33%) | 39/130 (30%) | - | - | - | 12/16 (75%) | 7/8 (88%) | 27/32 (84%) |
| Overall survival | 6/11 (55%) | 14/20 (70%) | - | 14/23 (93%) | 5/13 (38%) | 31/35 (89%) | 3/8 (38%) | 1/3 (33%) | 16/16 (100%) | 16/78 (21%) | 6/8 (75%) | 1/17 (6%) | 43/165 (26%) | 7/8 (88%) | 16/108 (15%) | - | 12/18 (67%) | 7/13 (54%) | 27/47 (57%) |
| Follow-up (mean, range) [mo.] | 24 (0.3–46) | 42 (6–104) | - | - | 41 (1–75) | - (?–67) | 60 (12–144) | 0.5 (0.3–0.6) | 64.8 (0.1–150) | - (?–72) | 12 (1–105) | 4 (2–8) | 103.2 (0.1–236) | - (?–1) | - | - | 72 (24–120) | 10 | 51.5 |
| Authors | Current Study | Abel et al. | Cox et al. |
Duke et al. | Gómez et al. | Korsuize et al. |
Lee et al. |
Mărgi-nean et al. | Sivan-danam et al. | Swanson et al. | v. Nissel-rooji et al. |
Volpe et al. | Wolter et al. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prenatal Diagnoses | 11 | 34 | 23 | 17 | 10 | 19 | 12 | 17 | 11 | 43 | 38 | 23 | 47 |
| Live births | 7 (64%) | 19 (56%) | 17 (74%) | 13 (76%) | 1 (10%) | 10 (53%) | 8 (67%) | 4 (24%) | 8 (73%) | 19 (44%) | 18 (47%) | 13 (57%) | 35 (74%) |
| NND/CHD | 1 (14%) | 5 (26%) | 3 (18%) | 8 (62%) | 0 | 0 | 2 (25%) | 3 (75%) | 1 (9%) | 8 (42%) | 6 (33%) | 5 (38%) | 8 |
| 0 | 0 | 2 (11%) | 5 | 0 | 0 | 1 | 0 | 0 | 2 | 3 | 3 | 3 (9%) |
| 1 | 5 | 1 (7%) | 3 | 0 | 0 | 1 | 2 | 1 | 4 | 1 | 2 | 5 (16%) |
| 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 |
| Surgeries total |
7 (100%) |
14 (74%) | 15 (88%) |
8 (62%) |
1 (100%) | 10 (100%) |
7 (88%) | 3 (75%) | - |
17 (89%) |
15 (83%) |
10 (77%) | 32 (91%) |
| Corrective Surgery | 6 | 13 | 15 | 7 | 1 | 10 | 7 | 3 (100%) | - | 17 | 16 | 6 | 32 (100%) |
| Palliative Care | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | - | 0 | 0 | 2 | 0 |
| Awaiting | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | - | 0 | 0 | 2 | 0 |
|
Intention-to- treat-survival |
6/7 (86%) | 14/20 (70%) | 14/17 (82%) | 5/12 (42%) | 1/1 (100%) | 10/10 (100%) |
6/8 (75%) | 1/3 (33%) |
7/8 (88%) | 11/19 (58%) | 12/16 (75%) |
7/8 (88%) | 27/32 (84%) |
| Overall survival | 6/11 (55%) | 14/34 (41%) | 14/23 (93%) | 5/17 (29%) | 1/10 (10%) | 10/19 (53%) | 6/12 (50%) | 1/17 (6%) | 7/11 (64%) | 11/43 (26%) | 12/18 (67%) | 8/23 (35%) | 27/47 (57%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wittek, A.; Plöger, R.; Walter, A.; Strizek, B.; Geipel, A.; Gembruch, U.; Neubauer, R.; Recker, F. Diagnosis, Management and Outcome of Truncus Arteriosus Communis Diagnosed during Fetal Life—Cohort Study and Systematic Literature Review. J. Clin. Med. 2024, 13, 6143. https://doi.org/10.3390/jcm13206143
Wittek A, Plöger R, Walter A, Strizek B, Geipel A, Gembruch U, Neubauer R, Recker F. Diagnosis, Management and Outcome of Truncus Arteriosus Communis Diagnosed during Fetal Life—Cohort Study and Systematic Literature Review. Journal of Clinical Medicine. 2024; 13(20):6143. https://doi.org/10.3390/jcm13206143
Chicago/Turabian StyleWittek, Agnes, Ruben Plöger, Adeline Walter, Brigitte Strizek, Annegret Geipel, Ulrich Gembruch, Ricarda Neubauer, and Florian Recker. 2024. "Diagnosis, Management and Outcome of Truncus Arteriosus Communis Diagnosed during Fetal Life—Cohort Study and Systematic Literature Review" Journal of Clinical Medicine 13, no. 20: 6143. https://doi.org/10.3390/jcm13206143
APA StyleWittek, A., Plöger, R., Walter, A., Strizek, B., Geipel, A., Gembruch, U., Neubauer, R., & Recker, F. (2024). Diagnosis, Management and Outcome of Truncus Arteriosus Communis Diagnosed during Fetal Life—Cohort Study and Systematic Literature Review. Journal of Clinical Medicine, 13(20), 6143. https://doi.org/10.3390/jcm13206143