

Exploring Chronic Hypocalcemia: Insights into Autoimmune Polyglandular Syndrome Type 1—A Case Study and Literature Review
 , ,
, ,
Abstract
:
1. Introduction
2. Case Report
2.1. Patient’s Medical History
2.2. Symptoms and Clinical Findings
2.3. Biological and Paraclinical Assessment
2.4. Therapeutic Intervention
2.5. Follow-Up and Outcome
3. Discussion
3.1. Literature Review
3.1.1. Definition of APS-1
3.1.2. Epidemiology
3.1.3. Pathogenesis and Autoimmunity of APS-1
3.1.4. Correlation between Genotype and Phenotype in APS-1
3.1.5. Clinical Features of APS-1
3.1.6. Therapeutic Approach in APS-1
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcucci, G.; Cianferotti, L.; Brandi, M.L. Clinical presentation and management of hypoparathyroidism. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, F.; Pepe, J.; Danese, V.C.; Fassino, V.; Cecchetti, V.; De Lucia, F.; Biamonte, F.; Colangelo, L.; Ferrazza, G.; Panzini, E.; et al. The relative influence of serum ionized calcium and 25- hydroxyvitamin D in regulating PTH secretion in healthy subjects. Bone 2019, 125, 200–206. [Google Scholar] [CrossRef]
- Bharill, S.; Wu, M. Hypocalcemia and Hypercalcemia in Children. Pediatr. Rev. 2023, 44, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Cusano, N.E.; Bilezikian, J.P. Signs and symptoms of hypoparathyroidism. Endocrinol. Metab. Clin. N. Am. 2018, 47, 759–770. [Google Scholar] [CrossRef]
- Underbjerg, L.; Sikjaer, T.; Mosekilde, L.; Rejnmark, L. Cardiovascular and renal complications to postsurgical hypoparathyroidism: A Danish nationwide controlled historic follow-up study. J. Bone Miner. Res. 2013, 28, 2277–2285. [Google Scholar] [CrossRef]
- Dittmar, M.; Kahaly, G.J. Polyglandular autoimmune syndromes: Immunogenetics and long-term follow-up. J. Clin. Endocrinol. Metab. 2003, 88, 2983–2992. [Google Scholar] [CrossRef]
- Neufeld, M.; Maclaren, N.; Blizzard, R. Autoimmune polyglandular syndromes. Pediatr. Ann. 1980, 9, 154–162. [Google Scholar] [CrossRef]
- Michels, A.W.; Gottlieb, P.A. Autoimmune polyglandular syndromes. Nat. Rev. Endocrinol. 2010, 6, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J.M.; Rojas-Villarraga, A.; Garcıa-Carrasco, M. The Autoimmune Tautology: From Polyautoimmunity and Familial Autoimmunity to the Autoimmune Genes. Autoimmune Dis. 2012, 2012, 297193. [Google Scholar] [CrossRef]
- Bousfiha, A.; Moundir, A.; Tangye, S.G.; Picard, C.; Jeddane, L.; Al-Herz, W.; Rundles, C.C.; Franco, J.L.; Holland, S.M.; Klein, C.; et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J. Clin. Immunol. 2022, 42, 1508–1520. [Google Scholar] [CrossRef]
- Guo, C.J.; Leung, P.S.C.; Zhang, W.; Ma, X.; Gershwin, M.E. The immunobiology and clinical features of type 1 autoimmune polyglandular syndrome (APS-1). Autoimmun. Rev. 2018, 17, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Husebye, E.S.; Perheentupa, J.; Rautemaa, R.; Kämpe, O. Clinical Manifestations and Management of Patients with Autoimmune Polyendocrine Syndrome Type I. J. Intern. Med. 2009, 265, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Nuralieva, N.; Yukina, M.; Sozaeva, L.; Donnikov, M.; Kovalenko, L.; Troshina, E.; Orlova, E.; Gryadunov, D.; Savvateeva, E.; Dedov, I. Diagnostic Accuracy of Methods for Detection of Antibodies against Type I Interferons in Patients with Endocrine Disorders. J. Pers. Med. 2022, 12, 1948. [Google Scholar] [CrossRef] [PubMed]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune polyendocrine syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Zlotogora, J.; Shapiro, M.S. Polyglandular autoimmune syndrome type I among Iranian Jews. J. Med. Genet. 1992, 29, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, P.; Myllarniemi, S.; Sipila, I.; Perheentupa, J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 1990, 322, 1829–1836. [Google Scholar] [CrossRef]
- Betterle, C.; Presotto, F. Autoimmune Polyendocrine Syndrome (APS) or multiple autoimmune syndrome (MAS). In Handbook of Systemic Autoimmune Diseases, Endocrine Manifestations of Systemic Autoimmune Diseases; Walker, S., Jara, L.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 135–148. [Google Scholar]
- Proust-Lemoine, E.; Saugier-Veber, P.; Wémeau, J.L. Polyglandular autoimmune syndrome type I. Presse Med. 2012, 41, e651–e662. [Google Scholar] [CrossRef] [PubMed]
- Sato, U.; Horikawa, R.; Katsumata, N.; Asakura, Y.; Kitanaka, S.; Tanaka, T. Novel compound heterozygous AIRE mutations in a Japanese patient with APECED. J. Pediatr. Endocrinol. Metab. 2004, 17, 917–921. [Google Scholar] [CrossRef]
- Capalbo, D.; Improda, N.; Esposito, A.; De Martino, L.; Barbieri, F.; Betterle, C.; Pignata, C.; Salerno, M. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy from the pediatric perspective. J. Endocrinol. Investig. 2013, 36, 903–912. [Google Scholar]
- Ferre, E.M.; Rose, S.R.; Rosenzweig, S.D.; Burbelo, P.D.; Romito, K.R.; Niemela, J.E.; Rosen, L.B.; Break, T.J.; Gu, W.; Hunsberger, S.; et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 2016, 1, e88782. [Google Scholar] [CrossRef]
- Pellegrino, M.; Bellacchio, E.; Dhamo, R.; Frasca, F.; Betterle, C.; Fierabracci, A. A novel homozygous mutation of the AIRE gene in an APECED patient from Pakistan: Case report and review of the literature. Front. Immunol. 2018, 9, 1835. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Strassburg, C.P.; Obermayer-Straub, P.; Brabant, G.; Manns, M.P. The genetic background of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy and its autoimmune disease components. J. Mol. Med. 2002, 80, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Perniola, R.; Musco, G. The biophysical and biochemical properties of the autoimmune regulator (AIRE) protein. Biochim. Biophys. Acta. 2014, 1842, 326–337. [Google Scholar] [CrossRef]
- Liiv, I.; Haljasorg, U.; Kisand, K.; Maslovskaja, J.; Laan, M.; Peterson, P. AIRE-induced apoptosis is associated with nuclear translocation of stress sensor protein GAPDH. Biochem. Biophys. Res. Commun. 2012, 423, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Rosatelli, M.C.; Meloni, A.; Meloni, A.; Devoto, M.; Cao, A.; Scott, H.S.; Peterson, P.; Heino, M.; Krohn, D.-M.N.; Nagamine, K.; et al. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum. Genet. 1998, 103, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Fierabracci, A. Type 1 diabetes in autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy syndrome (APECED): A “rare” manifestation in a “rare” disease. Int. J. Mol. Sci. 2016, 17, 1106. [Google Scholar] [CrossRef] [PubMed]
- Malchow, S.; Leventhal, D.S.; Nishi, S.; Fischer, B.I.; Shen, L.; Paner, G.P.; Amit, A.S.; Kang, C.; Geddes, J.E.; Allison, J.P.; et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science 2013, 339, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.D.; Gilmore, D.C.; Dileepan, T.; Nawrocka, W.I.; Chao, J.L.; Schoenbach, M.H.; Jenkins, M.K.; Adams, E.J.; Savage, P.A. Identification of Natural Regulatory T Cell Epitopes Reveals Convergence on a Dominant Autoantigen. Immunity 2017, 47, 107–117.e8. [Google Scholar] [CrossRef]
- Deleeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The Prognostic Value of FoxP3+ Tumor-Infiltrating Lymphocytes in Cancer: A Critical Review of the Literature. Clin. Cancer Res. 2012, 18, 3022. [Google Scholar] [CrossRef]
- Zhao, B.; Chang, L.; Fu, H.; Sun, G.; Yang, W. The Role of Autoimmune Regulator (AIRE) in Peripheral Tolerance. J. Immunol. Res. 2018, 2018, 3930750. [Google Scholar] [CrossRef] [PubMed]
- Philippot, Q.; Casanova, J.L.; Puel, A. Candidiasis in patients with APS-1: Low IL-17, high IFN-γ, or both? Curr. Opin. Immunol. 2021, 72, 318–323. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kashem, S.W.; Binstadt, B.A. Pathogenic and Protective Autoantibodies in Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED). Antibodies 2017, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Oftedal, B.E.; Hellesen, A.; Erichsen, M.M.; Bratland, E.; Vardi, A.; Perheentupa, J.; Kemp, E.H.; Fiskerstrand, T.; Viken, M.K.; Weetman, A.P.; et al. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity 2015, 42, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Cetani, F.; Barbesino, G.; Borsari, S.; Pardi, E.; Cianferotti, L.; Pinchera, A.; Marcocci, C. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J. Clin. Endocrinol. Metab. 2001, 86, 4747–4752. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.K.; Huoh, Y.S.; Reynolds, P.R.; Yu, L.; Rewers, M.; Reddy, M.; Anderson, M.S.; Hur, S.; Gelfand, E.W. Dominant-negative loss of function arises from a second, more frequent variant within the SAND domain of autoimmune regulator (AIRE). J. Autoimmun. 2018, 88, 114–120. [Google Scholar] [CrossRef]
- Constantine, G.M.; Lionakis, M.S. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 2019, 287, 103–112. [Google Scholar] [CrossRef]
- Oftedal, B.E.; Assing, K.; Baris, S.; Safgren, S.L.; Johansen, I.S.; Jakobsen, M.A.; Babovic-Vuksanovic, D.; Agre, K.; Klee, E.W.; Majcic, E.; et al. Dominant-negative heterozygous mutations in AIRE confer diverse autoimmune phenotypes. iScience 2023, 26, 106818. [Google Scholar] [CrossRef]
- Human Gene Mutation Database. Available online: https://www.hgmd.cf.ac.uk/ac/gene.php?gene=AIRE (accessed on 30 December 2023).
- Björses, P.; Halonen, M.; Palvimo, J.J.; Kolmer, M.; Aaltonen, J.; Ellonen, P.; Perheentupa, J.; Ulmanen, I.; Peltonen, L. Mutations in the AIRE gene: Effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am. J. Hum. Genet. 2000, 66, 378–392. [Google Scholar] [CrossRef]
- Cervato, S.; Mariniello, B.; Lazzarotto, F.; Morlin, L.; Zanchetta, R.; Radetti, G.; De Luca, F.; Valenzise, M.; Giordano, R.; Rizzo, D.; et al. Evaluation of the autoimmune regulator (AIRE) gene mutations in a cohort of Italian patients with autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy (APECED) and in their relatives. Clin. Endocrinol. 2009, 70, 421–428. [Google Scholar] [CrossRef]
- De Martino, L.; Capalbo, D.; Improda, N.; D’Elia, F.; Di Mase, R.; D’Assante, R.; D’Acunzo, I.; Pignata, C.; Salerno, M. APECED: A Paradigm of Complex Interactions between Genetic Background and Susceptibility Factors. Front. Immunol. 2013, 4, 331. [Google Scholar] [CrossRef] [PubMed]
- Perheentupa, J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab. 2006, 912, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.H.; Cheetham, T.; Imrie, H.; Vaidya, B.; Barnes, N.D.; Bilous, R.W.; Carr, D.; Meeran, K.; Shaw, N.J.; Smith, C.S.; et al. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am. J. Hum. Genet. 1998, 63, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Meloni, A.; Willcox, N.; Meager, A.; Atzeni, M.; Wolff, A.S.; Husebye, E.S.; Furcas, M.; Rosatelli, M.C.; Cao, A.; Congia, M. Autoimmune polyendocrine syndrome type 1: An extensive longitudinal study in Sardinian patients. J. Clin. Endocrinol. Metab. 2012, 97, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Fierabracci, A.; Lanzillotta, M.; Vorgučin, I.; Palma, A.; Katanić, D.; Betterle, C. Report of two siblings with APECED in Serbia: Is there a founder effect of c.769C>T AIRE genotype? Ital. J. Pediatr. 2021, 47, 126. [Google Scholar] [CrossRef] [PubMed]
- Kisand, K.; Bøe Wolff, A.S.; Podkrajsek, K.T.; Tserel, L.; Link, M.; Kisand, K.V.; Ersvaer, E.; Perheentupa, J.; Erichsen, M.M.; Bratanic, N.; et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010, 207, 299–308. [Google Scholar] [CrossRef]
- Ferré, E.M.N.; Break, T.J.; Burbelo, P.D.; Allgäuer, M.; Kleiner, D.E.; Jin, D.; Xu, Z.; Folio, L.R.; Mollura, D.J.; Swamydas, M.; et al. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci. Transl. Med. 2019, 11, eaav5597. [Google Scholar] [CrossRef] [PubMed]
- Chascsa, D.M.; Ferré, E.M.N.; Hadjiyannis, Y.; Alao, H.; Natarajan, M.; Quinones, M.; Kleiner, D.E.; Simcox, T.L.; Chitsaz, E.; Rose, S.R.; et al. APECED-Associated Hepatitis: Clinical, Biochemical, Histological and Treatment Data from a Large, Predominantly American Cohort. Hepatology 2021, 73, 1088–1104. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Røyrvik, E.C.; Aranda-Guillén, M.; Berger, A.H.; Landegren, N.; Artaza, H.; Hallgren, Å.; Grytaas, M.A.; Ström, S.; Bratland, E.; et al. GWAS for autoimmune Addison’s disease identifies multiple risk loci and highlights AIRE in disease susceptibility. Nat. Commun. 2021, 12, 959. [Google Scholar] [CrossRef]
- Laisk, T.; Lepamets, M.; Koel, M.; Abner, E.; Estonian Biobank Research, T.; Magi, R. Genome-wide association study identifies five risk loci for pernicious anemia. Nat. Commun. 2021, 12, 3761. [Google Scholar] [CrossRef]
- Kliegman, R.; Behrman, R.E.; Nelson, W.E. (Eds.) Nelson Textbook of Pediatrics, 20th ed.; Elsevier: Philadelphia, PA, USA, 2016; ISBN 978-1-4557-7566-8. [Google Scholar]
- Nambam, B.; Winter, W.E.; Schatz, D.A. IgG4 antibodies in autoimmune polyglandular disease and IgG4-related endocrinopathies: Pathophysiology and clinical characteristics. Curr. Opin. Pediatr. 2014, 26, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Naletto, L.; Frigo, A.C.; Ceccato, F.; Sabbadin, C.; Scarpa, R.; Presotto, F.; Dalla Costa, M.; Faggian, D.; Plebani, M.; Censi, S.; et al. The natural history of autoimmune Addison’s disease from the detection of autoantibodies to development of the disease: A long-term follow-up study on 143 patients. Eur. J. Endocrinol. 2019, 180, 223–234. [Google Scholar] [CrossRef]
- Garelli, S.; Dalla Costa, M.; Sabbadin, C.; Barollo, S.; Rubin, B.; Scarpa, R.; Masiero, S.; Fierabracci, A.; Bizzarri, C.; Crinò, A.; et al. Autoimmune polyendocrine syndrome type 1: An Italian survey on 158 patients. J. Endocrinol. Investig. 2021, 44, 2493–2510. [Google Scholar] [CrossRef] [PubMed]
- Proust-Lemoine, E.; Guyot, S. Polyendocrinopathies auto-immunes de type 1 et pathologies buccales. Presse Méd. 2017, 46, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Skrabic, V.; Skrabic, I.; Skrabic, R.; Roje, B.; Simunovic, M. Clinical Characteristics in the Longitudinal Follow-Up of APECED Syndrome in Southern Croatia-Case Series. Genes 2022, 13, 558. [Google Scholar] [CrossRef] [PubMed]
- Myhre, A.G.; Halonen, M.; Eskelin, P.; Ekwall, O.; Hedstrand, H.; Rorsman, F.; Kämpe, O.; Husebye, E.S. Autoimmune Polyendocrine Syndrome Type 1 (APS I) in Norway. Clin. Endocrinol. 2001, 54, 211–217. [Google Scholar] [CrossRef]
- Kang, M.S.; Sandhu, C.S.; Singh, N.; Evans, T. Initiation of levothyroxine in a patient with hypothyroidism inducing adrenal crisis requiring VA ECMO: A tale of preventable disaster. BMJ Case. Rep. 2019, 12, e230601. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, D.; Bossini, B.; Benvenuto, S.; Pellegrin, M.C.; Tornese, G. Pediatric Adrenal Insufficiency: Challenges and Solutions. Ther. Clin. Risk Manag. 2022, 18, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Abate, E.G.; Clarke, B.L. Review of Hypoparathyroidism. Front. Endocrinol. 2017, 7, 172. [Google Scholar] [CrossRef]
- Sanda, S.; Sclingmann, K.P.; Newfield, R.S. Autosomal dominant hypoparathyroidism with severe hypomagnesemia and hypocalcemia successfully treated with recombinant PTH and continuous subcutaneous magnesium infusion. J. Pediatr. Endocrinol. Metab. 2008, 21, 385–391. [Google Scholar] [CrossRef]
- Matarazzo, P.; Tuli, G.; Fiore, L.; Mussa, A.; Feyles, F.; Peiretti, V.; Lala, R. Teriparatide (rhPTH) treatment in children with syndromic hypoparathyroidism. J. Pediatr. Endocrinol. Metab. 2014, 27, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Tuli, G.; Buganza, R.; Tessaris, D.; Einaudi, S.; Matarazzo, P.; de Sanctis, L. Teriparatide (rhPTH 1-34) treatment in the pediatric age: Long-term efficacy and safety data in a cohort with genetic hypoparathyroidism. Endocrine 2020, 67, 457–465. [Google Scholar] [CrossRef]
- Winer, K.K.; Kelly, A.; Johns, B.S.A.; Zhang, B.; Dowdy, R.N.K.; Kim, L.; Reynolds, J.C.; Albert, P.S.; Cutler Jr, G.B. Long-Term Parathyroid Hormone 1-34 Replacement Therapy in Children with Hypoparathyroidism. J. Pediatr. 2018, 203, 391–399. [Google Scholar] [CrossRef]
- Winer, K.K.; Sinaii, N.; Peterson, D.; Sainz, B., Jr.; Cutler, G.B., Jr. Effects of once versus twice-daily parathyroid hormone 1–34 therapy in children with hypoparathyroidism. J. Clin. Endocrinol. Metab. 2008, 93, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Vahle, J.L.; Sato, M.; Long, G.G.; Young, J.K.; Francis, P.C.; Engelhardt, J.A.; Westmore, M.S.; Linda, Y.; Nold, J.B. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1–34) for 2 years and relevance to human safety. Toxicol. Pathol. 2002, 30, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Andrews, E.B.; Gilsenan, A.W.; Midkiff, K.; Sherrill, B.; Wu, Y.; Mann, B.H.; Masica, D. The US postmarketing surveillance study of adult osteosarcoma and teriparatide: Study design and findings from the first 7 years. J. Bone Miner. Res. 2012, 27, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Gilsenan, A.; Harding, A.; Kellier-Steele, N.; Harris, D.; Midkiff, K.; Andrews, E. The Forteo Patient Registry linkage to multiple state cancer registries: Study design and results from the first 8 years. Osteoporos. Int. 2018, 29, 2335–2343. [Google Scholar] [CrossRef]
- Humbert, L.; Cornu, M.; Proust-Lemoine, E.; Bayry, J.; Wemeau, J.L.; Vantyghem, M.C.; Sendid, B. Chronic Mucocutaneous Candidiasis in Autoimmune Polyendocrine Syndrome Type 1. Front. Immunol. 2018, 9, 2570. [Google Scholar] [CrossRef]
- Grace, M.L. Preventing infections in children and adults with asplenia. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 328–335. [Google Scholar]
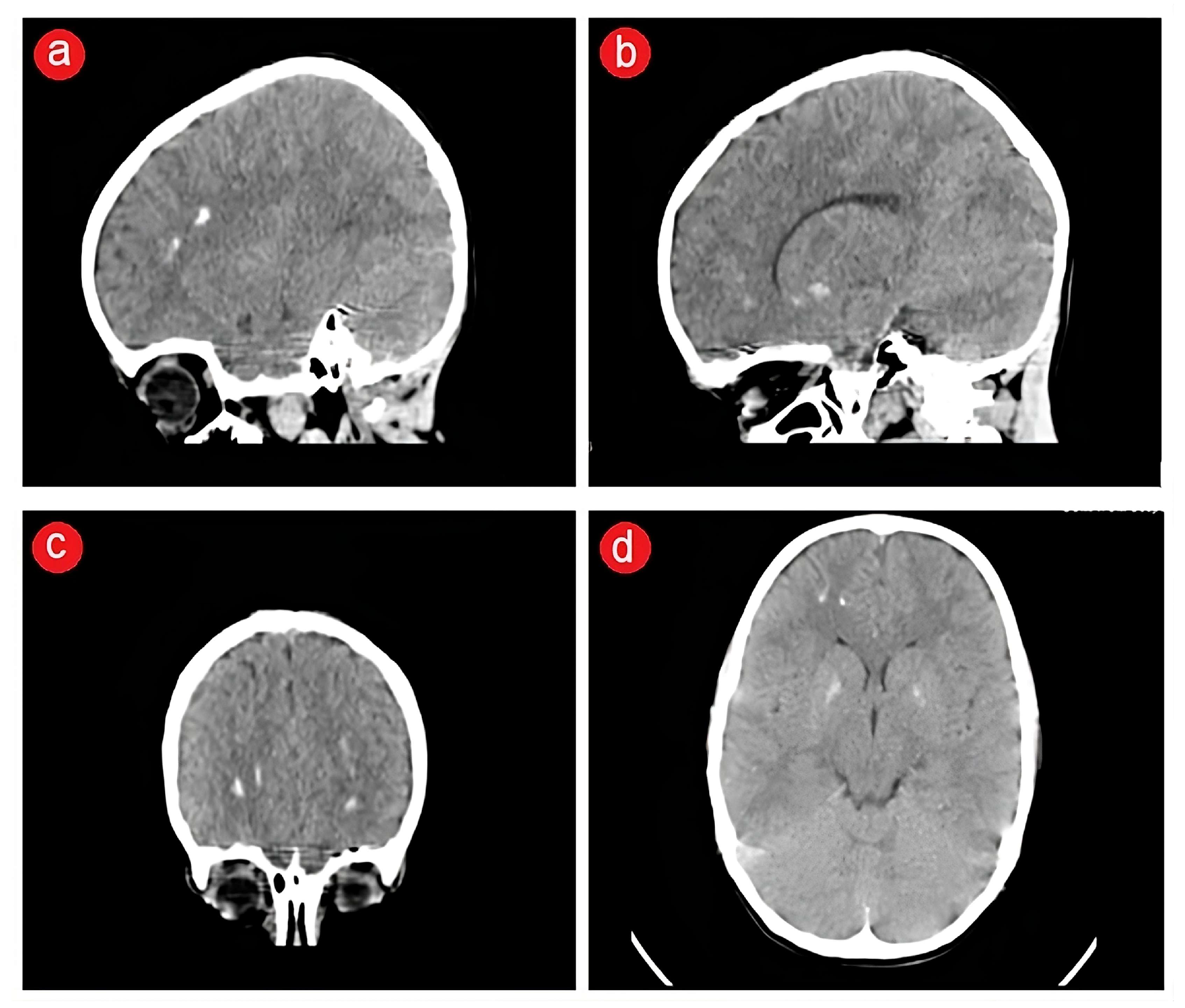

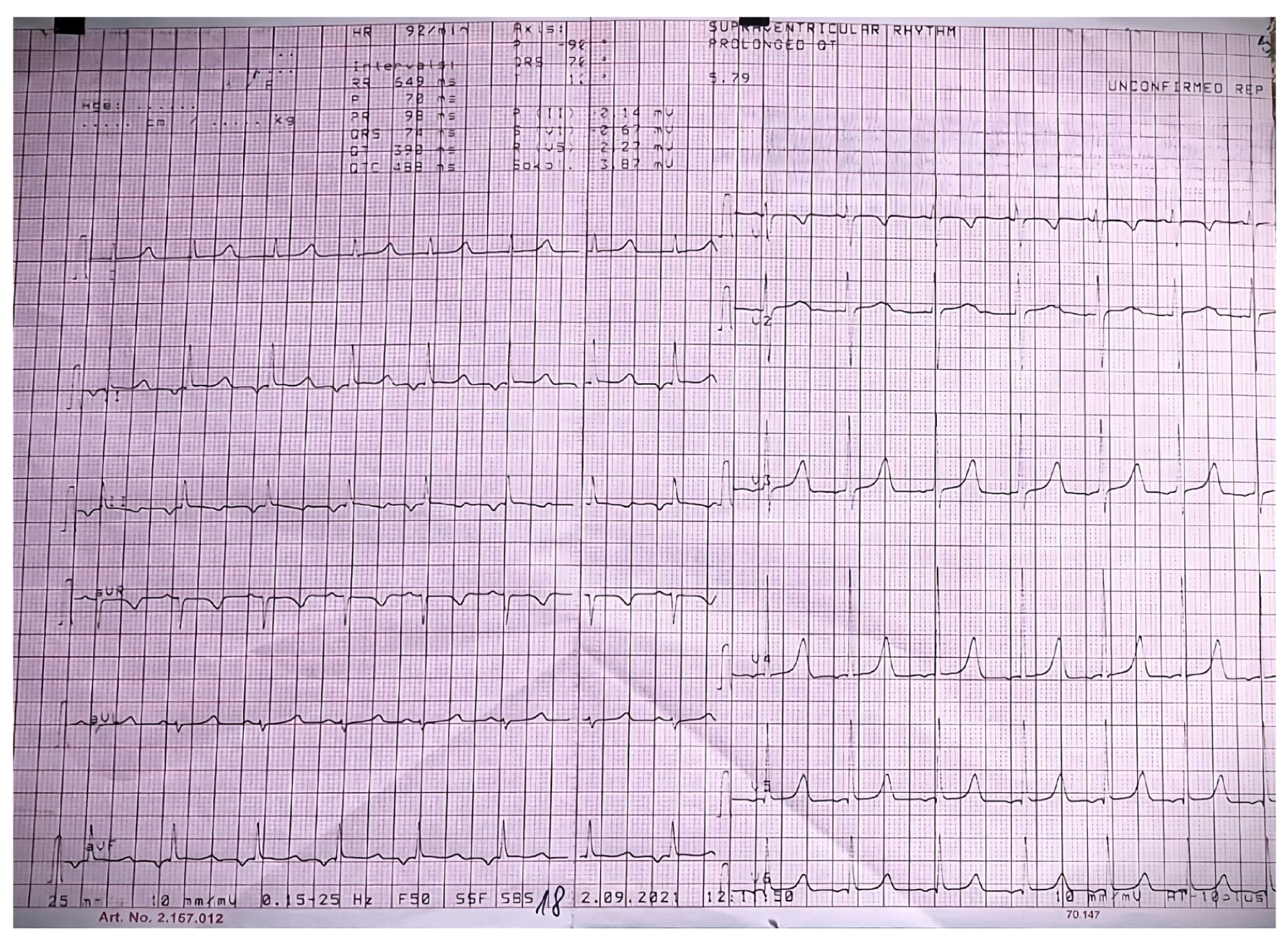

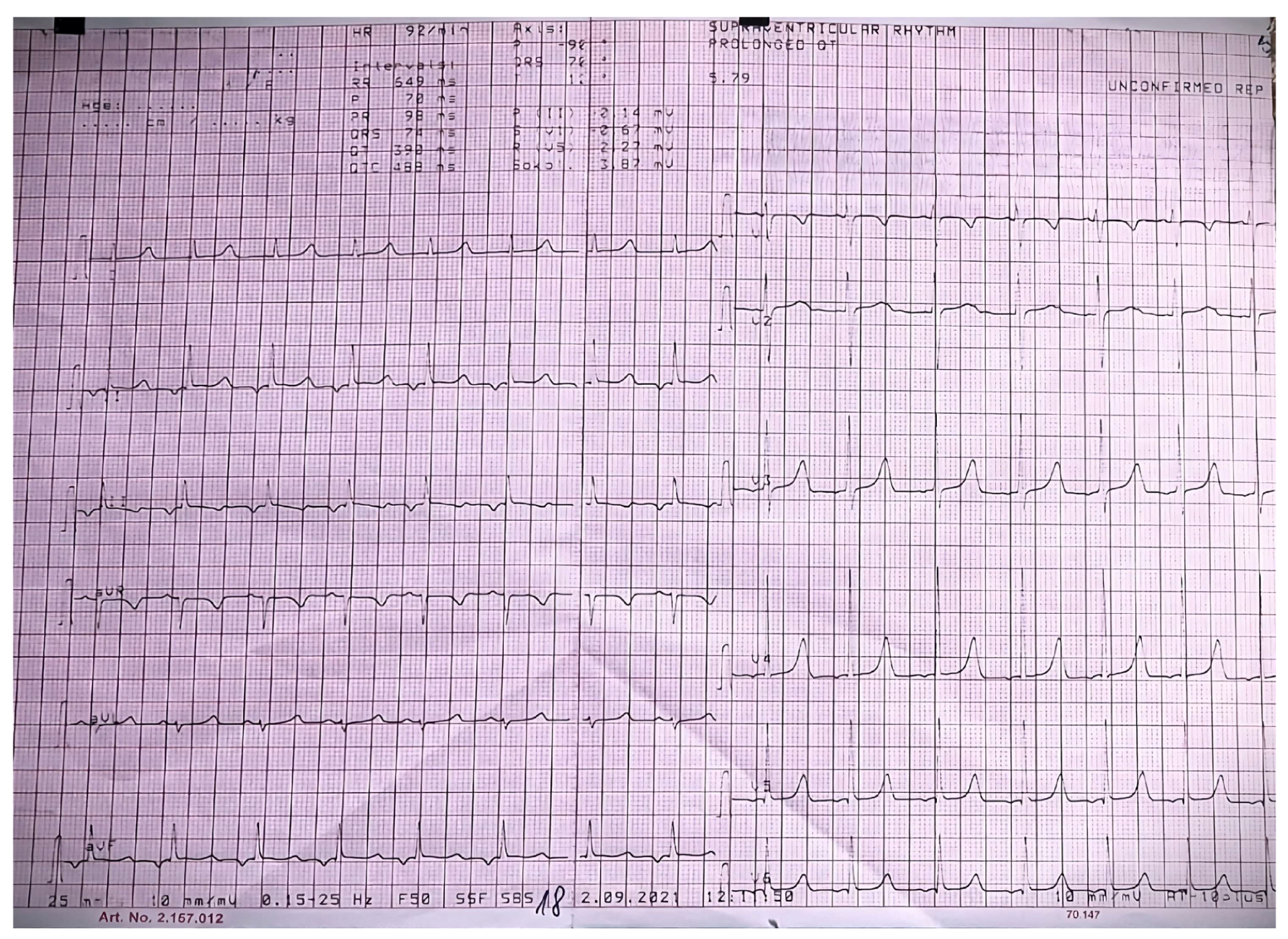
{kind=link}
{kind=link}
{kind=link}
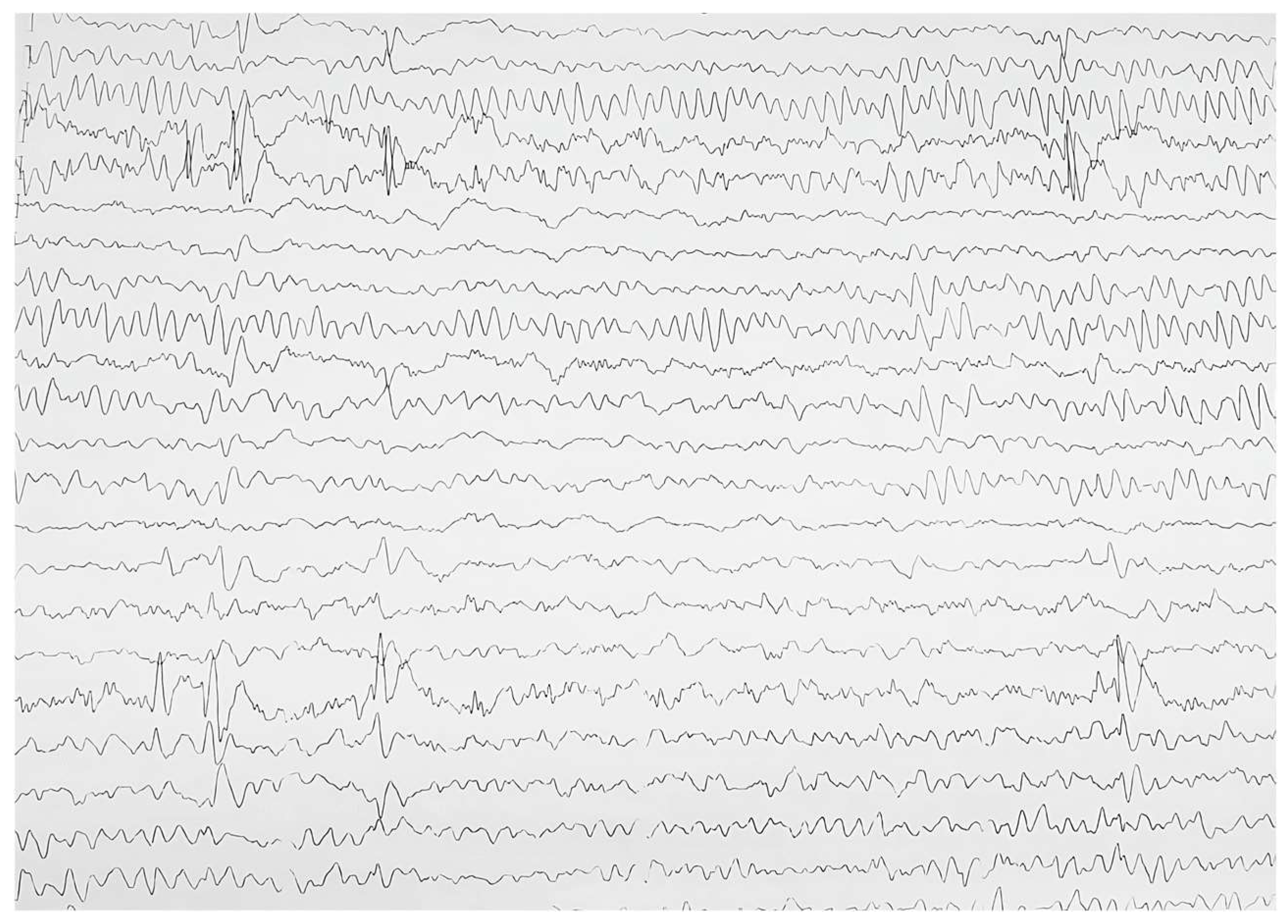
{kind=link}
{kind=link}
| Biological Investigation * | Result | Reference |
|---|---|---|
| TSH | 2.9 μIU/mL | 0.66–4.14 μIU/mL |
| FT4 | 18.91 pmol/L | 11.6–21.5 pmol/L |
| FT3 | 7.06 pmol/L | 4.1–7.9 pmol/L |
| Anti-thyroid peroxidase abs | <10 UI/mL | <35.0 UI/mL |
| Anti-thyroglobulin abs | <1.3 UI/mL | < 4.5 UI/mL |
| ACTH am | 29.50 pg/ml | ≤46.00 pg/ml |
| Cortisol am | 505.1 nmol/L | 171–536 nmol/L |
| Steroid 17-hydroxylase abs | 9.55 | <10, GZ < 15 |
| Steroid 21-hydroxylase abs | 6.43 | <10, GZ 10–15 |
| IgA tTg abs | 2.45 UI/mL | <20 UI/mL |
| IgG tTg abs | 1.64 UI/mL | <20 UI/mL |
| fecal calprotectin | 48.40 µg/g | <50.00 µg/g |
| IgG anti-Sm abs | 1.4 UI/mL | <15.0 UI/mL |
| IgG anti-LKM1 abs | negative | negative |
| ANA | negative | negative |
| pANCA | 3.5 U/mL | <7 U/mL |
| ASCA | 1.3 U/mL | <7 U/mL |
| IgG ab to H(+)/K(+) ATPase | 3.2 U/mL | <10.0 U/mL |
| IgG intrinsic factor abs | 0.7 U/mL | <6.0 U/mL |
| folate | 10.83 ng/mL | >5.38 ng/mL |
| ICA | 0.3 | <0.90 |
| anti-GAD 2 | 2.3 UI/mL | <10.0 UI/mL |
| IA2 | 3.5 UI/mL | <10.0 UI/mL |
| B12 vitamin | 763 pg/mL | 211–911 pg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brad, G.-F.; Nicoară, D.-M.; Scutca, A.-C.; Bugi, M.-A.; Asproniu, R.; Olariu, L.-G.; Jugănaru, I.; Cristun, L.-I.; Mărginean, O. Exploring Chronic Hypocalcemia: Insights into Autoimmune Polyglandular Syndrome Type 1—A Case Study and Literature Review. J. Clin. Med. 2024, 13, 2368. https://doi.org/10.3390/jcm13082368
Brad G-F, Nicoară D-M, Scutca A-C, Bugi M-A, Asproniu R, Olariu L-G, Jugănaru I, Cristun L-I, Mărginean O. Exploring Chronic Hypocalcemia: Insights into Autoimmune Polyglandular Syndrome Type 1—A Case Study and Literature Review. Journal of Clinical Medicine. 2024; 13(8):2368. https://doi.org/10.3390/jcm13082368
Chicago/Turabian StyleBrad, Giorgiana-Flavia, Delia-Maria Nicoară, Alexandra-Cristina Scutca, Meda-Ada Bugi, Raluca Asproniu, Laura-Gratiela Olariu, Iulius Jugănaru, Lucian-Ioan Cristun, and Otilia Mărginean. 2024. "Exploring Chronic Hypocalcemia: Insights into Autoimmune Polyglandular Syndrome Type 1—A Case Study and Literature Review" Journal of Clinical Medicine 13, no. 8: 2368. https://doi.org/10.3390/jcm13082368
APA StyleBrad, G.-F., Nicoară, D.-M., Scutca, A.-C., Bugi, M.-A., Asproniu, R., Olariu, L.-G., Jugănaru, I., Cristun, L.-I., & Mărginean, O. (2024). Exploring Chronic Hypocalcemia: Insights into Autoimmune Polyglandular Syndrome Type 1—A Case Study and Literature Review. Journal of Clinical Medicine, 13(8), 2368. https://doi.org/10.3390/jcm13082368