
Kenny–Caffey Syndrome Type 2 (KCS2): A New Case Report and Patient Follow-Up Optimization
 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Phenotype Analysis
2.2. Dataset Collection and Filtering
2.3. Patient Variant Analysis
3. Detailed Case Description
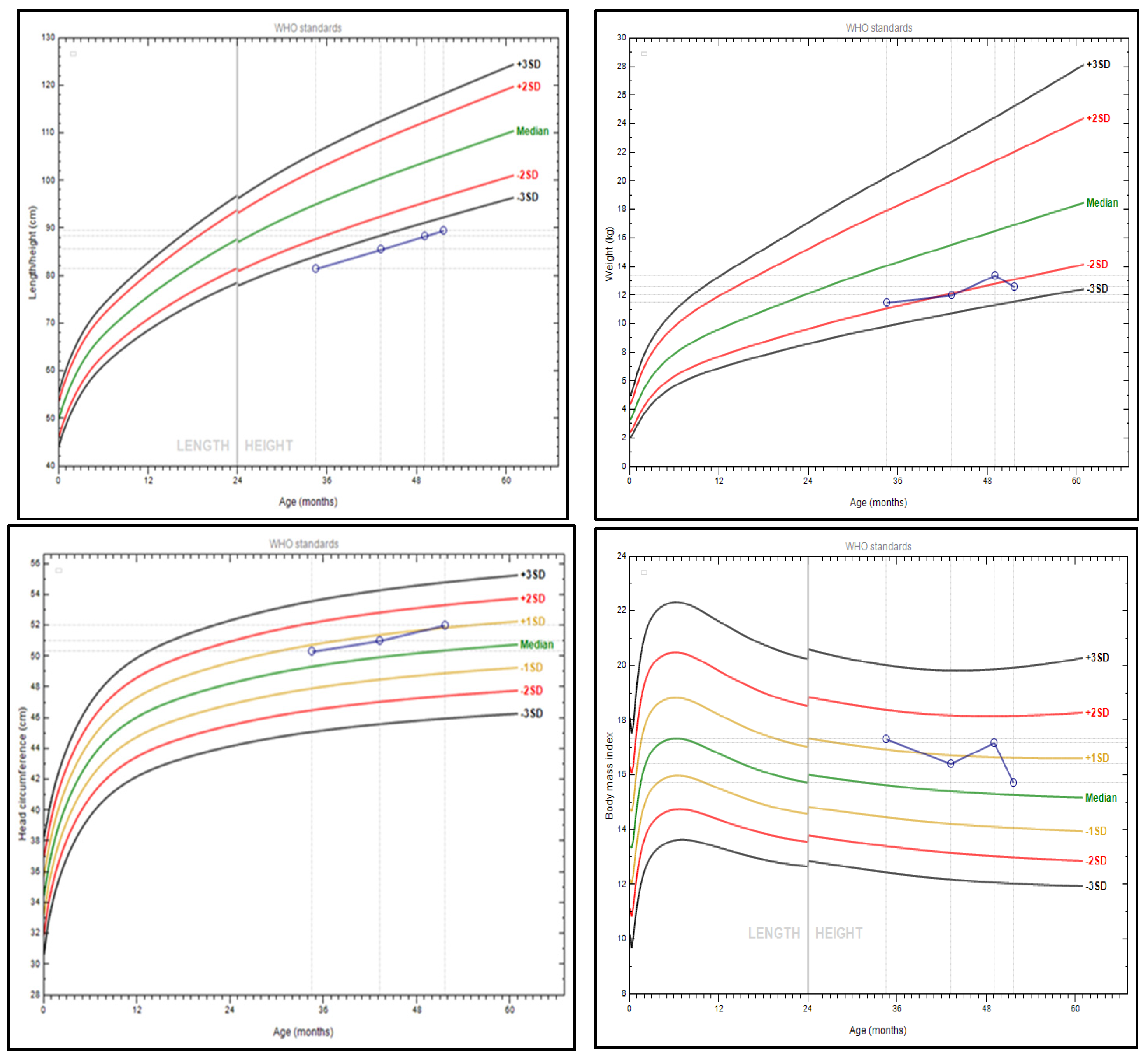
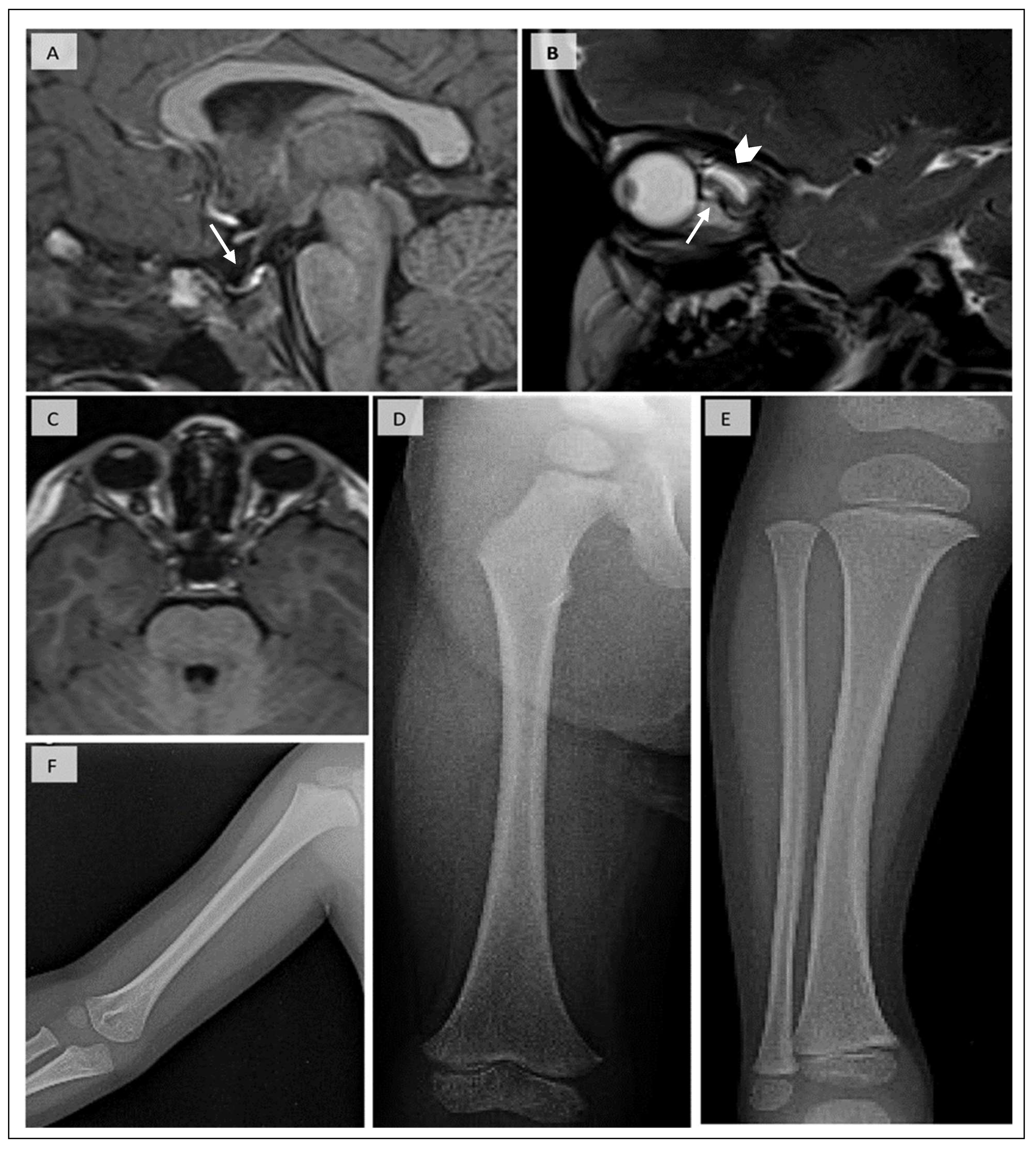
3.1. Patient’s Phenotype
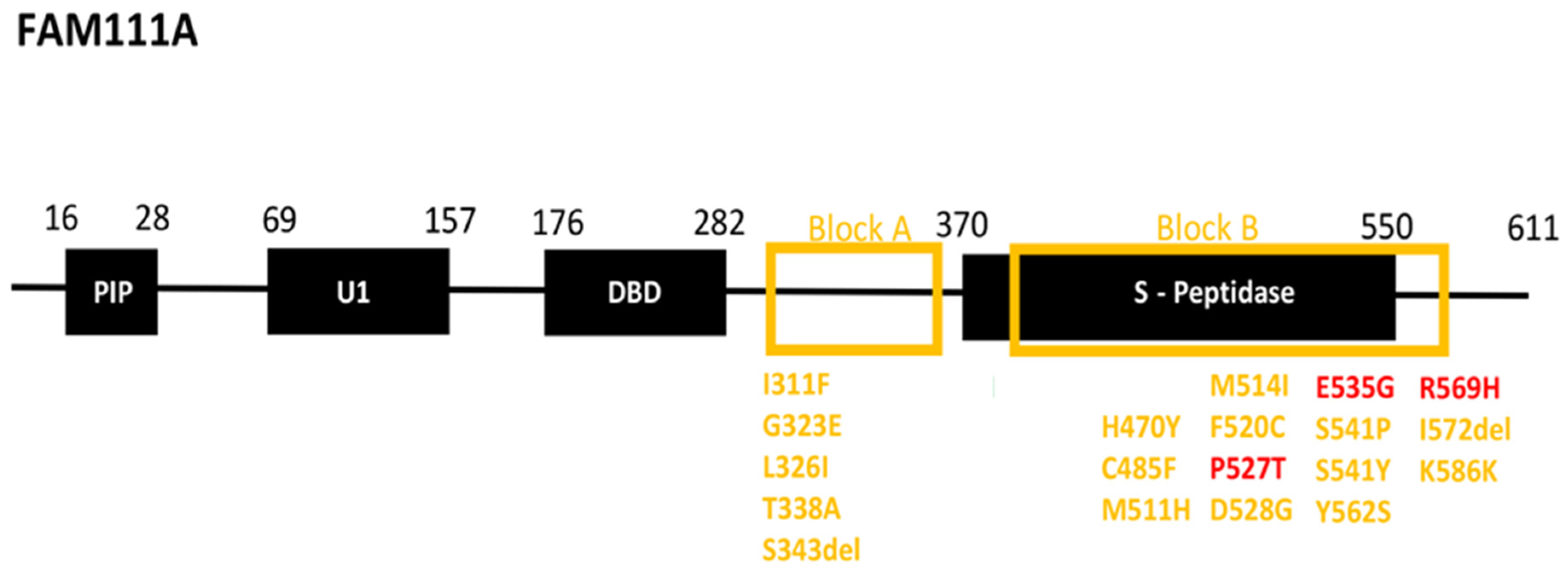
3.2. Patient Variant Analysis
3.3. Follow-Up
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abraham, M.B.; Li, D.; Tang, D.; O’Connell, S.M.; McKenzie, F.; Lim, E.M.; Hakonarson, H.; Levine, M.A.; Choong, C.S. Short stature and hypoparathyroidism in a child with Kenny-Caffey syndrome type 2 due to a novel mutation in FAM111A gene. Int. J. Pediatr. Endocrinol. 2017, 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Ohmachi, Y.; Urai, S.; Bando, H.; Yokoi, J.; Yamamoto, M.; Kanie, K.; Motomura, Y.; Tsujimoto, Y.; Sasaki, Y.; Oi, Y.; et al. Case report: Late middle-aged features of FAM111A variant, Kenny–Caffey syndrome type 2-suggestive symptoms during a long follow-up. Front. Endocrinol. 2023, 13, 1073173. [Google Scholar] [CrossRef] [PubMed]
- Nazar, A.; George, R.; Mathew, N. Oral rehabilitation of a patient with Kenny-Caffey syndrome using telescopic overdenture. J. Indian Prosthodont. Soc. 2021, 21, 204–207. [Google Scholar] [CrossRef]
- Frech, R.S.; McAlister, W.H. Medullary Stenosis of the Tubular Bones Associated with Hypocalcemic Convulsions and Short Stature. Radiology 1968, 91, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Fredriks, A.M.; van Buuren, S.; Van Heel, W.J.M.; Dijkman-Neerincx, R.H.M.; Verloove-Vanhorick, S.P.; Wit, J.M. Nationwide age references for sitting height, leg length, and sitting height/height ratio, and their diagnostic value for disproportionate growth disorders. Arch. Dis. Child. 2005, 90, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Rios-Szwed, D.O.; Alvarez, V.; Sanchez-Pulido, L.; Garcia-Wilson, E.; Jiang, H.; Bandau, S.; Lamond, A.; Alabert, C. FAM111A regulates replication origin activation and cell fitness. Life Sci. Alliance 2023, 6, e202302111. [Google Scholar] [CrossRef]
- Rosato, S.; Unger, S.; Campos-Xavier, B.; Caraffi, S.G.; Beltrami, L.; Pollazzon, M.; Ivanovski, I.; Castori, M.; Bonasoni, M.P.; Comitini, G.; et al. Clinical and Molecular Diagnosis of Osteocraniostenosis in Fetuses and Newborns: Prenatal Ultrasound, Clinical, Radiological and Pathological Features. Genes 2022, 13, 261. [Google Scholar] [CrossRef]
- Caffey, J. Congenital stenosis of medullary spaces in tubular bones and calvaria in two proportionate dwarfs—Mother and son; coupled with transitory hypocalcemic tetany. Am. J. Roentgenol. 1967, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kenny, F.M.; Linarelli, L. Dwarfism and cortical thickening of tubular bones. Transient hypocalcemia in a mother and son. Am. J. Dis. Child. 1966, 111, 201–207. [Google Scholar] [CrossRef]
- Chen, X.; Zou, C. Further delineation of phenotype and genotype of Kenny-Caffey syndrome type 2 (phenotype and genotype of KCS type 2). Mol. Genet. Genom. Med. 2024, 12, e2433. [Google Scholar] [CrossRef]
- Tonelli, L.; Sanchini, M.; Margutti, A.; Buldrini, B.; Superti-Furga, A.; Ferlini, A.; Selvatici, R.; Bigoni, S. Mother and daughter with Kenny-Caffey syndrome: The adult phenotype. Eur. J. Med. Genet. 2024, 69, 104943. [Google Scholar] [CrossRef] [PubMed]
- Schigt, H.; Bald, M.; van der Eerden, B.C.J.; Gal, L.; Ilenwabor, B.P.; Konrad, M.; Levine, M.A.; Li, D.; Mache, C.J.; Mackin, S.; et al. Expanding the Phenotypic Spectrum of Kenny-Caffey Syndrome. J. Clin. Endocrinol. Metab. 2023, 108, e754–e768. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, K.; Masel, J.; Sillence, D.O.; Arbuckle, S.; Juttnerova, V. Gracile bone dysplasias. Pediatr. Radiol. 2002, 32, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Unger, S.; Górna, M.W.; Le Béchec, A.; Do Vale-Pereira, S.; Bedeschi, M.F.; Geiberger, S.; Grigelioniene, G.; Horemuzova, E.; Lalatta, F.; Lausch, E.; et al. FAM111A mutations result in hypoparathyroidism and impaired skeletal development. Am. J. Hum. Genet. 2013, 92, 990–995. [Google Scholar] [CrossRef]
- Verloes, A.; Narcy, F.; Grattagliano, B.; Delezoide, A.L.; Guibaud, P.; Schaaps, J.P.; Merrer, M.L.; Maroteaux, P. Osteocraniostenosis. J. Med. Genet. 1994, 31, 772–778. [Google Scholar] [CrossRef]
- Korniszewski, L.; Arbuckle, S.; Kozlowski, K. Two familial cases with a lethal gracile bone dysplasia and intrauterine growth retardation. Am. J. Med. Genet. A 2003, 118A, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Fernandez, D.J.; Lal, M.; Buehler, E.; Moss, B. Triad of human cellular proteins, IRF2, FAM111A, and RFC3, restrict replication of orthopoxvirus SPI-1 host-range mutants. Proc. Natl. Acad. Sci. USA 2017, 114, 3720–3725. [Google Scholar] [CrossRef]
- Page, M.J.; Di Cera, E. Serine peptidases: Classification, structure and function. Cell. Mol. Life Sci. 2008, 65, 1220–1236. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Morton, F.R.; Kok, C.Y.; Kong, J.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2008, 36, D320–D325. [Google Scholar] [CrossRef]
- Welter, A.L.; Machida, Y.J. Functions and evolution of FAM111 serine proteases. Front. Mol. Biosci. 2022, 9, 1081166. [Google Scholar] [CrossRef]
- Cheng, S.S.W.; Chan, P.K.J.; Luk, H.-M.; Mok, M.T.-S.; Lo, I.F.M. Adult Chinese twins with Kenny–Caffey syndrome type 2: A potential age-dependent phenotype and review of literature. Am. J. Med. Genet. A 2021, 185, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.R.; Municchi, G. Six-hour and four-hour nocturnal sampling for growth hormone. J. Pediatr. Endocrinol. Metab. 1999, 12, 167–173. [Google Scholar] [CrossRef] [PubMed]
- WHO Multicentre Growth Reference Study Group; de Onis, M. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr. 2006, 450, 76–85. [Google Scholar] [CrossRef]
- Kliegman, R.M. Nelson Textbook of Pediatrics, 21st ed.; Elsevier: Philadelphia, MO, USA, 2019; p. 2466.e6-e12. [Google Scholar]
- Pagana, K.D.; Pagana, T.J.; Pagana, T.N. Mosby’s Diagnostic and Laboratory Test Reference, 16th ed.; Elsevier: St. Louis, MO, USA, 2023. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Eren, E.; Tezcan Ünlü, H.; Ceylaner, S.; Tarım, Ö. Compound Heterozygous Variants in FAM111A Cause Autosomal Recessive Kenny-Caffey Syndrome Type 2. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Isojima, T.; Doi, K.; Mitsui, J.; Oda, Y.; Tokuhiro, E.; Yasoda, A.; Yorifuji, T.; Horikawa, R.; Yoshimura, J.; Ishiura, H.; et al. A recurrent de novo FAM111A mutation causes Kenny-Caffey syndrome type 2. J. Bone Miner. Res. 2014, 29, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Koller, S.; Atac, D.; Pfäffli, O.A.; Hanson, J.V.M.; Feil, S.; Bähr, L.; Bahr, A.; Kottke, R.; Joset, P.; et al. Genotype-phenotype spectrum in isolated and syndromic nanophthalmos. Acta Ophthalmol. 2021, 99, e594–e607. [Google Scholar] [CrossRef]
- Yuan, N.; Lu, L.; Xing, X.P.; Wang, O.; Jiang, Y.; Wu, J.; He, M.H.; Wang, X.J.; Cao, L.W. Clinical and genetic features of Kenny-Caffey syndrome type 2 with multiple electrolyte disturbances: A case report. World J. Clin. Cases 2023, 11, 2290–2300. [Google Scholar] [CrossRef]
- Saha, S.; Sreenivas, V.; Goswami, R. Alfacalcidol vs Calcitriol in the Management of Patient With Hypoparathyroidism: A Randomized Controlled Trial. J. Clin. Endocrinol. Metab. 2021, 106, 2092–2102. [Google Scholar] [CrossRef]
- Kubodera, N. A New Look at the Most Successful Prodrugs for Active Vitamin D (D Hormone): Alfacalcidol and Doxercalciferol. Molecules 2009, 14, 3869–3880. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.M.; Regan, S.; Cooley, M.R.; Lauter, K.B.; Vrla, M.C.; Becker, C.B.; Burnett-Bowie, S.A.; Mannstadt, M. Long-term follow-up of patients with hypoparathyroidism. J. Clin. Endocrinol. Metab. 2012, 97, 4507–4514. [Google Scholar] [CrossRef]
- Díez, J.J.; Anda, E.; Pérez-Corral, B.; Paja, M.; Alcázar, V.; Sánchez-Ragnarsson, C.; Orois, A.; Romero-Lluch, A.R.; Sambo, M.; Oleaga, A.; et al. Incident comorbidities in patients with chronic hypoparathyroidism after thyroidectomy: A multicenter nationwide study. Front. Endocrinol. 2024, 15, 1348971. [Google Scholar] [CrossRef]
- Gosmanova, E.O.; Houillier, P.; Rejnmark, L.; Marelli, C.; Bilezikian, J.P. Renal complications in patients with chronic hypoparathyroidism on conventional therapy: A systematic literature review: Renal disease in chronic hypoparathyroidism. Rev. Endocr. Metab. Disord. 2021, 22, 297–316. [Google Scholar] [CrossRef]
- Khan, A.A.; Bilezikian, J.P.; Brandi, M.L.; Clarke, B.L.; Gittoes, N.J.; Pasieka, J.L.; Rejnmark, L.; Shoback, D.M.; Potts, J.T.; Guyatt, G.H.; et al. Evaluation and Management of Hypoparathyroidism Summary Statement and Guidelines from the Second International Workshop. J. Bone Miner. Res. 2022, 37, 2568–2585. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Gao, X.; Li, Y.; Zhang, Z.; Xie, S.; Ren, S.; Li, Y.; Li, H.; Niu, K.; Fu, S.; et al. Human FAM111A inhibits vaccinia virus replication by degrading viral protein I3 and is antagonized by poxvirus host range factor SPI-1. Proc. Natl. Acad. Sci. USA 2023, 120, e2304242120. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.; Oravcová, M.; Jami-Alahmadi, Y.; Wohlschlegel, J.A.; Lazzerini-Denchi, E.; Boddy, M.N. FAM111A induces nuclear dysfunction in disease and viral restriction. EMBO Rep. 2021, 22, e50803. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Bukowski-Wills, J.C.; Lee, S.B.; Kustatscher, G.; Nakamura, K.; de Lima Alves, F.; Menard, P.; Mejlvang, J.; Rappsilber, J.; Groth, A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014, 16, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Naicker, D.; Rhoda, C.; Sunda, F.; Arowolo, A. Unravelling the Intricate Roles of FAM111A and FAM111B: From Protease-Mediated Cellular Processes to Disease Implications. Int. J. Mol. Sci. 2024, 25, 2845. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Machida, Y.; Palani, S.; Caulfield, T.R.; Radisky, E.S.; Kaufmann, S.H.; Machida, Y.J. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 2020, 11, 1318. [Google Scholar] [CrossRef]
- Li, F.; Raczynska, J.E.; Chen, Z.; Yu, H. Structural Insight into DNA-Dependent Activation of Human Metalloprotease Spartan. Cell Rep. 2019, 26, 3336–3346.e4. [Google Scholar] [CrossRef]
- Tan, R.S.G.; Lee, C.H.L.; Pan, W.; Wohlgemuth, S.; Doschak, M.R.; Alexander, R.T. Disruption of the c-terminal serine protease domain of Fam111a does not alter calcium homeostasis in mice. Physiol. Rep. 2024, 12, e15977. [Google Scholar] [CrossRef] [PubMed]
- Nikkel, S.M.; Ahmed, A.; Smith, A.; Marcadier, J.; Bulman, D.E.; Boycott, K.M. Mother-to-daughter transmission of Kenny–Caffey syndrome associated with the recurrent, dominant FAM111A mutation p.Arg569His. Clin. Genet. 2014, 86, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.H.; Shen, Y.; Walvoord, E.C.; Miller, T.C.; Moon, J.E.; Hirschhorn, J.N.; Dauber, A. Whole exome sequencing to identify genetic causes of short stature. Horm. Res. Paediatr. 2014, 82, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Underbjerg, L.; Sikjaer, T.; Rejnmark, L. Long-Term Complications in Patients With Hypoparathyroidism Evaluated by Biochemical Findings: A Case-Control Study. J. Bone Miner. Res. 2018, 33, 822–831. [Google Scholar] [CrossRef]
- Levy, I.; Licht, C.; Daneman, A.; Sochett, E.; Harrington, J. The Impact of Hypoparathyroidism Treatment on the Kidney in Children: Long-Term Retrospective Follow-Up Study. J. Clin. Endocrinol. Metab. 2015, 100, 4106–4113. [Google Scholar] [CrossRef]
- Goswami, R.; Sharma, R.; Sreenivas, V.; Gupta, N.; Ganapathy, A.; Das, S. Prevalence and progression of basal ganglia calcification and its pathogenic mechanism in patients with idiopathic hypoparathyroidism. Clin. Endocrinol. 2012, 77, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Eom, T.H.; Kim, Y.H.; Kim, J.M. Recurrent seizures, mental retardation and extensive brain calcinosis related to delayed diagnosis of hypoparathyroidism in an adolescent boy. J. Clin. Neurosci. 2015, 22, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.H.; Kovacs, K.; Li, S.; Kulharya, A.S.; Johnson, B.L.; Eidson, M.S.; Cleveland, W.W. Kenny-Caffey syndrome and microorchidism. Am. J. Med. Genet. 1998, 80, 107–111. [Google Scholar] [CrossRef]
- Paterson, W.F.; Kelly, B.; Newman, W.; Savage, M.O.; Camacho-Hubner, C.; Dutton, G.N.; Tolmie, J.; Donaldson, M.D. Deterioration of visual acuity associated with growth hormone therapy in a child with extreme short stature and high hypermetropia. Horm. Res. Paediatr. 2007, 67, 67–72. [Google Scholar] [CrossRef]
- Kaleta, D.; Zapolnik, P.; Mazur, A.; Pyrkosz, A. A rare cause of short stature: Kenny-Caffey syndrome type 2—A case report and literature review. Pediatr. Pol. 2020, 95, 249–254. [Google Scholar] [CrossRef]
- Deconte, D.; Kreusch, T.C.; Salvaro, B.P.; Perin, W.F.; Ferreira, M.A.T.; Kopacek, C.; da Rosa, E.B.; Heringer, J.I.; Ligabue-Braun, R.; Zen, P.R.G.; et al. Ophthalmologic Impairment and Intellectual Disability in a Girl Presenting Kenny-Caffey Syndrome Type 2. J. Pediatr. Genet. 2020, 9, 263–269. [Google Scholar] [CrossRef]
- Lee, W.K.; Vargas, A.; Barnes, J.; Root, A.W. The Kenny-Caffey syndrome: Growth retardation and hypocalcemia in a young boy. Am. J. Med. Genet. 1983, 14, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Society, G.H.R. Consensus Guidelines for the Diagnosis and Treatment of Growth Hormone (GH) Deficiency in Childhood and Adolescence: Summary Statement of the GH Research Society1. J. Clin. Endocrinol. Metab. 2000, 85, 3990–3993. [Google Scholar] [CrossRef]
- Mauro, B.; Cristina, M. Growth Hormone Deficiency: Diagnosis and Therapy in Children. In Restricted Growth: Clinical, Genetic and Molecular Aspects; del Carmen Cardenas-Aguayo, M., Ed.; IntechOpen: Rijeka, Croatia, 2016; Chapter 2. [Google Scholar] [CrossRef]
- Yuen, K.C.J.; Johannsson, G.; Ho, K.K.Y.; Miller, B.S.; Bergada, I.; Rogol, A.D. Diagnosis and testing for growth hormone deficiency across the ages: A global view of the accuracy, caveats, and cut-offs for diagnosis. Endocr. Connect. 2023, 12, e220504. [Google Scholar] [CrossRef] [PubMed]
- Sodero, G.; Mariani, F.; Caprarelli, M.; Agazzi, C.; Quarta, L.; Benacquista, L.; Rigante, D.; Clelia, C. Growth hormone responses during arginine and clonidine stimulation test: Correlations with patients’ auxological and metabolic parameters in a single centre study. Growth Horm. IGF Res. 2023, 68, 101522. [Google Scholar] [CrossRef]
- Goyal, A.; Khadgawat, R. Diagnosis of childhood growth hormone deficiency: Controversies, consensus and the need for new diagnostic tools. Neurol. India 2018, 66, 1685–1686. [Google Scholar] [PubMed]
- Maghnie, M.; Labarta, J.I.; Koledova, E.; Rohrer, T.R. Short Stature Diagnosis and Referral. Front. Endocrinol. 2017, 8, 374. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.L.; Hollowell, J.G.; Pan, F.; Gifford, R.A.; Moore, W.V. Growth hormone secretory profiles: Variation on consecutive nights. J. Pediatr. 1989, 115, 51–56. [Google Scholar] [CrossRef]
- Yerawar, C.; Kabde, A.; Deokar, P. Kenny-Caffey syndrome type 2. QJM Int. J. Med. 2021, 114, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Cavole, T.R.; Perrone, E.; de Faria Soares, M.F.; Dias da Silva, M.R.; Maeda, S.S.; Lazaretti-Castro, M.; Alvarez Perez, A.B. Overlapping phenotype comprising Kenny-Caffey type 2 and Sanjad-Sakati syndromes: The first case report. Am. J. Med. Genet. A 2020, 182, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nie, M.; Wang, O.; Li, Y.; Jiang, Y.; Li, M.; Xia, W.; Xing, X. Genetic Screening in a Large Chinese Cohort of Childhood Onset Hypoparathyroidism by Next-Generation Sequencing Combined with TBX1-MLPA. J. Bone Miner. Res. 2019, 34, 2254–2263. [Google Scholar] [CrossRef]
- Grosse, G.; Hilger, A.; Ludwig, M.; Reutter, H.; Lorenzen, F.; Even, G.; Holterhus, P.M.; Woelfle, J. Targeted Resequencing of Putative Growth-Related Genes Using Whole Exome Sequencing in Patients with Severe Primary IGF-I Deficiency. Horm. Res. Paediatr. 2017, 88, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Boynton, J.R.; Pheasant, T.R.; Johnson, B.L.; Levin, D.B.; Streeten, B.W. Ocular findings in Kenny’s syndrome. Arch. Ophthalmol. 1979, 97, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Ally, N.; Ismail, S.; Alli, H.D. Prevalence of complications in eyes with nanophthalmos or microphthalmos: Protocol for a systematic review and meta-analysis. Syst. Rev. 2022, 11, 25. [Google Scholar] [CrossRef]
- Nouri-Mahdavi, K.; Nilforushan, N.; Razeghinejad, M.R.; Amini, H.; Perera, S.A. Glaucoma in a patient with nanophthalmos. J. Ophthalmic Vis. Res. 2011, 6, 208–214. [Google Scholar]
- Joseph, A.D.D.; Sirisena, N.D.; Kumanan, T.; Sujanitha, V.; Strelow, V.; Yamamoto, R.; Wieczorek, S.; Dissanayake, V.H.W. Hypoparathyroidism, Sensorineural deafness and renal disease (Barakat syndrome) caused by a reduced gene dosage in GATA3: A case report and review of literature. BMC Endocr. Disord. 2019, 19, 111. [Google Scholar] [CrossRef] [PubMed]
- Bashar, M.; Taimur, M.; Amreek, F.; Sayeed, K.A.; Tahir, A. Endocrinological Manifestations of Sanjad-Sakati Syndrome. Cureus 2020, 12, e8770. [Google Scholar] [CrossRef] [PubMed]
- Naguib, K.K.; Gouda, S.A.; Elshafey, A.; Mohammed, F.; Bastaki, L.; Azab, A.S.; Alawadi, S.A. Sanjad-Sakati syndrome/Kenny-Caffey syndrome type 1: A study of 21 cases in Kuwait. East. Mediterr. Health J. 2009, 15, 345–352. [Google Scholar] [CrossRef]
- Rafique, B.; Al-Yaarubi, S. Sanjad-Sakati Syndrome in Omani children. Oman Med. J. 2010, 25, 227–229. [Google Scholar] [CrossRef]
- Berkešová, B.A.; Borbély, Z. Barakat syndrome. Vnitr. Lek. 2023, 69, 16–19. [Google Scholar] [CrossRef]
- Alkaissi, H.R.; Banerji, M.A. Primary Hypoparathyroidism Presenting as Idiopathic Intracranial Hypertension in a Patient with Barakat Syndrome. Cureus 2022, 14, e24521. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.L.; Wang, J.J.; Tseng, S.H.; Chang, Y.S. Sociodemographic Factors and Comorbidities Including Hyperparathyroidism Are Associated with an Increased Risk of Band Keratopathy: A Population-Based Study in Taiwan. Front. Endocrinol. 2022, 13, 927513. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.F.; Jan, R.L.; Chang, C.; Wang, J.J.; Su, S.B.; Huang, C.C.; Tseng, S.H.; Chang, Y.S. Risk of Band Keratopathy in Patients with End-Stage Renal Disease. Sci. Rep. 2016, 6, 28675. [Google Scholar] [CrossRef]
- Tarrass, F.; Benjelloun, M.; Benghanem, M.G.; Ramdani, B. Calcareous degeneration of the eye—An unusual complication of uraemic hyperparathyroidism. Nephrol. Dial. Transplant. 2006, 21, 3330. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Cheong, H.I.; Kim, J.H.; Yu, Y.S.; Kwon, J.W. Presumed atypical HDR syndrome associated with Band Keratopathy and pigmentary retinopathy. J. Pediatr. Ophthalmol. Strabismus 2011, 48, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Elhusseiny, A.M.; Saeed, H.N. Corneal opacification in Sanjad-Sakati syndrome. Am. J. Ophthalmol. Case Rep. 2022, 26, 101503. [Google Scholar] [CrossRef]
- Moussaid, Y.; Griffiths, D.; Richard, B.; Dieux, A.; Lemerrer, M.; Léger, J.; Lacombe, D.; Bailleul-Forestier, I. Oral manifestations of patients with Kenny-Caffey Syndrome. Eur. J. Med. Genet. 2012, 55, 441–445. [Google Scholar] [CrossRef]
- Demir, T.; Kecik, D.; Cehreli, Z.C. Kenny-Caffey Syndrome: Oral findings and 4-year follow-up of overlay denture therapy. J. Dent. Child. 2007, 74, 236–240. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Age | Months | Years | Normal Values [24,25] | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 9 | 210/12 | 31/12 | 37/12 | 41/12 | 43/12 | 5 | ||
| Serum calcium (mmol/L) | 0.77 | 2.01 | 2.2 | 2.44 | 1.97 | 2.32 | 7 days–2 years: 2.3–2.65; 3 years–18 years: 2.25–2.7 | ||
| Calcium ionized (mmol/L) | 0.95 | 1.25 | 1.12 | 1.17 | 1.08 | 1.22 | Newborn 1.05–1.37 2 months–18 years 1.2–1.38 Adult 1.05–1.3 | ||
| Magnesium (mmol/L) | 0.58 | 0.8 | 0.71 | 0.75 | 1.02 | 0.63 | 0.7 | 0.69 | 0–6 days: 0.48–1.05 mmol/L; 7 days–2 years: 0.65–1.05 mmol/L; 2–14 years: 0.60–0.95 mmol/L) |
| Phosphorus (mmol/L) | 3.2 | 2.31 | 1.56 | 0.99 | 1.73 | 1.78 | 1.58 | 0–5 days: 1.55–2.65; 1–3 years: 1.25–2.10; 4–11 years: 1.20–1.80; 12–15 years: 0.95–1.75; 16–19 years: 0.9–1.5 | |
| Alkaline phosphatase (U/L) | 393 | 208 | 239 | 190 | 260 | 165 | <2 years: 85–235; 2–8 years: 65–210; 9–15 years: 60–300; 16–21 years: 30–200 | ||
| PTH (pg/mL) | 10.37 | 19.3 | 19.8 | 7.3 | 7.3 | 20.4 | 10.52 | 12–62 | |
| Vitamin D (ng/mL) | 59.4 | 22.4 | 16.4 | 43 | 15.1 | 20–80 | |||
| TSH (mIU/L) | 1.77 | 6.17 | 5.74 | 3.79 | 0–3 days: 1–20; 3–30 days: 0.5–6.5; 1–5 months 0.5–6; 6 months–18 years: 0.5–4.5 | ||||
| fT4 (pmol/L) | 14.7 | 15.4 | 15.4 | 2 weeks–20 years: 10.2–26 | |||||
| a–TPO IU/mL | <10 | <10 | |||||||
| a–Tg (IU/mL) | <20 | <20 | |||||||
| Cortisol (nmol/L) | 281 | 8 am: 82.7–579.3 | |||||||
| IGF-1 (ng/mL) | 15.4 | 21.4 | 45.5 | Males 0–11 months: 18–79 1 year: 20–108 2 years: 24–135 3 years: 28–148 4 years: 32–165 5 years: 37–196 | |||||
| IGFBP3 (mg/L) | 2.07 | 3 years: 1.6–4.5; 4 years: 1.8–4.9 | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatziagapiou, K.; Sertedaki, A.; Dermentzoglou, V.; Popović, N.Č.; Lambrou, G.I.; Papageorgiou, L.; Thireou, T.; Kanaka-Gantenbein, C.; Sakka, S.D. Kenny–Caffey Syndrome Type 2 (KCS2): A New Case Report and Patient Follow-Up Optimization. J. Clin. Med. 2025, 14, 118. https://doi.org/10.3390/jcm14010118
Hatziagapiou K, Sertedaki A, Dermentzoglou V, Popović NČ, Lambrou GI, Papageorgiou L, Thireou T, Kanaka-Gantenbein C, Sakka SD. Kenny–Caffey Syndrome Type 2 (KCS2): A New Case Report and Patient Follow-Up Optimization. Journal of Clinical Medicine. 2025; 14(1):118. https://doi.org/10.3390/jcm14010118
Chicago/Turabian StyleHatziagapiou, Kyriaki, Amalia Sertedaki, Vasiliki Dermentzoglou, Nataša Čurović Popović, George I. Lambrou, Louis Papageorgiou, Trias Thireou, Christina Kanaka-Gantenbein, and Sophia D. Sakka. 2025. "Kenny–Caffey Syndrome Type 2 (KCS2): A New Case Report and Patient Follow-Up Optimization" Journal of Clinical Medicine 14, no. 1: 118. https://doi.org/10.3390/jcm14010118
APA StyleHatziagapiou, K., Sertedaki, A., Dermentzoglou, V., Popović, N. Č., Lambrou, G. I., Papageorgiou, L., Thireou, T., Kanaka-Gantenbein, C., & Sakka, S. D. (2025). Kenny–Caffey Syndrome Type 2 (KCS2): A New Case Report and Patient Follow-Up Optimization. Journal of Clinical Medicine, 14(1), 118. https://doi.org/10.3390/jcm14010118