


Endothelial Dysfunction in Heart Failure: What Is Its Role?


Abstract
:1. Endothelial Functions
2. Methods for Measuring Endothelial Function
2.1. Intracoronary Artery Infusions Using Acetylcholine or Other Vasoactive Substances
2.2. Flow-Mediated Vasodilation (FMD)
2.3. Peripheral Arterial Tone (PAT)
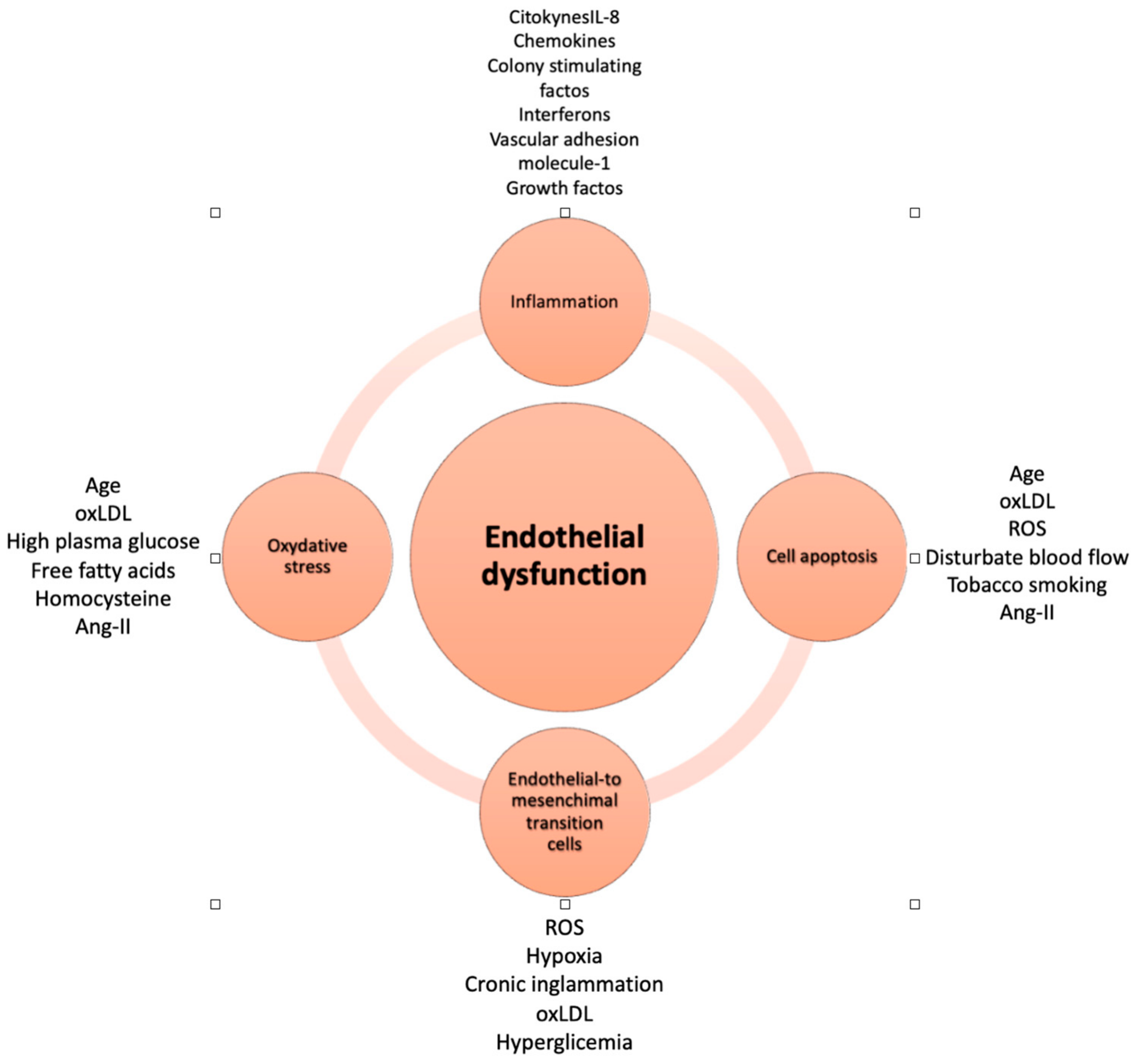
3. Endothelial Dysfunction
4. Endothelial Dysfunction and Heart Failure
5. Endothelial Dysfunction in Acute Heart Failure
6. Clinical Trials and Endothelial Function
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Krüger-Genge, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Cardillo, C. Obesity, blood vessels and metabolic syndrome. Acta Physiol. 2011, 203, 279–286. [Google Scholar] [CrossRef]
- Chatterjee, S. Endothelial Mechanotransduction, Redox Signaling and the Regulation of Vascular Inflammatory Pathways. Front. Physiol. 2018, 9, 524. [Google Scholar] [CrossRef]
- Shimokawa, H. 2014 Williams Harvey Lecture: Importance of coronary vasomotion abnormalities-from bench to bedside. Eur. Heart J. 2014, 35, 3180–3193. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Premer, C.; Kanelidis, A.J.; Hare, J.M.; Schulman, I.H. Rethinking Endothelial Dysfunction as a Crucial Target in Fighting Heart Failure. In Mayo Clinic Proceedings: Innovations, Quality & Outcomes; Elsevier BV: Amsterdam, The Netherlands, 2019; Volume 3, pp. 1–13. [Google Scholar]
- Newby, D.E.; Fox, K.A. Invasive assessment of the coronary circulation: Intravascular ultrasound and Doppler. Br. J. Clin. Pharmacol. 2002, 53, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Doucette, J.W.; Corl, P.D.; Payne, H.M.; E Flynn, A.; Goto, M.; Nassi, M.; Segal, J. Validation of a Doppler guide wire for intravascular measurement of coronary artery flow velocity. Circulation 1992, 85, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Schächinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Carrick, D.; Haig, C.; Rauhalammi, S.; Ahmed, N.; Mordi, I.; McEntegart, M.; Petrie, M.C.; Eteiba, H.; Hood, S.; Watkins, S.; et al. Prognostic significance of infarct core pathology revealed by quantitative non-contrast in comparison with contrast cardiac magnetic resonance imaging in reperfused ST-elevation myocardial infarction survivors. Eur. Heart J. 2016, 37, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Reriani, M.; Sara, J.D.; Flammer, A.J.; Gulati, R.; Li, J.; Rihal, C.; Lennon, R.; Lerman, L.O.; Lerman, A. Coronary endothelial function testing provides superior discrimination compared with standard clinical risk scoring in prediction of cardiovascular events. Coron. Artery Dis. 2016, 27, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Celermajer, D.S.; Sorensen, K.; Gooch, V.; Spiegelhalter, D.; Miller, O.; Sullivan, I.; Lloyd, J.; Deanfield, J. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Premer, C.; Blum, A.; Bellio, M.A.; Schulman, I.H.; Hurwitz, B.E.; Parker, M.; Dermarkarian, C.R.; DiFede, D.L.; Balkan, W.; Khan, A.; et al. Allogeneic Mesenchymal Stem Cells Restore Endothelial Function in Heart Failure by Stimulating Endothelial Progenitor Cells. EBioMedicine 2015, 2, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Gokce, N.; Keaney, J.F., Jr.; Hunter, L.M.; Watkins, M.T.; Nedeljkovic, Z.S.; Menzoian, J.O.; Vita, J.A. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J. Am. Coll. Cardiol. 2003, 41, 1769–1775. [Google Scholar] [CrossRef]
- Yeboah, J.; Folsom, A.R.; Burke, G.L.; Johnson, C.; Polak, J.F.; Post, W.; Lima, J.A.; Crouse, J.R.; Herrington, D.M. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: The multi-ethnic study of atherosclerosis. Circulation 2009, 120, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.M.; Gori, T.; Ghiadoni, L. Endothelial function testing and cardiovascular disease: Focus on peripheral arterial tonometry. Vasc. Health Risk Manag. 2014, 10, 577–584. [Google Scholar]
- Allan, R.B.; Vun, S.V.; Spark, J.I. A Comparison of Measures of Endothelial Function in Patients with Peripheral Arterial Disease and Age and Gender Matched Controls. Int. J. Vasc. Med. 2016, 2016, 2969740. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, N.M.; Palmisano, J.; Larson, M.G.; Sullivan, L.M.; Lehman, B.T.; Vasan, R.S.; Levy, D.; Mitchell, G.F.; Vita, J.A.; Benjamin, E.J. Relation of brachial and digital measures of vascular function in the community: The Framingham heart study. Hypertension 2011, 57, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Bass, A.; Ellis, K.; Tran, B.; Steele, S.; Caughey, M.; Stouffer, G.A.; Hinderliter, A.L. Relation between digital peripheral arterial tonometry and brachial artery ultrasound measures of vascular function in patients with coronary artery disease and in healthy volunteers. Am. J. Cardiol. 2012, 109, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Kuvin, J.T.; Patel, A.R.; A Sliney, K.; Pandian, N.G.; Sheffy, J.; Schnall, R.P.; Karas, R.H.; E Udelson, J. Assessment of peripheral vascular endothelial function with finger arterial pulse wave amplitude. Am. Heart J. 2003, 146, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Weisrock, F.; Fritschka, M.; Beckmann, S.; Litmeier, S.; Wagner, J.; Tahirovic, E.; Radenovic, S.; Zelenak, C.; Hashemi, D.; Busjahn, A.; et al. Reliability of peripheral arterial tonometry in patients with heart failure, diabetic nephropathy and arterial hypertension. Vasc. Med. 2017, 22, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Fujisue, K.; Sugiyama, S.; Matsuzawa, Y.; Akiyama, E.; Sugamura, K.; Matsubara, J.; Kurokawa, H.; Maeda, H.; Hirata, Y.; Kusaka, H.; et al. Prognostic Significance of Peripheral Microvascular Endothelial Dysfunction in Heart Failure with Reduced Left Ventricular Ejection Fraction. Circ. J. 2015, 79, 2623–2631. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Thuan, D.T.B.; Phu, H.T.; Nguyen, T.H.D.; Hasan, H.; Halabi, S.; Abdelhady, S.; Nasrallah, G.K.; Eid, A.H.; Pintus, G.; et al. Herbal Medicine for Cardiovascular Diseases: Efficacy, Mechanisms, and Safety. Front. Pharmacol. 2020, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Steyers, C.M.; Miller, F.J. Endothelial dysfunction in chronic inflammatory diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; El Yazbi, A.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. (Landmark Ed) 2022, 27, 105. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.B.; Punihaole, D.; Levine, T.B. Characterization of the role of nitric oxide and its clinical applications. Cardiology 2012, 122, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chang, Y.; Wei, W. Endothelial Dysfunction and Inflammation: Immunity in Rheumatoid Arthritis. Mediators Inflamm. 2016, 2016, 6813016. [Google Scholar] [CrossRef] [PubMed]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.; Lund, L.H. Microvascular endothelial dysfunction in heart failure with preserved ejection fraction. Heart 2016, 102, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; Shah, S.J.; Lindholm, D.; et al. Dapagliflozin in heart failure with preserved and mildly reduced ejection fraction: Rationale and design of the DELIVER trial. Eur. J. Heart Fail. 2021, 23, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.; Hayoz, D.; Münzel, T.; Hornig, B.; Just, H.; Brunner, H.R.; Zelis, R. Endothelial function in chronic congestive heart failure. Am. J. Cardiol. 1992, 69, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Olson, T.P.; Lam, C.S.; Flood, K.S.; Lerman, A.; Johnson, B.D.; Redfield, M.M. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol. 2010, 56, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, E.; Sugiyama, S.; Matsuzawa, Y.; Konishi, M.; Suzuki, H.; Nozaki, T.; Ohba, K.; Matsubara, J.; Maeda, H.; Horibata, Y.; et al. Incremental prognostic significance of peripheral endothelial dysfunction in patients with heart failure with normal left ventricular ejection fraction. J. Am. Coll. Cardiol. 2012, 60, 1778–1786. [Google Scholar] [CrossRef]
- Agnoletti, L.; Curello, S.; Bachetti, T.; Malacarne, F.; Gaia, G.; Comini, L.; Volterrani, M.; Bonetti, P.; Parrinello, G.; Cadei, M.; et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: Role of tumor necrosis factor-alpha. Circulation 1999, 100, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Hermann, C.; Zeiher, A.M.; Dimmeler, S. Shear stress inhibits H2O2-induced apoptosis of human endothelial cells by modulation of the glutathione redox cycle and nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3588–3592. [Google Scholar] [CrossRef] [PubMed]
- Rössig, L.; Haendeler, J.; Mallat, Z.; Hugel, B.; Freyssinet, J.-M.; Tedgui, A.; Dimmeler, S.; Zeiher, A.M. Congestive heart failure induces endothelial cell apoptosis: Protective role of carvedilol. J. Am. Coll. Cardiol. 2000, 36, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Glezeva, N.; Baugh, J.A. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail. Rev. 2014, 19, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Joannides, R.; Bizet-Nafeh, C.; Costentin, A.; Iacob, M.; Derumeaux, G.; Cribier, A.; Thuillez, C. Chronic ACE inhibition enhances the endothelial control of arterial mechanics and flow-dependent vasodilatation in heart failure. Hypertension 2001, 38, 1446–1450. [Google Scholar] [CrossRef]
- López Farré, A.; Casado, S. Heart failure, redox alterations, and endothelial dysfunction. Hypertension 2001, 38, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Hornig, B.; Landmesser, U.; Kohler, C.; Ahlersmann, D.; Spiekermann, S.; Christoph, A.; Tatge, H.; Drexler, H. Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: Role of superoxide dismutase. Circulation 2001, 103, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kusano, K.; Nakamura, Y.; Kakishita, M.; Ohta, K.; Nagase, S.; Yamamoto, M.; Miyaji, K.; Saito, H.; Morita, H.; et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 2002, 105, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Zepeda, R.J.; Castillo, R.; Rodrigo, R.; Prieto, J.C.; Aramburu, I.; Brugere, S.; Galdames, K.; Noriega, V.; Miranda, H.F. Effect of carvedilol and nebivolol on oxidative stress-related parameters and endothelial function in patients with essential hypertension. Basic. Clin. Pharmacol. Toxicol. 2012, 111, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Adji, A.; Vlachopoulos, C.; O’rourke, M.F. Effect of sildenafil on cardiac performance in patients with heart failure. Am. J. Cardiol. 2005, 96, 1436–1440. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.D.; Lachmann, J.; Camuso, J.; Lepore, J.J.; Shin, J.; Martinovic, M.E.; Systrom, D.M.; Bloch, K.D.; Semigran, M.J. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation 2007, 115, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Wilson, D.J.; Neutel, J.; Houston, M.; Weinberger, M.H.; Grimm, R.; Smith, D.H.; Sun, W. Coadministered amlodipine and atorvastatin produces early improvements in arterial wall compliance in hypertensive patients with dyslipidemia. Am. J. Hypertens. 2009, 22, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Mentz, R.J.; O’Connor, C.M. Pathophysiology and clinical evaluation of acute heart failure. Nat. Rev. Cardiol. 2016, 13, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.S.; Brutsaert, D.; Dickstein, K.; Drexler, H.; Follath, F.; Harjola, V.-P.; Hochadel, M.; Komajda, M.; Lassus, J.; Lopez-Sendon, J.L.; et al. EuroHeart Failure Survey II (EHFS II): A survey on hospitalized acute heart failure patients: Description of population. Eur. Heart J. 2006, 27, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Miró, Ò.; Sarasola, A.G.; Fuenzalida, C.; Calderón, S.; Jacob, J.; Aguirre, A.; Wu, D.M.; Rizzi, M.A.; Malchair, P.; Haro, A.; et al. Departments involved during the first episode of acute heart failure and subsequent emergency department revisits and rehospitalisations: An outlook through the NOVICA cohort. Eur. J. Heart Fail. 2019, 21, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Butt, J.H.; Fosbøl, E.L.; Gerds, T.A.; Andersson, C.; McMurray, J.J.; Petrie, M.C.; Gustafsson, F.; Madelaire, C.; Kristensen, S.L.; Gislason, G.H.; et al. Readmission and death in patients admitted with new-onset versus worsening of chronic heart failure: Insights from a nationwide cohort. Eur. J. Heart Fail. 2020, 22, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Javaloyes, P.; Miró, Ò.; Gil, V.; Martín-Sánchez, F.J.; Jacob, J.; Herrero, P.; Takagi, K.; Alquézar-Arbé, A.; Díez, M.P.L.; Martín, E.; et al. Clinical phenotypes of acute heart failure based on signs and symptoms of perfusion and congestion at emergency department presentation and their relationship with patient management and outcomes. Eur. J. Heart Fail. 2019, 21, 1353–1365. [Google Scholar] [CrossRef]
- Campanile, A.; Ciccarelli, M.; Galasso, G.; Dell’Aquila, F.; Procaccini, V.; Vigorito, F.; Vecchione, C.; Ravera, A. Predictors of complications in initially haemodynamically stable patients admitted in a modern coronary care unit. J. Cardiovasc. Med. 2021, 22, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Chioncel, O.; Ciccarelli, M.; Galasso, G.; Dell’aquila, F.; Procaccini, V.; Vigorito, F.; Vecchione, C.; Ravera, A. Clinical phenotypes and outcome of patients hospitalized for acute heart failure: The ESC Heart Failure Long-Term Registry. Eur. J. Heart Fail. 2017, 19, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Mebazaa, A.; Harjola, V.; Coats, A.J.; Piepoli, M.F.; Crespo-Leiro, M.G.; Laroche, C.; Seferovic, P.M.; Anker, S.D.; Ferrari, R.; et al. Time-sensitive approach in the management of acute heart failure. ESC Heart Fail. 2021, 8, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, M.; Pires, I.F.; Bauersachs, J.; Bertrand, L.; Beauloye, C.; Dawson, D.; Hamdani, N.; Hilfiker-Kleiner, D.; van Laake, L.W.; Lezoualc’h, F.; et al. Acute heart failure: Mechanisms and pre-clinical models-a Scientific Statement of the ESC Working Group on Myocardial Function. Cardiovasc. Res. 2023, 119, 2390–2404. [Google Scholar] [CrossRef] [PubMed]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Unger, R.E.; Brunner, J.; Kirkpatrick, C. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc. Res. 2003, 60, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Viau, D.M.; A Sala-Mercado, J.; Spranger, M.D.; O’Leary, D.S.; Levy, P.D. The pathophysiology of hypertensive acute heart failure. Heart 2015, 101, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Cordwin, D.J.; Berei, T.J.; Pogue, K.T. The Role of sGC Stimulators and Activators in Heart Failure With Reduced Ejection Fraction. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Voors, A.A.; Kremer, D.; Geven, C.; Maaten, J.M.t.; Struck, J.; Bergmann, A.; Pickkers, P.; Metra, M.; Mebazaa, A.; Düngen, H.D.; et al. Adrenomedullin in heart failure: Pathophysiology and therapeutic application. Eur. J. Heart Fail. 2019, 21, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Kremer, D.; Geven, C.; ter Maaten, J.M.; Struck, J.; Bergmann, A.; Pickkers, P.; Metra, M.; Mebazaa, A.; Düngen, H.; et al. Mesenchymal stem cells overexpressing adrenomedullin improve heart function through antifibrotic action in rats experiencing heart failure. Mol. Med. Rep. 2018, 17, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Looi, Y.H.; Kane, K.A.; McPhaden, A.R.; Wainwright, C.L. Adrenomedullin acts via nitric oxide and peroxynitrite to protect against myocardial ischaemia-induced arrhythmias in anaesthetized rats. Br. J. Pharmacol. 2006, 148, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Okumura, H.; Nagaya, N.; Itoh, T.; Okano, I.; Hino, J.; Mori, K.; Tsukamoto, Y.; Ishibashi-Ueda, H.; Miwa, S.; Tambara, K.; et al. Adrenomedullin infusion attenuates myocardial ischemia/reperfusion injury through the phosphatidylinositol 3-kinase/Akt-dependent pathway. Circulation 2004, 109, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, N.; Nishikimi, T.; Uematsu, M.; Yoshitomi, Y.; Miyao, Y.; Miyazaki, S.; Goto, Y.; Kojima, S.; Kuramochi, M.; Matsuo, H.; et al. Plasma adrenomedullin as an indicator of prognosis after acute myocardial infarction. Heart 1999, 81, 483–487. [Google Scholar] [CrossRef]
- Nishikimi, T.; Karasawa, T.; Inaba, C.; Ishimura, K.; Tadokoro, K.; Koshikawa, S.; Yoshihara, F.; Nagaya, N.; Sakio, H.; Kangawa, K.; et al. Effects of long-term intravenous administration of adrenomedullin (AM) plus hANP therapy in acute decompensated heart failure: A pilot study. Circ. J. 2009, 73, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, N.; Satoh, T.; Nishikimi, T.; Uematsu, M.; Furuichi, S.; Sakamaki, F.; Oya, H.; Kyotani, S.; Nakanishi, N.; Goto, Y.; et al. Hemodynamic, renal, and hormonal effects of adrenomedullin infusion in patients with congestive heart failure. Circulation 2000, 101, 498–503. [Google Scholar] [CrossRef]
- Nakamura, M.; Yoshida, H.; Makita, S.; Arakawa, N.; Niinuma, H.; Hiramori, K. Potent and long-lasting vasodilatory effects of adrenomedullin in humans. Comparisons between normal subjects and patients with chronic heart failure. Circulation 1997, 95, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Pollesello, P.; Haikala, H.; Végh, Á.; Sorsa, T.; Levijoki, J.; Szilágyi, S.; Édes, I.; Tóth, A.; Papp, Z.; et al. ORM-3819 promotes cardiac contractility through Ca(2+) sensitization in combination with selective PDE III inhibition, a novel approach to inotropy. Eur. J. Pharmacol. 2016, 775, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Márton, Z.; Pataricza, J.; Pollesello, P.M.; Varró, A.M.; Papp, J.G.M. The Novel Inodilator ORM-3819 Relaxes Isolated Porcine Coronary Arteries: Role of Voltage-Gated Potassium Channel Activation. J. Cardiovasc. Pharmacol. 2019, 74, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Metra, M.; Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Pang, P.S.; Ponikowski, P.; Voors, A.A.; et al. Effects of Serelaxin in Patients with Acute Heart Failure. N. Engl. J. Med. 2019, 381, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, M.; Du, X.; Dschietzig, T.B.; Summers, R.J. The actions of relaxin on the human cardiovascular system. Br. J. Pharmacol. 2017, 174, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Difede, D.L.; Rieger, A.C.; Florea, V.; Landin, A.; El-Khorazaty, J.; Khan, A.; Mushtaq, M.; Lowery, M.; Byrnes, J.; et al. Randomized Comparison of Allogeneic Versus Autologous Mesenchymal Stem Cells for Nonischemic Dilated Cardiomyopathy: POSEIDON-DCM Trial. J. Am. Coll. Cardiol. 2017, 69, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Legallois, D.; Belin, A.; Nesterov, S.V.; Milliez, P.; Parienti, J.-J.; Knuuti, J.; Abbas, A.; Tirel, O.; Agostini, D.; Manrique, A. Cardiac rehabilitation improves coronary endothelial function in patients with heart failure due to dilated cardiomyopathy: A positron emission tomography study. Eur. J. Prev. Cardiol. 2016, 23, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Siasos, G.; Zaromitidou, M.; Hatzis, G.; Mourouzis, K.; Chrysohoou, C.; Zisimos, K.; Mazaris, S.; Tourikis, P.; Athanasiou, D.; et al. Atorvastatin treatment improves endothelial function through endothelial progenitor cells mobilization in ischemic heart failure patients. Atherosclerosis 2015, 238, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Erbs, S.; Beck, E.B.; Linke, A.; Adams, V.; Gielen, S.; Kränkel, N.; Möbius-Winkler, S.; Höllriegel, R.; Thiele, H.; Hambrecht, R.; et al. High-dose rosuvastatin in chronic heart failure promotes vasculogenesis, corrects endothelial function, and improves cardiac remodeling--results from a randomized, double-blind, and placebo-controlled study. Int. J. Cardiol. 2011, 146, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Leeson, P.; Guzik, T.J.; Zhang, M.-H.; Tousoulis, D.; Antonopoulos, A.S.; Demosthenous, M.; Marinou, K.; Hale, A.; et al. Rapid, direct effects of statin treatment on arterial redox state and nitric oxide bioavailability in human atherosclerosis via tetrahydrobiopterin-mediated endothelial nitric oxide synthase coupling. Circulation 2011, 124, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.V.; Rahman, S.T.; Haque, T.; Merchant, N.; Bhaheetharan, S.; Harris, J., 3rd; Umar, K.; Wahi, J.; Ferdinand, K.C. Vascular effects of nebivolol added to hydrochlorothiazide in African Americans with hypertension and echocardiographic evidence of diastolic dysfunction: The NASAA study. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 291–297. [Google Scholar] [CrossRef]
- Falskov, B.; Hermann, T.S.; Raunsø, J.; Christiansen, B.; Rask-Madsen, C.; Major-Pedersen, A.; Køber, L.; Torp-Pedersen, C.; Dominguez, H. Endothelial function is unaffected by changing between carvedilol and metoprolol in patients with heart failure—A randomized study. Cardiovasc. Diabetol. 2011, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Amore, L.; Alghisi, F.; Pancaldi, E.; Pascariello, G.; Cersosimo, A.; Cimino, G.; Bernardi, N.; Calvi, E.; Lombardi, C.M.; Sciatti, E.; et al. Study of endothelial function and vascular stiffness in patients affected by dilated cardiomyopathy on treatment with sacubitril/valsartan. Am. J. Cardiovasc. Dis. 2022, 12, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Cassano, V.; Armentaro, G.; Magurno, M.; Aiello, V.; Borrello, F.; Miceli, S.; Maio, R.; Perticone, M.; Marra, A.M.; Cittadini, A.; et al. Short-term effect of sacubitril/valsartan on endothelial dysfunction and arterial stiffness in patients with chronic heart failure. Front. Pharmacol. 2022, 13, 1069828. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Methods | Advantage | Disadvantage | Uncertain |
|---|---|---|---|
| Quantitative Coronary Angiography ± IVUS | Most direct Gold standard of early detection of endothelial dysfunction Heavily validated | Invasive procedure with connected risks Not feasible when screening large population Expensive Skilled technicians | None |
| Flow-Mediated Vasodilation (FMD) | Heavily validated Rapidity Noninvasive Easy to use Potentially feasible when screening large population | Expensive Skilled technicians | Many confounding factors due to ambience and diet (Food intake, drugs, vitamins, tobacco use...) |
| Peripheral Arterial Tonometry (PAT) | Operator independent Internal control Rapidity Noninvasive Easy to use Potentially feasible when screening large population Inexpensive | Microvascular measurement Unclear physiology | Correlation with FMD not clear Validity Does not measure endothelial disfunction directly |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drera, A.; Rodella, L.; Brangi, E.; Riccardi, M.; Vizzardi, E. Endothelial Dysfunction in Heart Failure: What Is Its Role? J. Clin. Med. 2024, 13, 2534. https://doi.org/10.3390/jcm13092534
Drera A, Rodella L, Brangi E, Riccardi M, Vizzardi E. Endothelial Dysfunction in Heart Failure: What Is Its Role? Journal of Clinical Medicine. 2024; 13(9):2534. https://doi.org/10.3390/jcm13092534
Chicago/Turabian StyleDrera, Andrea, Luca Rodella, Elisa Brangi, Mauro Riccardi, and Enrico Vizzardi. 2024. "Endothelial Dysfunction in Heart Failure: What Is Its Role?" Journal of Clinical Medicine 13, no. 9: 2534. https://doi.org/10.3390/jcm13092534
APA StyleDrera, A., Rodella, L., Brangi, E., Riccardi, M., & Vizzardi, E. (2024). Endothelial Dysfunction in Heart Failure: What Is Its Role? Journal of Clinical Medicine, 13(9), 2534. https://doi.org/10.3390/jcm13092534