

Current Approaches and Innovations in Managing Preeclampsia: Highlighting Maternal Health Disparities


{kind=link}
{kind=link}
{kind=link}
Abstract
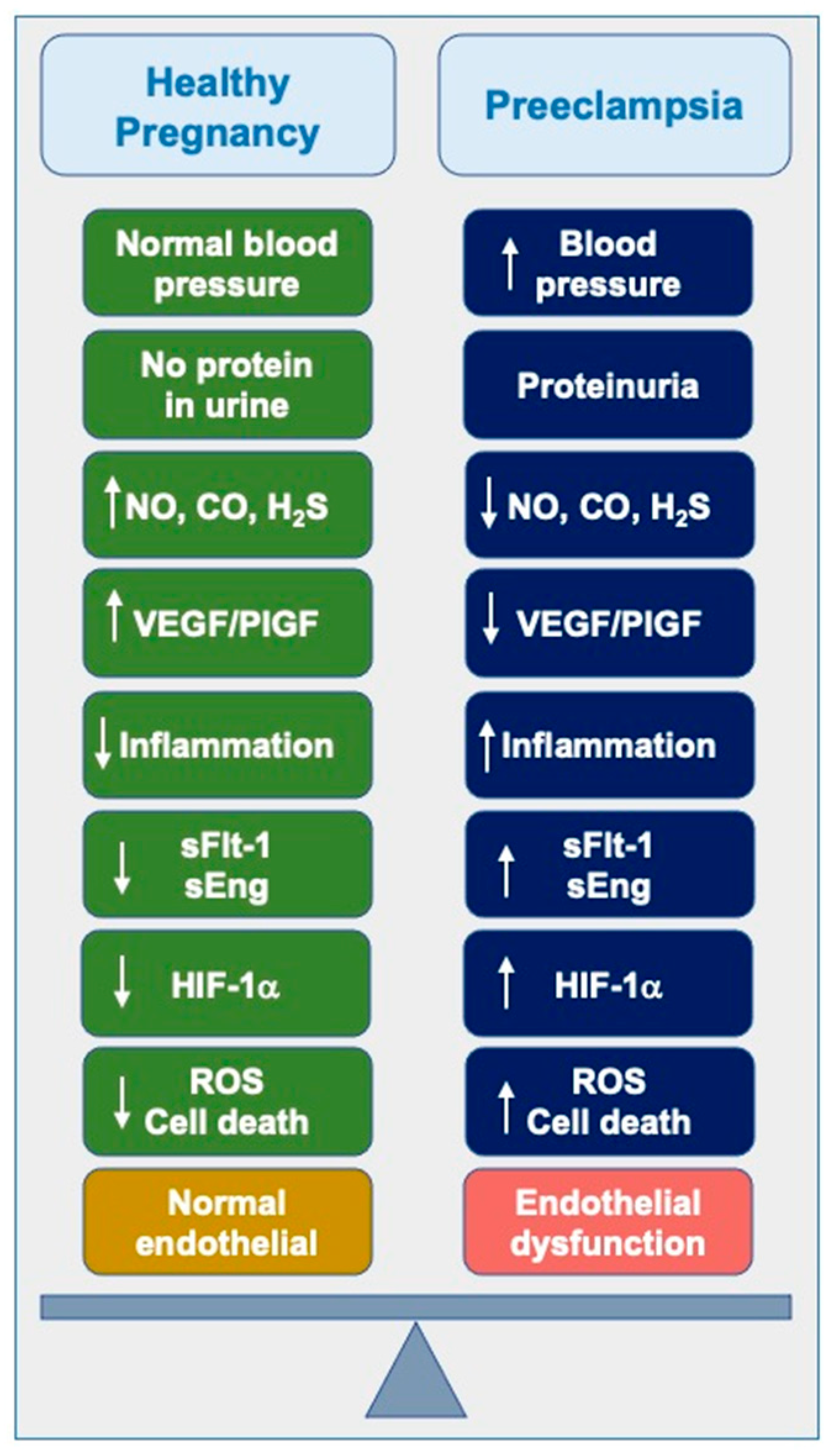
1. Introduction
2. Diagnostic Measures
2.1. Historical Perspectives
2.2. Diagnostic Measures and Current Management
2.2.1. The Role of Aspirin in Managing Preeclampsia
2.2.2. Magnesium Sulfate in Managing Preeclampsia
2.3. Inhibition of COMT and Suppression of Hypoxia-Inducible Factors (HIFs)
2.4. CoQ10 Supplementation
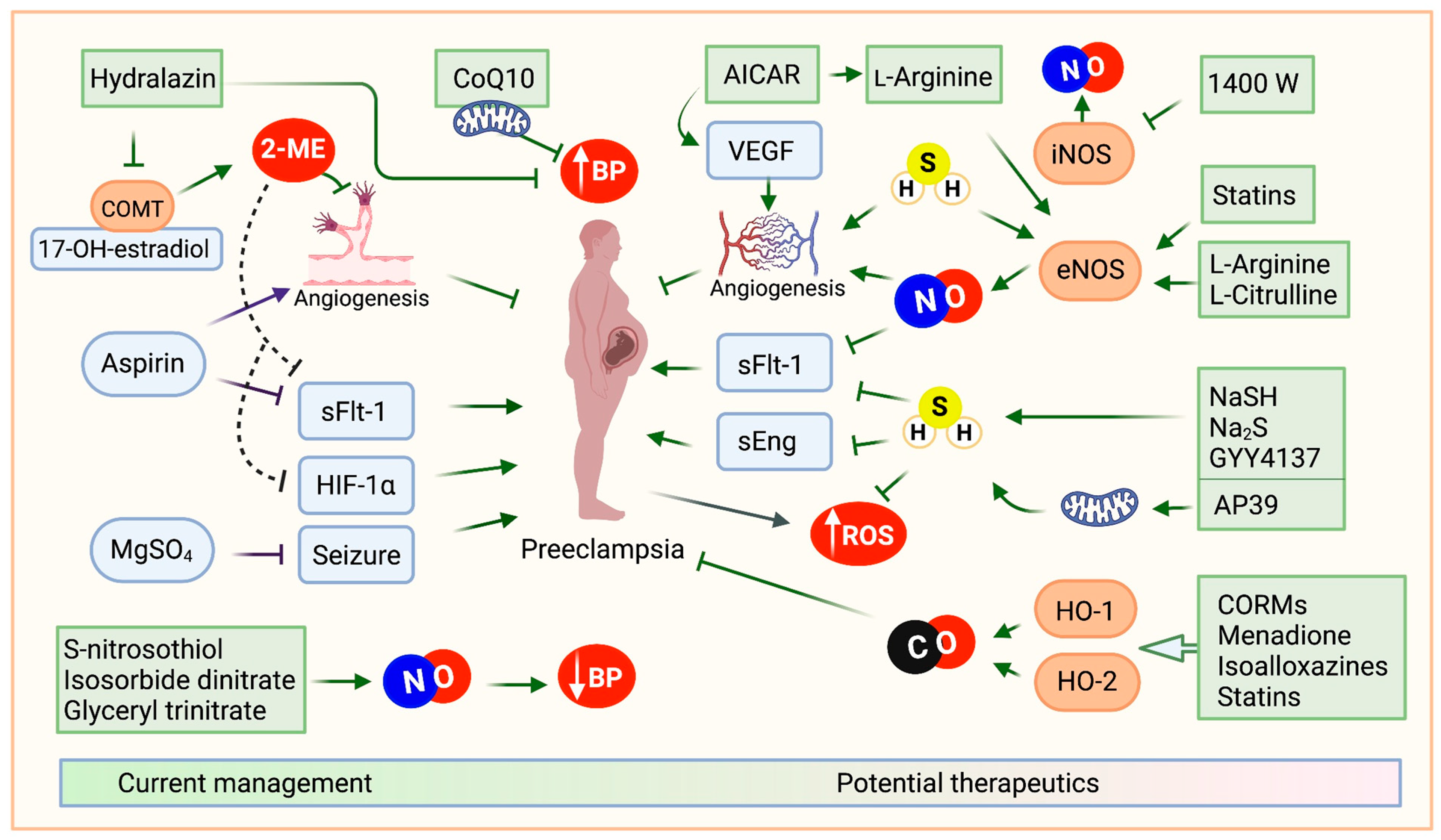
3. Experimental Treatments of Interest
3.1. Restoration of Angiogenic Balance
3.2. Nitric Oxide-Donors
3.3. Modulation of Carbon Monoxide Levels
3.4. H2S-Donors
3.5. Statins
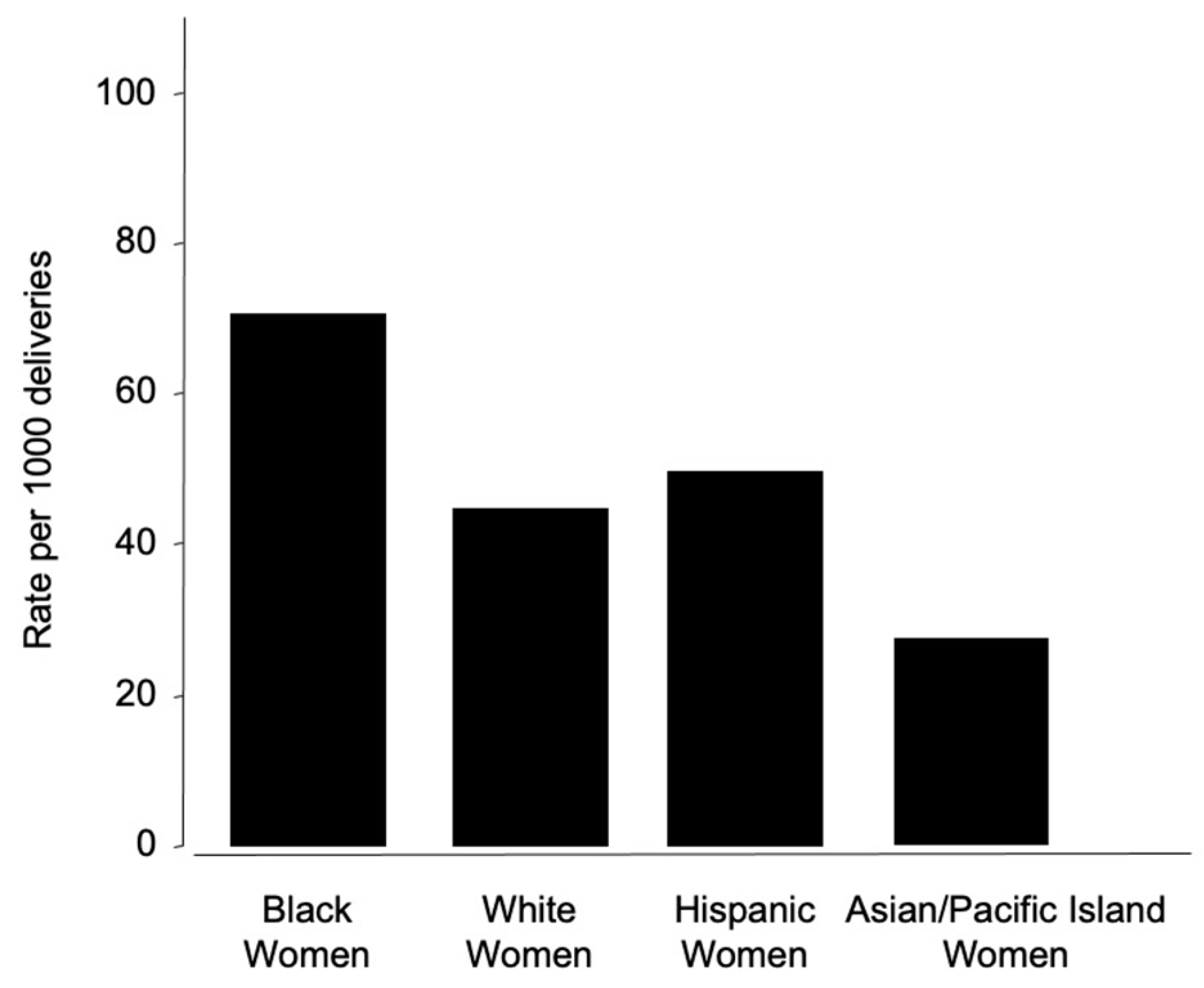
4. Preeclampsia and Racial Disparities
4.1. Maternal Mortality and PE Disparities
4.2. Genetic and Environmental Factors
4.3. Systemic Racism in Medicine
4.4. Addressing Racial Disparities
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-ME | 2-methoxyestradiol |
| ACOG | American Congress of Obstetricians and Gynecologists |
| ADMA | asymmetric dimethylarginine |
| AICAR | 5-aminoimidazole-4-carboxamide-3-ribonucleoside |
| AMPK | AMP-activated protein kinase |
| APOL1 | Apolipoprotein L1 |
| CBS | Cystathionine-β-synthase |
| COMT | Catechol-O-Methyltransferase |
| CoQ10 | Coenzyme Q10 |
| CSE | Cystathionine γ-lyase |
| eNOS | endothelial NOS/Type 3 |
| FIGO | Federation of Gynecology and Obstetrics |
| HIF-1 | Hypoxia-inducible factor 1 |
| HIF-2 | Hypoxia-inducible factor 2 |
| HIF-3 | Hypoxia-inducible factor 3 |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| HRE | Hypoxia responsive elements |
| iNOS | inducible NOS/Type 2, |
| IUGR | Intrauterine growth |
| Na2S | Sodium sulfide |
| NaSH | Sodium hydrosulfide |
| NEC | Necrotizing enterocolitis |
| nNOS | neuronal NOS/Type 1, |
| NO | Nitric oxide |
| PIGF | Placental growth factor |
| ROS | Reactive oxygen species |
| sEng | Soluble endoglobin |
| sFlt-1 | Soluble fms-like tyrosine kinase 1 |
| VEGF | Vascular endothelial growth factor |
References
- Dymara-Konopka, W.; Laskowska, M. The Role of Nitric Oxide, ADMA, and Homocysteine in The Etiopathogenesis of Preeclampsia-Review. Int. J. Mol. Sci. 2019, 20, 2757. [Google Scholar] [CrossRef]
- Shih, T.; Peneva, D.; Xu, X.; Sutton, A.; Triche, E.; Ehrenkranz, R.A.; Paidas, M.; Stevens, W. The Rising Burden of Preeclampsia in the United States Impacts Both Maternal and Child Health. Am. J. Perinatol. 2016, 33, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Duhig, K.E.; Shennan, A.H. Recent advances in the diagnosis and management of pre-eclampsia. F1000Prime Rep. 2015, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Peracoli, J.C.; Borges, V.T.M.; Ramos, J.G.L.; Cavalli, R.C.; Costa, S.; Oliveira, L.G.; Souza, F.L.P.; Korkes, H.A.; Brum, I.R.; Costa, M.L.; et al. Pre-eclampsia/Eclampsia. Rev. Bras. Ginecol. Obstet. 2019, 41, 318–332. [Google Scholar] [CrossRef]
- Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222. Obstet. Gynecol. 2020, 135, e237–e260. [CrossRef] [PubMed]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef]
- LaMarca, B.; Amaral, L.M.; Harmon, A.C.; Cornelius, D.C.; Faulkner, J.L.; Cunningham, M.W., Jr. Placental Ischemia and Resultant Phenotype in Animal Models of Preeclampsia. Curr. Hypertens. Rep. 2016, 18, 38. [Google Scholar] [CrossRef]
- El-Sayed, A.A.F. Preeclampsia: A review of the pathogenesis and possible management strategies based on its pathophysiological derangements. Taiwan. J. Obstet. Gynecol. 2017, 56, 593–598. [Google Scholar] [CrossRef]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef]
- Covarrubias, A.E.; Lecarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengellér, Z.K. AP39, a Modulator of Mitochondrial Bioenergetics, Reduces Antiangiogenic Response and Oxidative Stress in Hypoxia-Exposed Trophoblasts: Relevance for Preeclampsia Pathogenesis. Am. J. Pathol. 2019, 189, 104–114. [Google Scholar] [CrossRef]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W.; et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine γ-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Alpoim, P.N.; Perucci, L.O.; Godoi, L.C.; Goulart, C.O.L.; Dusse, L.M.S. Oxidative stress markers and thrombomodulin plasma levels in women with early and late severe preeclampsia. Clin. Chim. Acta 2018, 483, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Tjoa, M.L.; Oudejans, C.B.M.; van Vugt, J.M.G.; Blankenstein, M.A.; van Wijk, I.J. Markers for presymptomatic prediction of preeclampsia and intrauterine growth restriction. Hypertens. Pregnancy 2004, 23, 171–189. [Google Scholar] [CrossRef]
- Kanasaki, K.; Kalluri, R. The biology of preeclampsia. Kidney Int. 2009, 76, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Teran, E.; Hernández, I.; Tana, L.; Teran, S.; Galaviz-Hernandez, C.; Sosa-Macías, M.; Molina, G.; Calle, A. Mitochondria and Coenzyme Q10 in the Pathogenesis of Preeclampsia. Front. Physiol. 2018, 9, 1561. [Google Scholar] [CrossRef]
- Poon, L.C.; Shennan, A.; Hyett, J.A.; Kapur, A.; Hadar, E.; Divakar, H.; McAuliffe, F.; da Silva Costa, F.; von Dadelszen, P.; McIntyre, H.D.; et al. The International Federation of Gynecology and Obstetrics (FIGO) initiative on pre-eclampsia: A pragmatic guide for first-trimester screening and prevention. Int. J. Gynaecol. Obstet. 2019, 145 (Suppl. S1), 1–33. [Google Scholar] [CrossRef]
- Tanner, M.S.; Davey, M.A.; Mol, B.W.; Rolnik, D.L. The evolution of the diagnostic criteria of preeclampsia-eclampsia. Am. J. Obstet. Gynecol. 2022, 226, S835–s843. [Google Scholar] [CrossRef]
- Davey, D.A.; MacGillivray, I. The classification and definition of the hypertensive disorders of pregnancy. Am. J. Obstet. Gynecol. 1988, 158, 892–898. [Google Scholar] [CrossRef]
- Brown, M.; Hague, W.; Higgins, J.; Lowe, S.; McCowan, L.; Oats, J.; Peek, M.; Rowan, J.; Walters, B. The detection, investigation and management of hypertension in pregnancy: Full consensus statement. Aust. N. Z. J. Obstet. Gynaecol. 2000, 40, 139–155. [Google Scholar] [CrossRef]
- ACOG Committee on Obstetric Practice. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. American College of Obstetricians and Gynecologists. Int. J. Gynaecol. Obstet. 2002, 77, 67–75. [Google Scholar]
- Roberts, J.M.; August, P.A.; Bakris, G.; Barton, J.R.; Bernstein, I.M.; Druzin, M.; Gaiser, R.R.; Granger, J.R.; Jeyabalan, A.; Johnson, D.D.; et al. Hypertension in Pregnancy: Executive Summary. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Karrar, S.A.; Hong, P.L. Preeclampsia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Wadhwani, P.; Saha, P.K.; Kalra, J.K.; Gainder, S.; Sundaram, V. A study to compare maternal and perinatal outcome in early vs. late onset preeclampsia. Obstet. Gynecol. Sci. 2020, 63, 270–277. [Google Scholar] [CrossRef] [PubMed]
- The Fetal Medicine Foundation. Risk for Preeclampsia. Available online: https://fetalmedicine.org/research/assess/preeclampsia/first-trimester (accessed on 18 December 2024).
- Mendoza, M.; Garcia-Manau, P.; Arevalo, S.; Aviles, M.; Serrano, B.; Sanchez-Duran, M.A.; Garcia-Ruiz, I.; Bonacina, E.; Carreras, E. Diagnostic accuracy of first-trimester combined screening for early-onset and preterm pre-eclampsia at 8–10 compared with 11–13 weeks’ gestation. Ultrasound Obstet. Gynecol. 2021, 57, 84–90. [Google Scholar] [CrossRef]
- Pedroso, M.A.; Palmer, K.R.; Hodges, R.J.; Costa, F.D.S.; Rolnik, D.L. Uterine Artery Doppler in Screening for Preeclampsia and Fetal Growth Restriction. Rev. Bras. Ginecol. Obstet. 2018, 40, 287–293. [Google Scholar] [CrossRef]
- Crovetto, F.; Figueras, F.; Triunfo, S.; Crispi, F.; Rodriguez-Sureda, V.; Dominguez, C.; Llurba, E.; Gratacós, E. First trimester screening for early and late preeclampsia based on maternal characteristics, biophysical parameters, and angiogenic factors. Prenat. Diagn. 2015, 35, 183–191. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Karumanchi, S.A.; Epstein, F.H. Placental ischemia and soluble fms-like tyrosine kinase 1: Cause or consequence of preeclampsia? Kidney Int. 2007, 71, 959–961. [Google Scholar] [CrossRef]
- Lu, F.; Longo, M.; Tamayo, E.; Maner, W.; Al-Hendy, A.; Anderson, G.D.; Hankins, G.D.; Saade, G.R. The effect of over-expression of sFlt-1 on blood pressure and the occurrence of other manifestations of preeclampsia in unrestrained conscious pregnant mice. Am. J. Obstet. Gynecol. 2007, 196, 396.e1–396.e7. [Google Scholar] [CrossRef]
- Cim, N.; Kurdoglu, M.; Ege, S.; Yoruk, I.; Yaman, G.; Yildizhan, R. An analysis on the roles of angiogenesis-related factors including serum vitamin D, soluble endoglin (sEng), soluble fms-like tyrosine kinase 1 (sFlt1), and vascular endothelial growth factor (VEGF) in the diagnosis and severity of late-onset preeclampsia. J. Matern. Fetal Neonatal Med. 2017, 30, 1602–1607. [Google Scholar] [CrossRef]
- Haggerty, C.L.; Seifert, M.E.; Tang, G.; Olsen, J.; Bass, D.C.; Karumanchi, S.A.; Ness, R.B. Second trimester anti-angiogenic proteins and preeclampsia. Pregnancy Hypertens. 2012, 2, 158–163. [Google Scholar] [CrossRef]
- Maynard, S.E.; Karumanchi, S.A. Angiogenic factors and preeclampsia. Semin. Nephrol. 2011, 31, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Stepan, H.; Herraiz, I.; Schlembach, D.; Verlohren, S.; Brennecke, S.; Chantraine, F.; Klein, E.; Lapaire, O.; Llurba, E.; Ramoni, A.; et al. Implementation of the sFlt-1/PlGF ratio for prediction and diagnosis of pre-eclampsia in singleton pregnancy: Implications for clinical practice. Ultrasound Obstet. Gynecol. 2015, 45, 241–246. [Google Scholar] [CrossRef]
- Vatten, L.J.; Eskild, A.; Nilsen, T.I.; Jeansson, S.; Jenum, P.A.; Staff, A.C. Changes in circulating level of angiogenic factors from the first to second trimester as predictors of preeclampsia. Am. J. Obstet. Gynecol. 2007, 196, 239.e1–239.e6. [Google Scholar] [CrossRef] [PubMed]
- Verlohren, S.; Herraiz, I.; Lapaire, O.; Schlembach, D.; Zeisler, H.; Calda, P.; Sabria, J.; Markfeld-Erol, F.; Galindo, A.; Schoofs, K.; et al. New gestational phase-specific cutoff values for the use of the soluble fms-like tyrosine kinase-1/placental growth factor ratio as a diagnostic test for preeclampsia. Hypertension 2014, 63, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.-H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating Angiogenic Factors and the Risk of Preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef]
- Vatish, M.; Strunz-McKendry, T.; Hund, M.; Allegranza, D.; Wolf, C.; Smare, C. sFlt-1/PlGF ratio test for pre-eclampsia: An economic assessment for the UK. Ultrasound Obstet. Gynecol. 2016, 48, 765–771. [Google Scholar] [CrossRef]
- Rana, S.; Burke, S.D.; Karumanchi, S.A. Imbalances in circulating angiogenic factors in the pathophysiology of preeclampsia and related disorders. Am. J. Obstet. Gynecol. 2022, 226, S1019–S1034. [Google Scholar] [CrossRef]
- Han, L.; Holland, O.J.; Da Silva Costa, F.; Perkins, A.V. Potential biomarkers for late-onset and term preeclampsia: A scoping review. Front. Physiol. 2023, 14, 1143543. [Google Scholar] [CrossRef]
- Rana, S.; Powe, C.E.; Salahuddin, S.; Verlohren, S.; Perschel, F.H.; Levine, R.J.; Lim, K.H.; Wenger, J.B.; Thadhani, R.; Karumanchi, S.A. Angiogenic factors and the risk of adverse outcomes in women with suspected preeclampsia. Circulation 2012, 125, 911–919. [Google Scholar] [CrossRef]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-eclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef]
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk Factors, Diagnosis, Management, and the Cardiovascular Impact on the Offspring. J. Clin. Med. 2019, 8, 1625. [Google Scholar] [CrossRef]
- Chappell, L.C.; Brocklehurst, P.; Green, M.E.; Hunter, R.; Hardy, P.; Juszczak, E.; Linsell, L.; Chiocchia, V.; Greenland, M.; Placzek, A.; et al. Planned early delivery or expectant management for late preterm pre-eclampsia (PHOENIX): A randomised controlled trial. Lancet 2019, 394, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Hauspurg, A.; Jeyabalan, A. Postpartum preeclampsia or eclampsia: Defining its place and management among the hypertensive disorders of pregnancy. Am. J. Obstet. Gynecol. 2022, 226, S1211–S1221. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, R.; Mukhtarova, N.; Flyke, I.P.; Lasarev, M.; Kim, K.; Hennekens, C.H.; Hoppe, K.K. Dose of aspirin to prevent preterm preeclampsia in women with moderate or high-risk factors: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0247782. [Google Scholar] [CrossRef] [PubMed]
- ACOG Committee Opinion No. 743: Low-Dose Aspirin Use During Pregnancy. Obstet. Gynecol. 2018, 132, e44–e52. [CrossRef]
- Euser, A.G.; Cipolla, M.J. Magnesium Sulfate for the Treatment of Eclampsia. Stroke 2009, 40, 1169–1175. [Google Scholar] [CrossRef]
- Hybiak, J.; Broniarek, I.; Kiryczyński, G.; Los, L.D.; Rosik, J.; Machaj, F.; Sławiński, H.; Jankowska, K.; Urasińska, E. Aspirin and its pleiotropic application. Eur. J. Pharmacol. 2020, 866, 172762. [Google Scholar] [CrossRef]
- Richards, E.M.F.; Giorgione, V.; Stevens, O.; Thilaganathan, B. Low-dose aspirin for the prevention of superimposed preeclampsia in women with chronic hypertension: A systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2023, 228, 395–408. [Google Scholar] [CrossRef]
- Smith, J.M.; Lowe, R.F.; Fullerton, J.; Currie, S.M.; Harris, L.; Felker-Kantor, E. An integrative review of the side effects related to the use of magnesium sulfate for pre-eclampsia and eclampsia management. BMC Pregnancy Childbirth 2013, 13, 34. [Google Scholar] [CrossRef]
- Doyle, L.W.; Crowther, C.A.; Middleton, P.; Marret, S.; Rouse, D. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Costantine, M.M.; Weiner, S.J. Effects of antenatal exposure to magnesium sulfate on neuroprotection and mortality in preterm infants: A meta-analysis. Obstet. Gynecol. 2009, 114, 354–364. [Google Scholar] [CrossRef]
- Conde-Agudelo, A.; Romero, R. Antenatal magnesium sulfate for the prevention of cerebral palsy in preterm infants less than 34 weeks’ gestation: A systematic review and metaanalysis. Am. J. Obstet. Gynecol. 2009, 200, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Padda, J.; Khalid, K.; Colaco, L.B.; Padda, S.; Boddeti, N.L.; Khan, A.S.; Cooper, A.C.; Jean-Charles, G. Efficacy of Magnesium Sulfate on Maternal Mortality in Eclampsia. Cureus 2021, 13, e17322. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, W.; Haymond, M.W.; McKay, S.; Karaviti, L.P. Osteopenic effects of MgSO4 in multiple pregnancies. J. Pediatr. Endocrinol. Metab. 2006, 19, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef]
- Kietzmann, T. Liver Zonation in Health and Disease: Hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters. Int. J. Mol. Sci. 2019, 20, 2347. [Google Scholar] [CrossRef]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef]
- Rajakumar, A.; Brandon, H.M.; Daftary, A.; Ness, R.; Conrad, K.P. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 2004, 25, 763–769. [Google Scholar] [CrossRef]
- Akhilesh, M.; Mahalingam, V.; Nalliah, S.; Ali, R.M.; Ganesalingam, M.; Haleagrahara, N. Hypoxia-inducible factor-1α as a predictive marker in pre-eclampsia. Biomed. Rep. 2013, 1, 257–258. [Google Scholar] [CrossRef]
- Kanasaki, K.; Palmsten, K.; Sugimoto, H.; Ahmad, S.; Hamano, Y.; Xie, L.; Parry, S.; Augustin, H.G.; Gattone, V.H.; Folkman, J.; et al. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 2008, 453, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Parchem, J.G.; Kanasaki, K.; Kanasaki, M.; Sugimoto, H.; Xie, L.; Hamano, Y.; Lee, S.B.; Gattone, V.H.; Parry, S.; Strauss, J.F.; et al. Loss of placental growth factor ameliorates maternal hypertension and preeclampsia in mice. J. Clin. Investig. 2018, 128, 5008–5017. [Google Scholar] [CrossRef] [PubMed]
- Ellershaw, D.C.; Gurney, A.M. Mechanisms of hydralazine induced vasodilation in rabbit aorta and pulmonary artery. Br. J. Pharmacol. 2001, 134, 621–631. [Google Scholar] [CrossRef]
- Magee, L.A.; Cham, C.; Waterman, E.J.; Ohlsson, A.; von Dadelszen, P. Hydralazine for treatment of severe hypertension in pregnancy: Meta-analysis. BMJ 2003, 327, 955–960. [Google Scholar] [CrossRef]
- Symoens, J. Ketanserin: A novel cardiovascular drug. Blood Coagul. Fibrinolysis 1990, 1, 219–224. [Google Scholar] [CrossRef]
- Teran, E.; Hernandez, I.; Nieto, B.; Tavara, R.; Ocampo, J.E.; Calle, A. Coenzyme Q10 supplementation during pregnancy reduces the risk of pre-eclampsia. Int. J. Gynaecol. Obstet. 2009, 105, 43–45. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Llanos-Gonzalez, E.; Alcain, F.J. The Use of Coenzyme Q10 in Cardiovascular Diseases. Antioxidants 2021, 10, 755. [Google Scholar] [CrossRef]
- Raizner, A.E.; Quinones, M.A. Coenzyme Q(10) for Patients With Cardiovascular Disease: JACC Focus Seminar. J. Am. Coll. Cardiol. 2021, 77, 609–619. [Google Scholar] [CrossRef]
- Zhao, D.; Liang, Y.; Dai, S.; Hou, S.; Liu, Z.; Liu, M.; Dong, X.; Zhan, Y.; Tian, Z.; Yang, Y. Dose-Response Effect of Coenzyme Q10 Supplementation on Blood Pressure among Patients with Cardiometabolic Disorders: A Grading of Recommendations Assessment, Development, and Evaluation (GRADE)-Assessed Systematic Review and Meta-Analysis of Randomized Controlled Trials. Adv. Nutr. 2022, 13, 2180–2194. [Google Scholar] [CrossRef]
- Yonekura Collier, A.R.; Zsengeller, Z.; Pernicone, E.; Salahuddin, S.; Khankin, E.V.; Karumanchi, S.A. Placental sFLT1 is associated with complement activation and syncytiotrophoblast damage in preeclampsia. Hypertens. Pregnancy 2019, 38, 193–199. [Google Scholar] [CrossRef]
- Cindrova-Davies, T. The therapeutic potential of antioxidants, ER chaperones, NO and H2S donors, and statins for treatment of preeclampsia. Front. Pharmacol. 2014, 5, 119. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L.; Eis, A.L.; Brockman, D.E.; Greer, I.A.; Lyall, F. Endothelial nitric oxide synthase in placental villous tissue from normal, pre-eclamptic and intrauterine growth restricted pregnancies. Hum. Reprod. 1997, 12, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Dillon, K.M.; Carrazzone, R.J.; Matson, J.B.; Kashfi, K. The evolving landscape for cellular nitric oxide and hydrogen sulfide delivery systems: A new era of customized medications. Biochem. Pharmacol. 2020, 176, 113931. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.F.; Gemmel, M.; Powers, R.W. Nitric oxide signaling in pregnancy and preeclampsia. Nitric Oxide 2020, 95, 55–62. [Google Scholar] [CrossRef]
- Li, F.; Hagaman, J.R.; Kim, H.S.; Maeda, N.; Jennette, J.C.; Faber, J.E.; Karumanchi, S.A.; Smithies, O.; Takahashi, N. eNOS deficiency acts through endothelin to aggravate sFlt-1-induced pre-eclampsia-like phenotype. J. Am. Soc. Nephrol. 2012, 23, 652–660. [Google Scholar] [CrossRef]
- Du, L.; He, F.; Kuang, L.; Tang, W.; Li, Y.; Chen, D. eNOS/iNOS and endoplasmic reticulum stress-induced apoptosis in the placentas of patients with preeclampsia. J. Human. Hypertens. 2017, 31, 49–55. [Google Scholar] [CrossRef]
- Amaral, L.M.; Pinheiro, L.C.; Guimaraes, D.A.; Palei, A.C.; Sertório, J.T.; Portella, R.L.; Tanus-Santos, J.E. Antihypertensive effects of inducible nitric oxide synthase inhibition in experimental pre-eclampsia. J. Cell Mol. Med. 2013, 17, 1300–1307. [Google Scholar] [CrossRef]
- Johal, T.; Lees, C.C.; Everett, T.R.; Wilkinson, I.B. The nitric oxide pathway and possible therapeutic options in pre-eclampsia. Br. J. Clin. Pharmacol. 2014, 78, 244–257. [Google Scholar] [CrossRef]
- Kulandavelu, S.; Dulce, R.A.; Murray, C.I.; Bellio, M.A.; Fritsch, J.; Kanashiro-Takeuchi, R.; Arora, H.; Paulino, E.; Soetkamp, D.; Balkan, W.; et al. S-Nitrosoglutathione Reductase Deficiency Causes Aberrant Placental S-Nitrosylation and Preeclampsia. J. Am. Heart Assoc. 2022, 11, e024008. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Maas, R.; Freese, R.; Jung, D.; Lukacs, Z.; Jambrecina, A.; Spickler, W.; Schulze, F.; Böger, R.H. Pharmacokinetic and pharmacodynamic properties of oral L-citrulline and L-arginine: Impact on nitric oxide metabolism. Br. J. Clin. Pharmacol. 2008, 65, 51–59. [Google Scholar] [CrossRef]
- Weckman, A.M.; McDonald, C.R.; Baxter, J.-A.B.; Fawzi, W.W.; Conroy, A.L.; Kain, K.C. Perspective: L-arginine and L-citrulline Supplementation in Pregnancy: A Potential Strategy to Improve Birth Outcomes in Low-Resource Settings. Adv. Nutr. 2019, 10, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality—An update on patient populations with a wide range of cardiovascular risk. Pharmacol. Res. 2009, 60, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Banek, C.T.; Bauer, A.J.; Needham, K.M.; Dreyer, H.C.; Gilbert, J.S. AICAR administration ameliorates hypertension and angiogenic imbalance in a model of preeclampsia in the rat. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1159–H1165. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.L.; Hsu, C.N.; Tain, Y.L. Whether AICAR in Pregnancy or Lactation Prevents Hypertension Programmed by High Saturated Fat Diet: A Pilot Study. Nutrients 2020, 12, 448. [Google Scholar] [CrossRef]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [CrossRef]
- Motterlini, R.; Mann, B.E.; Foresti, R. Therapeutic applications of carbon monoxide-releasing molecules. Expert. Opin. Investig. Drugs 2005, 14, 1305–1318. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Choi, A.M. Heme oxygenase: Colors of defense against cellular stress. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1029–L1037. [Google Scholar] [CrossRef]
- Applegate, L.A.; Luscher, P.; Tyrrell, R.M. Induction of heme oxygenase: A general response to oxidant stress in cultured mammalian cells. Cancer Res. 1991, 51, 974–978. [Google Scholar]
- Foresti, R.; Motterlini, R. The heme oxygenase pathway and its interaction with nitric oxide in the control of cellular homeostasis. Free Radic. Res. 1999, 31, 459–475. [Google Scholar] [CrossRef]
- Maines, M.D. The Heme Oxygenase System: A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Mann, B.E.; Motterlini, R. CO and NO in medicine. Chem. Commun. 2007, 4197–4208. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, R. Carbon monoxide: Endogenous production, physiological functions, and pharmacological applications. Pharmacol. Rev. 2005, 57, 585–630. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2016, 15, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Peterson, N.; McRae, K.E.; Pudwell, J.; Tayade, C.; Smith, G.N. Carbon monoxide increases utero-placental angiogenesis without impacting pregnancy specific adaptations in mice. Reprod. Biol. Endocrinol. 2020, 18, 49. [Google Scholar] [CrossRef]
- Vukomanovic, D.; Jia, Z.; Nakatsu, K.; Smith, G.N.; Ozolinš, T.R.S. Riboflavin and pyrroloquinoline quinone generate carbon monoxide in the presence of tissue microsomes or recombinant human cytochrome P-450 oxidoreductase: Implications for possible roles in gasotransmission. Can. J. Physiol. Pharmacol. 2020, 98, 336–342. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Wikstrom, A.K.; Stephansson, O.; Cnattingius, S. Tobacco use during pregnancy and preeclampsia risk: Effects of cigarette smoking and snuff. Hypertension 2010, 55, 1254–1259. [Google Scholar] [CrossRef]
- Venditti, C.C.; Smith, G.N. Involvement of the heme oxygenase system in the development of preeclampsia and as a possible therapeutic target. Womens Health 2014, 10, 623–643. [Google Scholar] [CrossRef]
- Ahmed, A.; Rezai, H.; Broadway-Stringer, S. Evidence-Based Revised View of the Pathophysiology of Preeclampsia. Adv. Exp. Med. Biol. 2017, 956, 355–374. [Google Scholar] [CrossRef]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, S.A.; Sidle, E.H.; Smith, G.N. Direct placental effects of cigarette smoke protect women from pre-eclampsia: The specific roles of carbon monoxide and antioxidant systems in the placenta. Med. Hypotheses 2005, 64, 17–27. [Google Scholar] [CrossRef]
- Ahmed, A.; Rahman, M.; Zhang, X.; Acevedo, C.H.; Nijjar, S.; Rushton, I.; Bussolati, B.; St John, J. Induction of placental heme oxygenase-1 is protective against TNFalpha-induced cytotoxicity and promotes vessel relaxation. Mol. Med. 2000, 6, 391–409. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.; Robson, S.C.; Myatt, L.; Bulmer, J.N.; Lyall, F. Heme oxygenase expression in human placenta and placental bed: Reduced expression of placenta endothelial HO-2 in preeclampsia and fetal growth restriction. FASEB J. 2001, 15, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Zenclussen, A.C.; Lim, E.; Knoeller, S.; Knackstedt, M.; Hertwig, K.; Hagen, E.; Klapp, B.F.; Arck, P.C. Heme oxygenases in pregnancy II: HO-2 is downregulated in human pathologic pregnancies. Am. J. Reprod. Immunol. 2003, 50, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Kaitu’u-Lino, T.J.; Onda, K.; Beard, S.; Hastie, R.; Binder, N.K.; Cluver, C.; Tuohey, L.; Whitehead, C.; Brownfoot, F.; et al. Heme Oxygenase-1 Is Not Decreased in Preeclamptic Placenta and Does Not Negatively Regulate Placental Soluble fms-Like Tyrosine Kinase-1 or Soluble Endoglin Secretion. Hypertension 2015, 66, 1073–1081. [Google Scholar] [CrossRef]
- George, E.M.; Cockrell, K.; Arany, M.; Stec, D.E.; Rimoldi, J.M.; Gadepalli, R.S.V.; Granger, J.P. Carbon Monoxide Releasing Molecules Blunt Placental Ischemia-Induced Hypertension. Am. J. Hypertens. 2017, 30, 931–937. [Google Scholar] [CrossRef]
- Bainbridge, S.A.; Farley, A.E.; McLaughlin, B.E.; Graham, C.H.; Marks, G.S.; Nakatsu, K.; Brien, J.F.; Smith, G.N. Carbon monoxide decreases perfusion pressure in isolated human placenta. Placenta 2002, 23, 563–569. [Google Scholar] [CrossRef]
- Venditti, C.C.; Casselman, R.; Murphy, M.S.; Adamson, S.L.; Sled, J.G.; Smith, G.N. Chronic carbon monoxide inhalation during pregnancy augments uterine artery blood flow and uteroplacental vascular growth in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R939–R948. [Google Scholar] [CrossRef]
- Venditti, C.C.; Casselman, R.; Young, I.; Karumanchi, S.A.; Smith, G.N. Carbon monoxide prevents hypertension and proteinuria in an adenovirus sFlt-1 preeclampsia-like mouse model. PLoS ONE 2014, 9, e106502. [Google Scholar] [CrossRef]
- Vukomanovic, D.; Rahman, M.N.; Bilokin, Y.; Golub, A.G.; Brien, J.F.; Szarek, W.A.; Jia, Z.; Nakatsu, K. In vitroActivation of heme oxygenase-2 by menadione and its analogs. Med. Gas Res. 2014, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Vukomanovic, D.; Rahman, M.N.; Jia, Z.; Nakatsu, K. Drug-enhanced carbon monoxide production from heme by cytochrome P450 reductase. Med. Gas Res. 2017, 7, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Odozor, C.U.; Peterson, N.; Pudwell, J.; Smith, G.N. Endogenous carbon monoxide production by menadione. Placenta 2018, 71, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.B.; Chowanadisai, W.; Mishchuk, D.O.; Satre, M.A.; Slupsky, C.M.; Rucker, R.B. Dietary pyrroloquinoline quinone (PQQ) alters indicators of inflammation and mitochondrial-related metabolism in human subjects. J. Nutr. Biochem. 2013, 24, 2076–2084. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, J. Recent Advances on Carbon Monoxide Releasing Molecules for Antibacterial Applications. ChemMedChem 2021, 16, 3628–3634. [Google Scholar] [CrossRef]
- Sela, S.; Natanson-Yaron, S.; Zcharia, E.; Vlodavsky, I.; Yagel, S.; Keshet, E. Local retention versus systemic release of soluble VEGF receptor-1 are mediated by heparin-binding and regulated by heparanase. Circ. Res. 2011, 108, 1063–1070. [Google Scholar] [CrossRef]
- McRae, K.E.; Peterson, N.; Dickson, M.A.; Smith, G.N. CORM-A1 treatment leads to increased carbon monoxide in pregnant mice. Pregnancy Hypertens. 2018, 14, 97–104. [Google Scholar] [CrossRef]
- Kashfi, K. The dichotomous role of H(2)S in cancer cell biology? Déjà vu all over again. Biochem. Pharmacol. 2018, 149, 205–223. [Google Scholar] [CrossRef]
- Kashfi, K.; Olson, K.R. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem. Pharmacol. 2013, 85, 689–703. [Google Scholar] [CrossRef]
- Patel, P.; Vatish, M.; Heptinstall, J.; Wang, R.; Carson, R.J. The endogenous production of hydrogen sulphide in intrauterine tissues. Reprod. Biol. Endocrinol. 2009, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, K.M.; Bos, E.M.; Rajakumar, A.; Ris-Stalpers, C.; van Pampus, M.G.; Timmer, A.; Erwich, J.J.H.M.; Faas, M.M.; van Goor, H.; Lely, A.T. Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta 2012, 33, 518–521. [Google Scholar] [CrossRef]
- Holwerda, K.M.; Burke, S.D.; Faas, M.M.; Zsengeller, Z.; Stillman, I.E.; Kang, P.M.; van Goor, H.; McCurley, A.; Jaffe, I.Z.; Karumanchi, S.A.; et al. Hydrogen sulfide attenuates sFlt1-induced hypertension and renal damage by upregulating vascular endothelial growth factor. J. Am. Soc. Nephrol. 2014, 25, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H(2)S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Cheung, N.S.; Zhu, Y.Z.; Chu, S.H.; Siau, J.L.; Wong, B.S.; Armstrong, J.S.; Moore, P.K. Hydrogen sulphide: A novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun. 2005, 326, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Calvert, J.W.; Duranski, M.R.; Ramachandran, A.; Lefer, D.J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H801–H806. [Google Scholar] [CrossRef]
- Li, H.; Xu, F.; Gao, G.; Gao, X.; Wu, B.; Zheng, C.; Wang, P.; Li, Z.; Hua, H.; Li, D. Hydrogen sulfide and its donors: Novel antitumor and antimetastatic therapies for triple-negative breast cancer. Redox Biol. 2020, 34, 101564. [Google Scholar] [CrossRef]
- Zheng, Y.; Ji, X.; Ji, K.; Wang, B. Hydrogen sulfide prodrugs—A review. Acta Pharm. Sin. B 2015, 5, 367–377. [Google Scholar] [CrossRef]
- Drucker, N.A.; Jensen, A.R.; Te Winkel, J.P.; Markel, T.A. Hydrogen Sulfide Donor GYY4137 Acts Through Endothelial Nitric Oxide to Protect Intestine in Murine Models of Necrotizing Enterocolitis and Intestinal Ischemia. J. Surg. Res. 2019, 234, 294–302. [Google Scholar] [CrossRef]
- Marín, R.; Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Sobrevia, L. Oxidative stress and mitochondrial dysfunction in early-onset and late-onset preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165961. [Google Scholar] [CrossRef]
- Ahmad, A.; Olah, G.; Szczesny, B.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, A Mitochondrially Targeted Hydrogen Sulfide Donor, Exerts Protective Effects in Renal Epithelial Cells Subjected to Oxidative Stress in Vitro and in Acute Renal Injury in Vivo. Shock 2016, 45, 88–97. [Google Scholar] [CrossRef]
- Szczesny, B.; Módis, K.; Yanagi, K.; Coletta, C.; Le Trionnaire, S.; Perry, A.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 2014, 41, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Kashfi, K. Lipoproteins and cancer: The role of HDL-C, LDL-C, and cholesterol-lowering drugs. Biochem. Pharmacol. 2022, 196, 114654. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Statins: Pros and cons. Med. Clin. 2018, 150, 398–402. [Google Scholar] [CrossRef]
- Kato, S.; Smalley, S.; Sadarangani, A.; Chen-Lin, K.; Oliva, B.; Branes, J.; Carvajal, J.; Gejman, R.; Owen, G.I.; Cuello, M. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J. Cell Mol. Med. 2010, 14, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, T.W.; Scalia, R.; Murohara, T.; Guo, J.P.; Lefer, A.M. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1652–1659. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef]
- Ahmadi, Y.; Ghorbanihaghjo, A.; Argani, H. The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: A review of molecular mechanisms. Chem. Biol. Interact. 2017, 273, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Singh, J.; Khan, Y.; Seshan, S.V.; Girardi, G. A new mouse model to explore therapies for preeclampsia. PLoS ONE 2010, 5, e13663. [Google Scholar] [CrossRef]
- Costantine, M.M.; Tamayo, E.; Lu, F.; Bytautiene, E.; Longo, M.; Hankins, G.D.V.; Saade, G.R. Using pravastatin to improve the vascular reactivity in a mouse model of soluble fms-like tyrosine kinase-1-induced preeclampsia. Obstet. Gynecol. 2010, 116, 114–120. [Google Scholar] [CrossRef]
- Lefkou, E.; Mamopoulos, A.; Dagklis, T.; Vosnakis, C.; Rousso, D.; Girardi, G. Pravastatin improves pregnancy outcomes in obstetric antiphospholipid syndrome refractory to antithrombotic therapy. J. Clin. Investig. 2016, 126, 2933–2940. [Google Scholar] [CrossRef] [PubMed]
- Kumasawa, K.; Iriyama, T.; Nagamatsu, T.; Osuga, Y.; Fujii, T. Pravastatin for preeclampsia: From animal to human. J. Obstet. Gynaecol. Res. 2020, 46, 1255–1262. [Google Scholar] [CrossRef]
- Brownfoot, F.C.; Tong, S.; Hannan, N.J.; Hastie, R.; Cannon, P.; Kaitu’u-Lino, T.J. Effects of simvastatin, rosuvastatin and pravastatin on soluble fms-like tyrosine kinase 1 (sFlt-1) and soluble endoglin (sENG) secretion from human umbilical vein endothelial cells, primary trophoblast cells and placenta. BMC Pregnancy Childbirth 2016, 16, 117. [Google Scholar] [CrossRef]
- Henry, C. HUSL LIbrary: Social Justice: Racial Disparity. Available online: https://library.law.howard.edu/socialjustice/disparity (accessed on 18 December 2024).
- Liese, K.L.; Mogos, M.; Abboud, S.; Decocker, K.; Koch, A.R.; Geller, S.E. Racial and Ethnic Disparities in Severe Maternal Morbidity in the United States. J. Racial Ethn. Health Disparities 2019, 6, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Grewal, J.; Männistö, T.; Mendola, P.; Chen, Z.; Xie, Y.; Laughon, S.K. Racial/ethnic differences in pregnancy-related hypertensive disease in nulliparous women. Ethn. Dis. 2014, 24, 283–289. [Google Scholar]
- Shahul, S.; Tung, A.; Minhaj, M.; Nizamuddin, J.; Wenger, J.; Mahmood, E.; Mueller, A.; Shaefi, S.; Scavone, B.; Kociol, R.D.; et al. Racial Disparities in Comorbidities, Complications, and Maternal and Fetal Outcomes in Women with Preeclampsia/eclampsia. Hypertens. Pregnancy 2015, 34, 506–515. [Google Scholar] [CrossRef]
- Tanaka, M.; Jaamaa, G.; Kaiser, M.; Hills, E.; Soim, A.; Zhu, M.; Shcherbatykh, I.Y.; Samelson, R.; Bell, E.; Zdeb, M.; et al. Racial disparity in hypertensive disorders of pregnancy in New York State: A 10-year longitudinal population-based study. Am. J. Public. Health 2007, 97, 163–170. [Google Scholar] [CrossRef]
- Gyamfi-Bannerman, C.; Pandita, A.; Wright, J.D.; Siddiq, Z.; D’Alton, M.E.; Friedman, A.M. 434: Racial disparities in preeclampsia outcomes at delivery. Am. J. Obstet. Gynecol. 2019, 220, S294. [Google Scholar] [CrossRef]
- Lisonkova, S.; Joseph, K.S. Incidence of preeclampsia: Risk factors and outcomes associated with early- versus late-onset disease. Am. J. Obstet. Gynecol. 2013, 209, 544.e1–544.e12. [Google Scholar] [CrossRef]
- Mogos, M.F.; Liese, K.L.; Thornton, P.D.; Manuck, T.A.; O’Brien, W.D., Jr.; McFarlin, B.L. Inpatient Maternal Mortality in the United States, 2002-2014. Nurs Res 2020, 69, 42–50. [Google Scholar] [CrossRef]
- Johnson, J.D.; Louis, J.M. Does race or ethnicity play a role in the origin, pathophysiology, and outcomes of preeclampsia? An expert review of the literature. Am. J. Obstet. Gynecol. 2020, 226, S876–S885. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi-Bannerman, C.; Pandita, A.; Miller, E.C.; Boehme, A.K.; Wright, J.D.; Siddiq, Z.; D’Alton, M.E.; Friedman, A.M. Preeclampsia outcomes at delivery and race. J. Matern. Fetal Neonatal Med. 2020, 33, 3619–3626. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Shah, A.; Jimenez-Kimble, R.; Sauk, J.; Ecker, J.L.; Thadhani, R. Differential Risk of Hypertensive Disorders of Pregnancy among Hispanic Women. J. Am. Soc. Nephrol. 2004, 15, 1330–1338. [Google Scholar] [CrossRef]
- Zamora-Kapoor, A.; Nelson, L.A.; Buchwald, D.S.; Walker, L.R.; Mueller, B.A. Pre-eclampsia in American Indians/Alaska Natives and Whites: The Significance of Body Mass Index. Matern. Child. Health J. 2016, 20, 2233–2238. [Google Scholar] [CrossRef]
- Miller, A.K.; Azhibekov, T.; O’Toole, J.F.; Sedor, J.R.; Williams, S.M.; Redline, R.W.; Bruggeman, L.A. Association of preeclampsia with infant APOL1 genotype in African Americans. BMC Med. Genet. 2020, 21, 110. [Google Scholar] [CrossRef]
- Reidy, K.J.; Hjorten, R.C.; Simpson, C.L.; Rosenberg, A.Z.; Rosenblum, S.D.; Kovesdy, C.P.; Tylavsky, F.A.; Myrie, J.; Ruiz, B.L.; Haque, S.; et al. Fetal-Not Maternal-APOL1 Genotype Associated with Risk for Preeclampsia in Those with African Ancestry. Am. J. Hum. Genet. 2018, 103, 367–376. [Google Scholar] [CrossRef]
- Osungbade, K.O.; Ige, O.K. Public health perspectives of preeclampsia in developing countries: Implication for health system strengthening. J. Pregnancy 2011, 2011, 481095. [Google Scholar] [CrossRef]
- Malik, A.; Jee, B.; Gupta, S.K. Preeclampsia: Disease biology and burden, its management strategies with reference to India. Pregnancy Hypertens. 2019, 15, 23–31. [Google Scholar] [CrossRef]
- Ayala-Ramírez, P.; Serrano, N.; Barrera, V.; Bejarano, J.P.; Silva, J.L.; Martínez, R.; Gil, F.; Olaya, C.M.; García-Robles, R. Risk factors and fetal outcomes for preeclampsia in a Colombian cohort. Heliyon 2020, 6, e05079. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, E.C. Impact of socioeconomic status and subjective social class on overall and health-related quality of life. BMC Public. Health 2015, 15, 783. [Google Scholar] [CrossRef] [PubMed]
- Sones, J.L.; Davisson, R.L. Preeclampsia, of mice and women. Physiol. Genomics 2016, 48, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Ghulmiyyah, L.; Sibai, B. Maternal mortality from preeclampsia/eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef]
- Majumder, S.; Moriarty, K.L.; Lee, Y.; Crombleholme, T.M. Placental Gene Therapy for Fetal Growth Restriction and Preeclampsia: Preclinical Studies and Prospects for Clinical Application. J. Clin. Med. 2024, 13, 5647. [Google Scholar] [CrossRef]
- Holdt Somer, S.J.; Sinkey, R.G.; Bryant, A.S. Epidemiology of racial/ethnic disparities in severe maternal morbidity and mortality. Semin. Perinatol. 2017, 41, 258–265. [Google Scholar] [CrossRef]
- Hardeman, R.R.; Medina, E.M.; Kozhimannil, K.B. Structural Racism and Supporting Black Lives—The Role of Health Professionals. N. Engl. J. Med. 2016, 375, 2113–2115. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dickerson, A.G.; Joseph, C.A.; Kashfi, K. Current Approaches and Innovations in Managing Preeclampsia: Highlighting Maternal Health Disparities. J. Clin. Med. 2025, 14, 1190. https://doi.org/10.3390/jcm14041190
Dickerson AG, Joseph CA, Kashfi K. Current Approaches and Innovations in Managing Preeclampsia: Highlighting Maternal Health Disparities. Journal of Clinical Medicine. 2025; 14(4):1190. https://doi.org/10.3390/jcm14041190
Chicago/Turabian StyleDickerson, Alexis G., Christiana A. Joseph, and Khosrow Kashfi. 2025. "Current Approaches and Innovations in Managing Preeclampsia: Highlighting Maternal Health Disparities" Journal of Clinical Medicine 14, no. 4: 1190. https://doi.org/10.3390/jcm14041190
APA StyleDickerson, A. G., Joseph, C. A., & Kashfi, K. (2025). Current Approaches and Innovations in Managing Preeclampsia: Highlighting Maternal Health Disparities. Journal of Clinical Medicine, 14(4), 1190. https://doi.org/10.3390/jcm14041190