
Clinical, Genetic, EEG, Neuroimaging Insights and Conservative Treatment in Pediatric Focal Epilepsy: A Retrospective Observational Study

Abstract
1. Introduction
2. Materials and Methods
2.1. Inclusion and Exclusion Criteria
2.2. Follow-Up and Diagnostic Approach
2.3. Seizure Etiology, Classification, and Characteristics
2.4. Clinical and Developmental Data Collection
2.5. Neurophysiology Study
2.6. Neuroimaging
2.7. Genetics
2.8. Statistical Analysis
3. Results
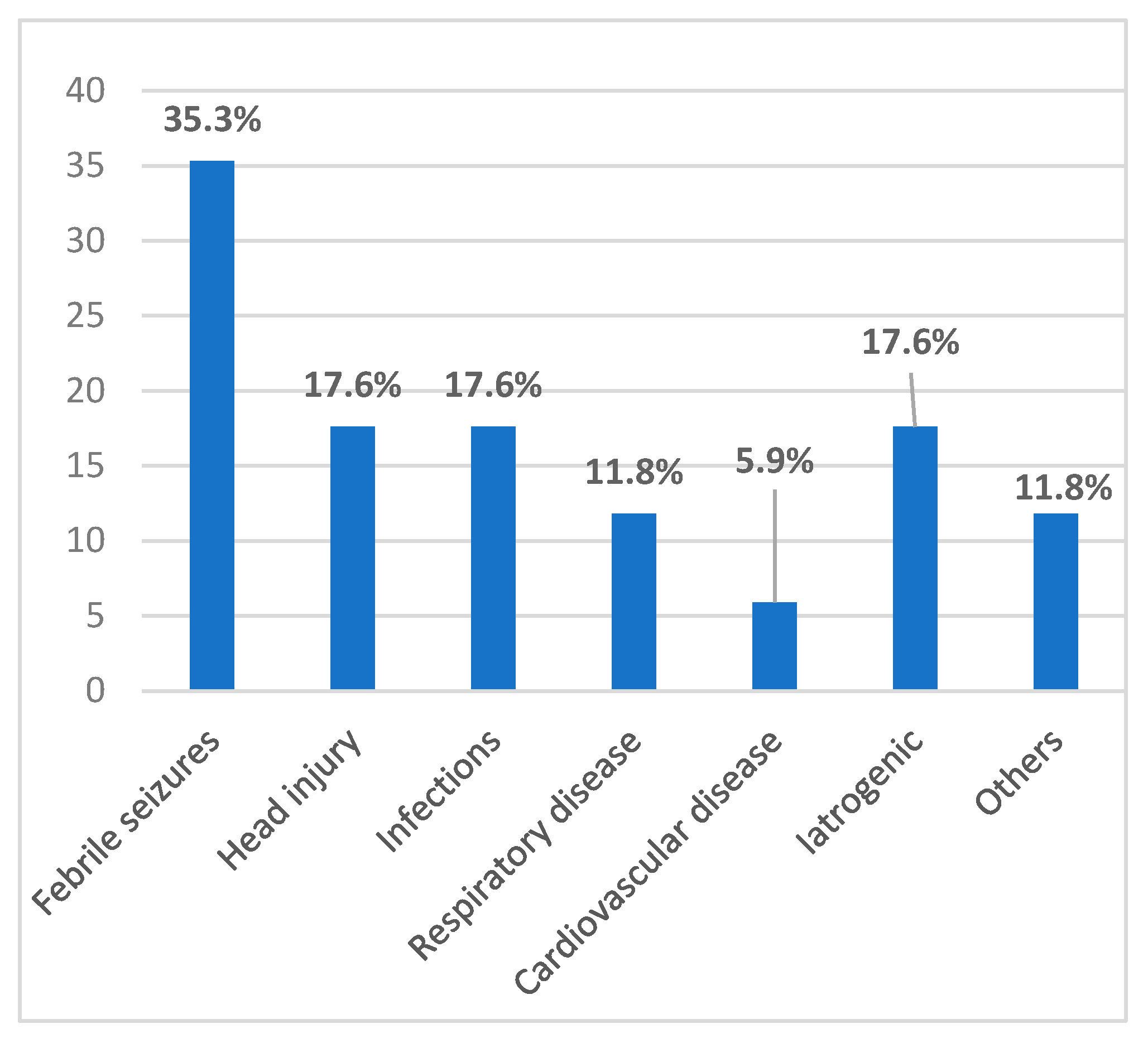
3.1. Prenatal, Perinatal, and Postnatal Risk Factors
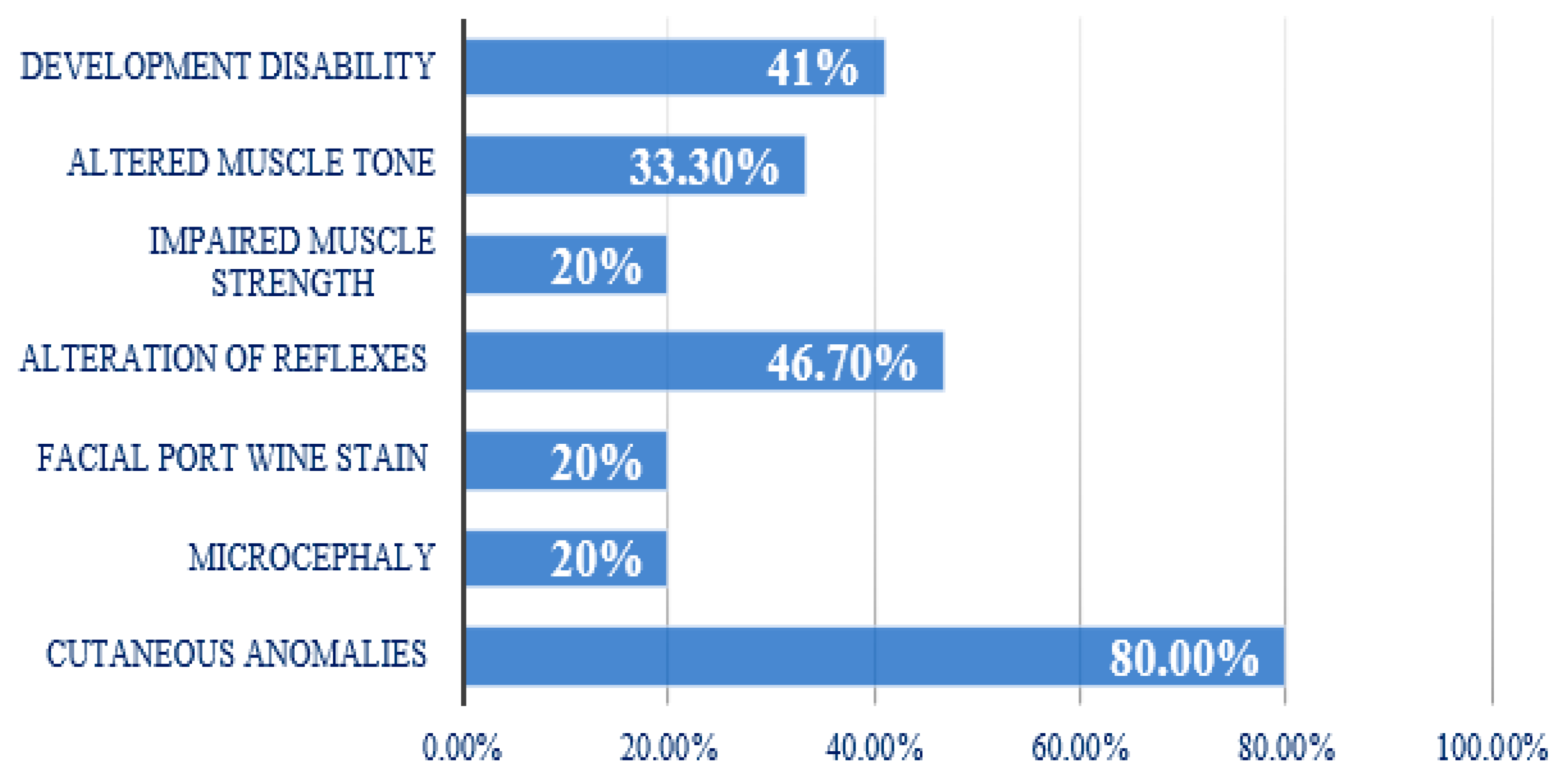
3.2. Clinical Aspects
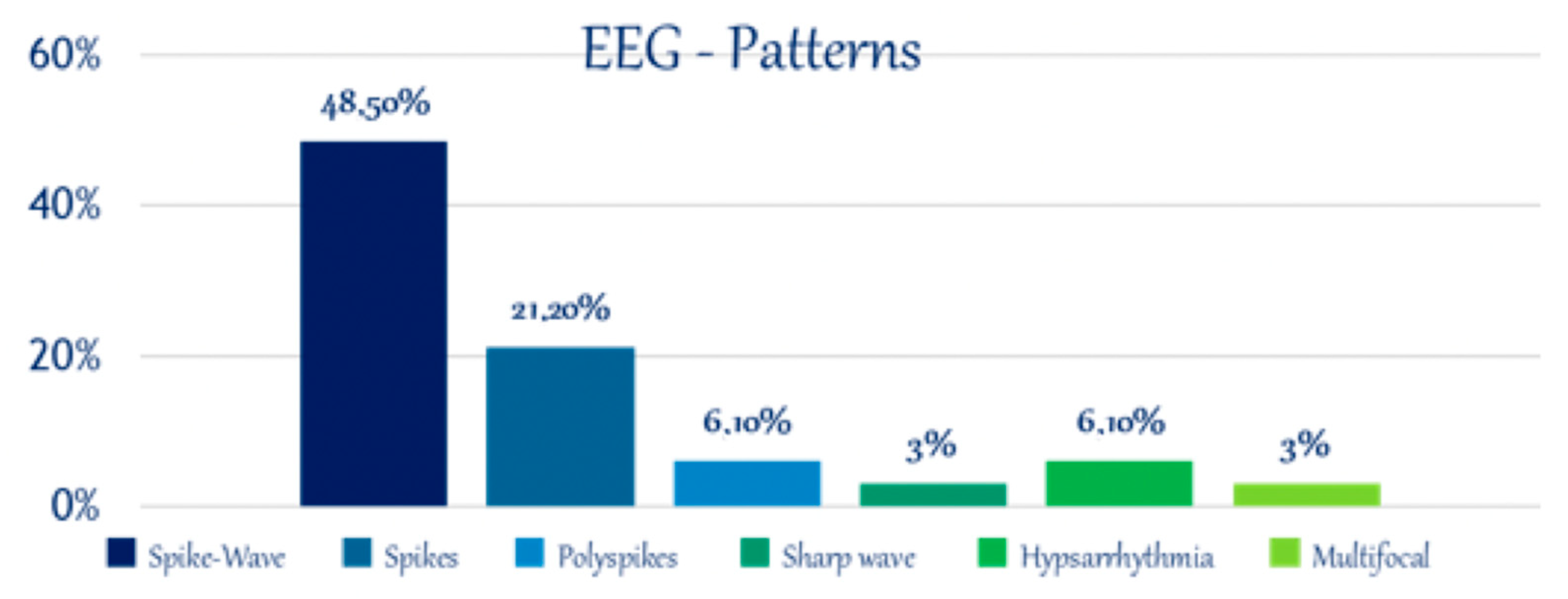
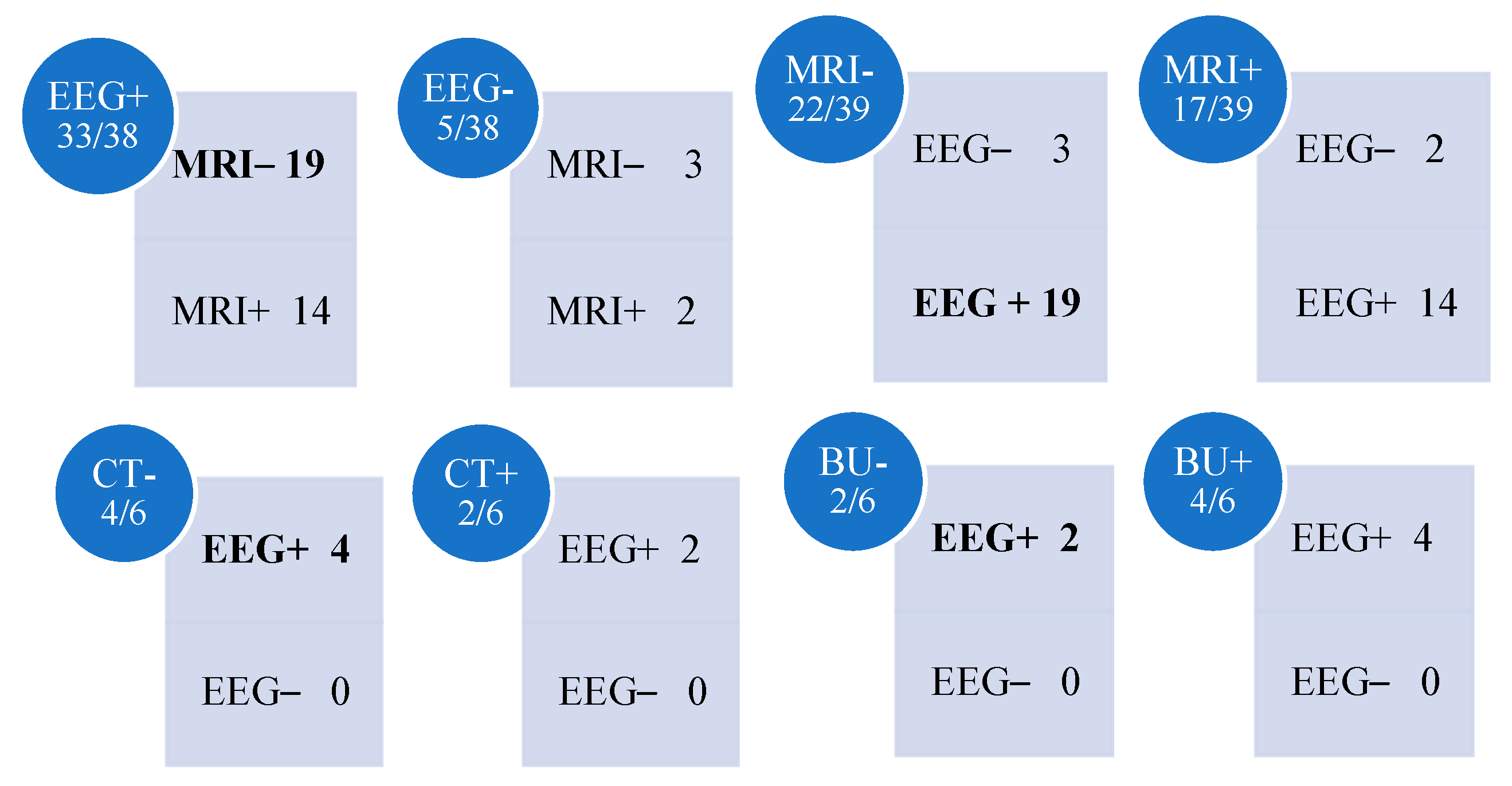
3.3. Electroencephalography

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | EEG Findings | MRI Findings | CT Findings |
|---|---|---|---|
| 1 | Focal spikes (R, central) | Non-significant findings | Not performed |
| 2 | Normal | Normal | Not performed |
| 3 | Focal spike–wave discharges (R, temporal) | Subcortical tubers (R, temporal and frontal) | Not performed |
| 4 | Focal spikes (R, central) | Enlarged subarachnoid spaces | Not performed |
| 5 | Focal spike–wave (R, temporal) | Normal | Not performed |
| 6 | Normal | Normal | Not performed |
| 7 | Multifocal paroxysmal anomalies | Normal | Not performed |
| 8 | Focal spikes with secondary generalization (central and temporal) | Normal | Not performed |
| 9 | Sharp waves (L, central and temporal) | Normal | Not performed |
| 10 | Spike–wave (L, central and temporal) | Normal | Not performed |
| 11 | Generalized spike–wave (L, temporal and occipital; bilateral frontal) | Right mesial temporal sclerosis | Not performed |
| 12 | Generalized spike–wave (R, central and temporal) | Normal | Not performed |
| 13 | Spike–wave and polyspike–wave (R); hypsarrhythmia | Diffuse polymicrogyria, thickened and dysplastic right fronto-parietal cortex; hypoplasia of the corpus callosum (Figure 7) | Not performed |
| 14 | Normal | Enlarged subarachnoid space, cortical atrophy, microcalcifications, reduced vascularization (L) | Not performed |
| 15 | Spike–wave (L, central and temporal) | White matter damage | Not performed |
| 16 | Interhemispheric asymmetry | Right parietal and temporal leptomeningeal capillary malformation | Large cerebrospinal fluid spaces (R frontal) |
| 17 | No available EEG | Left volume loss, asymmetric myelination, parenchymal dystrophic calcifications, pial angioma | Not performed |
| 18 | Slow background activity (L, occipital) | Leptomeningeal capillary malformation (R, occipital and parietal) | Occipital calcifications (R) |
| 19 | Generalized spike–wave (R, occipital and parietal) | Normal | Not performed |
| 20 | Focal spikes (R, occipital) | Cortical–subcortical signal alteration (L) | Not performed |
| 21 | Focal spikes (R, occipital) | Normal | Not performed |
| 22 | Spikes | Multicystic encephalomalacia (L, temporal and frontal); cross atrophy; left Sylvian artery thrombosis | Significant findings |
| 23 | Generalized spike–wave (L–R, frontal and temporal) | Diffuse white matter hyperintensities; small cerebrospinal fluid cyst (L); corpus callosum hypoplasia | Not performed |
| 24 | Hypsarrhythmia and multifocal paroxysmal anomalies | Normal | Intraventricular hemorrhage (Grade II); periventricular hyperechogenicity |
| 25 | Normal | Cerebellar atrophy and hypoplasia | Not performed |
| 26 | Spikes | Normal | Not performed |
| 27 | Generalized spike–wave (L, temporal) | Normal | Not performed |
| 28 | Generalized spike–wave (L, temporal) | Normal | Not performed |
| 29 | Generalized spike–wave (L, occipital) | Normal | Not performed |
| 30 | Focal spikes (central and temporal) | Normal | Not performed |
| 31 | Multifocal paroxysmal anomalies | Dysmorphic ventricles; small corpus callosum; Galen vein malformation | Not performed |
| 32 | Focal spikes (L, central and temporal) | Normal | Not performed |
| 33 | Focal spikes (R, central and temporal) | Asymmetric lateral ventricles and temporal volume (L) | Not performed |
| 34 | Generalized spike–wave (L–R, central and temporal) | Cerebellar atrophy | Not performed |
| 35 | Generalized spike–wave (L, temporal) | Reduced lateral ventricle size | Increased ventricular spaces; left lateral ventricle mass |
| 36 | Generalized spike–wave (occipital) | Normal | Not performed |
| 37 | Generalized spike–wave (L–R, occipital) | Normal | Not performed |
| 38 | Normal | Normal | Not performed |
| 39 | Generalized spike–wave in all leads | Normal | Not performed |
3.4. Neuroimaging
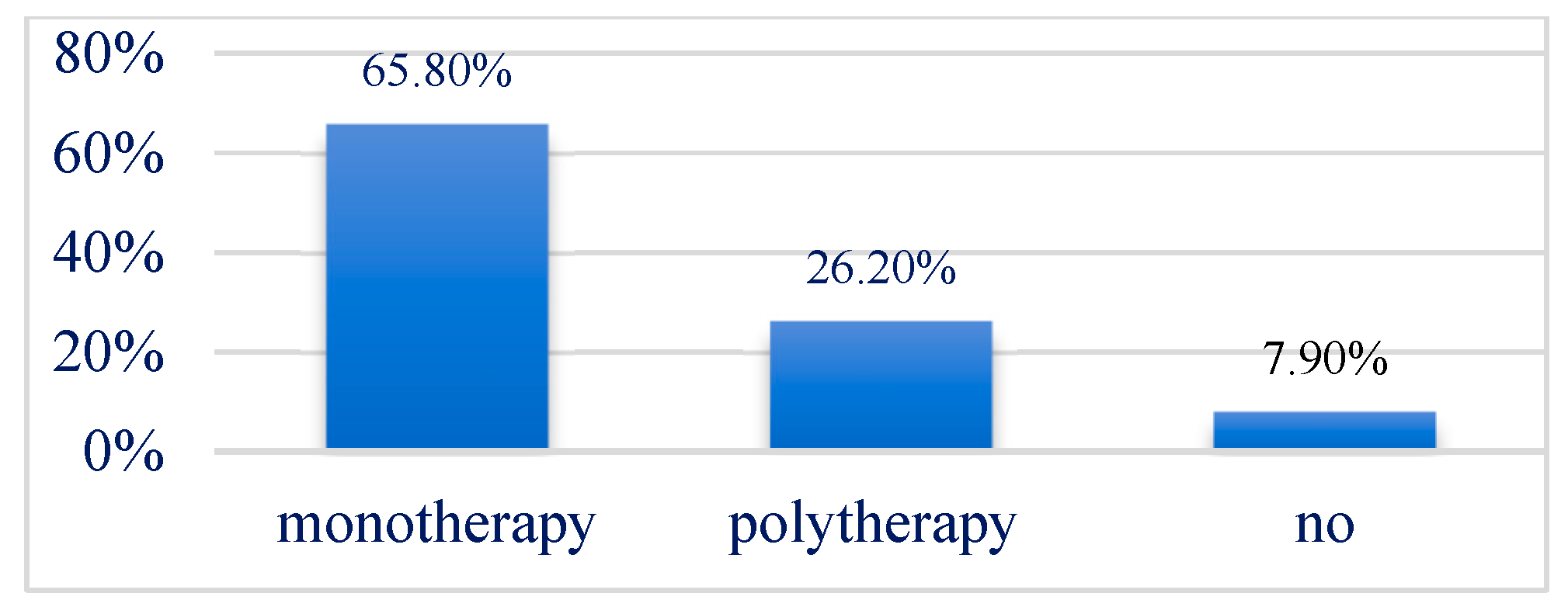
3.5. Treatment
4. Discussion
5. Study Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [PubMed]
- Forsgren, L.; Beghi, E.; Õun, A.; Sillanpää, M. The epidemiology of epilepsy in Europe—A systematic review. Eur. J. Neurol. 2005, 12, 245–253. [Google Scholar] [CrossRef]
- Laoprasert, P. Atlas of Pediatric EEG; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Reddy, D.S. The neuroendocrine basis of sex differences in epilepsy. Pharmacol. Biochem. Behav. 2017, 152, 97–104. [Google Scholar] [PubMed]
- Carlson, C.; Dugan, P.; Kirsch, H.E.; Friedman, D.; EPGP Investigators. Sex differences in seizure types and symptoms. Epilepsy Behav. 2014, 41, 103–108. [Google Scholar] [PubMed]
- Ochoa-Gómez, L.; López-Laso, E.; Luna-Muñoz, E.; Rodrigo, C.F.; Martínez, R.F.; Samper-Villagrasa, P.; Monge-Galindo, L.; Peña-Segura, J.L.; García-Jiménez, M.C. A study of epilepsy according to the age at onset and monitored for 3 years in a regional reference paediatric neurology unit. An. Pediatr. 2017, 86, 11–19. [Google Scholar]
- Boelen, S.; Lodder, S.S.; Berends, A.; Veldwijk, H.; van de Ven-Verest, M.; Tan, I.Y.; Aldenkamp, A.P. Effect of epilepsy on psychomotor function in children with uncomplicated epilepsy. Dev. Med. Child Neurol. 2005, 47, 546–550. [Google Scholar]
- Vlooswijk, M.C.G.; Jansen, J.F.A.; Majoie, H.J.M.; de Krom, M.C.; Majoie, H.J.; Hofman, P.A.; Aldenkamp, A.P.; Backes, W.H. Loss of network efficiency associated with cognitive decline in chronic epilepsy. Neurology 2011, 77, 938–944. [Google Scholar]
- Helmstaedter, C.; Witt, J.A. Epilepsy and cognition—A bidirectional relationship? Seizure 2017, 49, 83–89. [Google Scholar]
- Ottman, R.; Hirose, S.; Jain, S.; Vasoli, V.M.; Hauser, W.A.; Buchhalter, J.R. Accuracy of family history information on epilepsy and other seizure disorders. Neurology 2011, 76, 390–396. [Google Scholar]
- Demissie, K.; Breckenridge, M.B.; Rhoads, G.G.; Balasubramanian, B.A.; Gandhi, K.; Joseph, K.S.; Kramer, M. Operative vaginal delivery and neonatal and infant adverse outcomes: Population-based retrospective analysis. Br. Med. J. 2004, 329, 24–26. [Google Scholar]
- Shakeshaft, A.; Panjwani, N.; McDowall, R.; Crudgington, H.; Peña Ceballos, J.; Andrade, D.M.; Beier, C.P.; Fong, C.Y.; Gesche, J.; Greenberg, D.A.; et al. Trait impulsivity in Juvenile Myoclonic Epilepsy. Ann. Clin. Transl. Neurol. 2021, 8, 138–152. [Google Scholar] [PubMed]
- Laughon, S.K.; Berghella, V.; Reddy, U.M.; Sundaram, R.; Lu, Z.; Hoffman, M.K. Neonatal and maternal outcomes with prolonged second stage of labor. Obstet. Gynecol. 2014, 124, 57–67. [Google Scholar] [PubMed]
- Ruggieri, M.; Polizzi, A.; Catanzaro, S.; Bianco, M.L.; Praticò, A.D.; Di Rocco, C. Neurocutaneous melanocytosis (melanosis). Childs Nerv Syst. 2020, 36, 2571–2596. [Google Scholar] [PubMed]
- Kakkar, S.; Mendiratta, V.; Sharma, N.; Aneja, S.; Harjai, B. Cutaneous manifestations of seizure disorder in children—A study of 100 seizure patients. Pediatr. Dermatol. 2007, 24, 579–581. [Google Scholar]
- Noachtar, S.; Rémi, J. The role of EEG in epilepsy: A critical review. Epilepsy Behav. 2009, 15, 22–33. [Google Scholar]
- Vecchi, M.; Baroni, G.; Brinciotti, M.; Stivala, M.; Guerrini, R.; Albamonte, E.; Ranalli, D.; Battaglia, D.; Lunardi, G.; Boniver, C.; et al. Symptomatic and presumed symptomatic focal epilepsies in childhood: An observational, prospective multicentre study. Epilepsia 2016, 57, 1808–1816. [Google Scholar] [CrossRef]
- Coryell, J.; Gaillard, W.D.; Shellhaas, R.A.; Grinspan, Z.M.; Wirrell, E.C.; Knupp, K.G.; Wusthoff, C.J.; Keator, C.; Sullivan, J.E.; Loddenkemper, T.; et al. Neuroimaging of early life epilepsy. Pediatrics 2018, 142, e20180652. [Google Scholar]
- Berg, A.T.; Vickrey, B.G.; Langfitt, J.T.; Fulbright, R.K.; DiMario, F.; Testa, F.M.; Levy, S.R. Frequency, prognosis, and surgical treatment of structural abnormalities seen with magnetic resonance imaging in childhood epilepsy. Brain 2009, 132, 2785–2797. [Google Scholar]
- Eltze, C.M.; Chong, W.K.; Cox, T.; Whitney, A.; Cortina-Borja, M.; Chin, R.F.; Scott, R.C.; Cross, J.H. A population-based study of newly diagnosed epilepsy in infants. Epilepsia 2013, 54, 437–445. [Google Scholar]
- Wang, P.J. Magnetic resonance imaging in symptomatic/cryptogenic partial epilepsies of infants and children. Acta Paediatr. Sin. 1997, 38, 127–136. [Google Scholar]
- Kim, D.W.; Lee, S.Y.; Chung, S.E.; Cheong, H.K.; Jung, K.Y.; Korean Epilepsy Society. Clinical characteristics of patients with treated epilepsy in Korea: A nationwide epidemiologic study. Epilepsia 2014, 55, 67–75. [Google Scholar] [PubMed]
- Kramer, U. Atypical presentations of benign childhood epilepsy with centrotemporal spikes: A review. J. Child Neurol. 2008, 23, 785–790. [Google Scholar] [PubMed]
- Ben Ameur, S.; Bouguila, H.; Mhirsi, A.; Yaich, S.; Mnif, Z.; Kamoun, T.; Hachicha, M. Cerebral imaging in epileptic children: Study of 140 cases. Tunis. Med. 2014, 92, 24–28. [Google Scholar] [PubMed]
- Bronen, R.A.; Fulbright, R.K.; Spencer, D.D.; Spencer, S.S.; Kim, J.H.; Lange, R.C.; Sutilla, C. Refractory epilepsy: Comparison of MR imaging, CT, and histopathologic findings in 117 patients. Radiology 1996, 201, 97–105. [Google Scholar]
- Bradley, W.G.; Shey, R.B. MR imaging evaluation of seizures. Radiology 2000, 214, 651–656. [Google Scholar]
- Ali, A.; Akram, F.; Khan, G.; Hussain, S. Paediatrics brain imaging in epilepsy: Common presenting symptoms and spectrum of abnormalities detected on MRI. J. Ayub Med. Coll. Abbottabad 2017, 29, 215–218. [Google Scholar]
- Muhlhofer, W.; Tan, Y.L.; Mueller, S.G.; Knowlton, R. MRI-negative temporal lobe epilepsy—What do we know? Epilepsia 2017, 58, 727–742. [Google Scholar]
- Lee, Y.J.; Lee, J.S. Temporal lobe epilepsy surgery in children versus adults: From etiologies to outcomes. Korean J. Pediatr. 2013, 56, 275–281. [Google Scholar]
- Wong-Kisiel, L.C.; Britton, J.W.; Patterson, M.C.; Witte, R.J.; Santana-Almansa, A.; Worrell, G.A.; Britton, J.; Brinkmann, B.H. Morphometric analysis on T1-weighted MRI complements visual MRI review in focal cortical dysplasia. Neurology 2018, 140, 184–191. [Google Scholar]
- Wang, Z.I.; Alexopoulos, A.V.; Jones, S.E.; Jaisani, Z.; Najm, I.M.; Prayson, R.A. The pathology of magnetic-resonance-imaging-negative epilepsy. Mod. Pathol. 2013, 26, 1051–1058. [Google Scholar]
- Boutzoukas, E.M.; Stein, M.A.; Luberto, C.M.; Sepeta, L.N.; You, X.; Gaillard, W.D.; Wallace, G.L.; Berl, M.M. Cortical thickness in childhood left focal epilepsy: Thinning beyond the seizure focus. Epilepsy Behav. 2020, 102, 106825. [Google Scholar] [CrossRef] [PubMed]
- Madan, N.; Grant, P.E. New directions in clinical imaging of cortical dysplasias. Epilepsia 2009, 50, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, L.; Aronica, E.; Baayen, J.C.; Sepeta, L.N.; Oluigbo, C.O.; Havens, K.; Freilich, E.R.; Schreiber, J.M.; Gaillard, W.D. Temporal lobe epilepsy and focal cortical dysplasia in children: A tip to find the abnormality. Epilepsia 2017, 58, 113–122. [Google Scholar] [CrossRef]
- Alarcón, G.; Binnie, C.D.; Elwes, R.D.; Selway, R.P.; Lacruz, M.E.; Elwes, R.D.; Jarosz, J.M.; Honavar, M.; Brunhuber, F.; Mullatti, N.; et al. Is it worth pursuing surgery for epilepsy in patients with normal neuroimaging? J. Neurol. Neurosurg. Psychiatry 2006, 77, 474–480. [Google Scholar] [CrossRef]
- Fong, J.S.; Englot, D.J.; Yang, L.; Prayson, R.A.; Busch, R.; Bingaman, W. Seizure outcome and its predictors after temporal lobe epilepsy surgery in patients with normal MRI. Epilepsia 2011, 52, 1393–1401. [Google Scholar] [CrossRef]
- Cukiert, A.; Burattini, J.A.; Marquez, R.M.; Sousa, A.; Vieira, J.O.; Argentoni, M.; Forster, C.; Baldauf, C. Results of surgery in patients with refractory extratemporal epilepsy with normal or nonlocalizing magnetic resonance findings investigated with subdural grids. Epilepsia 2001, 42, 889–894. [Google Scholar] [CrossRef]
- Bien, C.G.; Schulze-Bonhage, A.; Deckert, J.; Clusmann, H.; Becker, A.J.; Urbach, H. Characteristics and surgical outcomes of patients with refractory magnetic resonance imaging-negative epilepsies. Arch. Neurol. 2009, 66, 1491–1499. [Google Scholar] [CrossRef]
- Widjaja, E.; Raybaud, C. Advances in neuroimaging in patients with epilepsy. Neurosurg. Focus 2008, 25, E3. [Google Scholar] [CrossRef]
- Jay, V.; Becker, L.E.; Blaser, S.; Hwang, P.A.; Hoffman, H.J.; Harwood-Nash, D. Pathology of temporal lobectomy for refractory seizures in children. J. Neurosurg. 1993, 79, 53–61. [Google Scholar] [CrossRef]
- Farrell, M.A.; DeRosa, M.J.; Curran, J.G.; Secor, D.L.; Cornford, M.E.; Comair, Y.G.; Peacock, W.J.; Shields, W.D.; Vinters, H.V. Neuropathologic findings in cortical resections (including hemispherectomies) performed for the treatment of intractable childhood epilepsy. Acta Neuropathol. 1992, 19, 463–466. [Google Scholar] [CrossRef]
- Barisano, G.; Fattori, S.; Castellaro, M.; Jann, K.; Cabeen, R.; Wang, D.J.; Toga, A.W.; Law, M. Clinical 7T MRI: Are we there yet? A review about magnetic resonance imaging at ultra-high field. Magn. Reason. Imaging 2006, 92, 20180492. [Google Scholar]
- Trattnig, S.; Bogner, W.; Chmelik, J.; Hangel, G.; Strasser, B.; Dymerska, B.; Cardoso, P.L.; Robinson, S.D. Key clinical benefits of neuroimaging at 7 T. Neuroimage 2018, 168, 477–489. [Google Scholar] [PubMed]
- Feng, X.; Piper, R.J.; Prentice, F.; Clayden, J.D.; Baldeweg, T. Functional brain connectivity in children with focal epilepsy: A systematic review of functional MRI studies. Seizure 2024, 117, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Nosadini, M.; Eyre, M.; Giacomini, T.; Valeriani, M.; Della Corte, M.; Praticò, A.D.; Annovazzi, P.; Cordani, R.; Cordelli, D.M.; Crichiutti, G.; et al. Early Immunotherapy and Longer Corticosteroid Treatment Are Associated With Lower Risk of Relapsing Disease Course in Pediatric MOGAD. Neurol. Neuroimmunol. Neuroinflammation 2022, 29, e200065. [Google Scholar]
- Nevitt, S.J.; Sudell, M.; Cividini, S.; Marson, A.G.; Smith, C.T. Antiepileptic drug monotherapy for epilepsy: A network meta-analysis of individual participant data. Cochrane Database Syst. Rev. 2017, 12, CD011412. [Google Scholar] [CrossRef]
- Rosati, A.; Ilvento, L.; Lucenteforte, E.; Pugi, A.; Crescioli, G.; McGreevy, K.S.; Virgili, G.; Mugelli, A.; De Masi, S.; Guerrini, R. Comparative efficacy of antiepileptic drugs in children and adolescents: A network meta-analysis. Epilepsia 2018, 59, 297–314. [Google Scholar]
- Nascimento, F.A.; Friedman, D.; Peters, J.M.; Bensalem-Owen, M.K.; Cendes, F.; Rampp, S.; Wirrell, E.; Blümcke, I.; Tatum, W.; Beniczky, S. Focal epilepsies: Update on diagnosis and classification. Epileptic Disord. 2023, 25, 1–17. [Google Scholar]





| Patient | Age at Onset | Physical Features | Seizure Semiology | Psychomotor Development |
|---|---|---|---|---|
| 1 | 4 years | None | Focal impaired awareness seizures with LoC, Si, VF | Normal |
| 2 | 4 months | Single café-au-lait macule (lumbar region) | Focal seizures with head deviation and bilateral eyelid ptosis | Normal |
| 3 | 3 years | Hypopigmented macules (back and limbs) | Focal seizures with eyelid and perioral myoclonus, gelastic seizures, leading to LoC | Delayed, ADHD |
| 4 | 5 months | Single café-au-lait macule, hypotonia of lower limbs, unilateral hypertonia (R) | Clonic seizures affecting the upper limbs | Normal |
| 5 | 3 years | None | Generalized seizures with visual fixation, atonic features, and clonic jaw movements (~30 min) | Normal |
| 6 | 1 year | None | Focal seizures with VF, recurrent sialorrhea, dysarthria, hypoesthesia of limbs, urinary incontinence (~1 min) | Delayed |
| 7 | 3 years | None | Autonomic seizures with pallor, diaphoresis, hypotonia (~1 min), swallowing difficulties, and impaired awareness | Normal |
| 8 | 5 years | Hyperreflexia | Focal seizures with impaired awareness, uncertain gait, VF, focal tonic seizures in the right upper limb, progressing to GTCS | Normal |
| 9 | 9 years | Single café-au-lait macule (left hemithorax) | Focal clonic seizures of the lower limb (R), evolving into GTCS, ocular version, and LoC | Normal |
| 10 | 10 years | None | Focal seizures with impaired awareness, buccal paresthesia, unilateral clonus, ocular version, recurrent vomiting, and postictal headache | Normal |
| 11 | 12 years | Three café-au-lait macules | Generalized seizures with left deviation, sialorrhea, and clonic activity of all four limbs, impaired awareness | Delayed |
| 12 | 9 years | None | Generalized tonic–clonic seizures (GTCS), hypertonia, postictal headache, and vomiting | Normal |
| 13 | 3 months | Mild hypotonia | Head and eye deviation (R), buccal automatisms, and tonic movements of the right upper limb | Delayed |
| 14 | 11 years | None | Focal clonic seizures affecting the upper limb, VF | Normal |
| 15 | 12 years | Two café-au-lait macules | Gelastic seizures, LoC, head deviation, sialorrhea, clonic movements in four limbs, impaired awareness | Normal |
| 16 | 5 months | Angioma in trigeminal branches and right trunk | Head and eye deviation (L), transient ischemic attack (TIA) with hemiparesis, prolonged tonic–clonic seizures | Delayed |
| 17 | 2 years | Eyelid and left-sided facial angioma, iris angioma | No seizures reported | Normal |
| 18 | 3 years | Facial angioma, nasal root (R) | TIA with lameness in lower limb (R), headache in orbital and temporal region (R), vomiting | Delayed |
| 19 | 6 years | None | Déjà-vu aura tachycardia, headache, gastralgia, confusion | Normal |
| 20 | 4 years | Single café-au-lait macule, reduced reflexes bilaterally, dysarthria, lunar facies | Paroxysmal chewing movements, sialorrhea, buccal deviation (L), generalized hypertonia (R), LoC, VF | Delayed |
| 21 | 3 years | Subareolar café-au-lait macule (R) | Hypertonia, sialorrhea, LoC, clonus | Delayed |
| 22 | 4 years | Hemiplegia (R) | Unknown | Delayed |
| 23 | 1 year | Hypotonic muscle mass | Tremors, buccal deviation, LoC, clonic jaw movements, sialorrhea | Delayed |
| 24 | 9 months | Microcephaly, axial hypotonia, arthrogryposis, single palmar sulcus, hyperreflexia | Perioral cyanosis, ocular version (~30 sec) | Delayed |
| 25 | 10 months | Hemimegalencephaly (R), reduced reflexes | Hypotonia, VF | Delayed |
| 26 | 11 months | Two café-au-lait macules, bilateral cavus foot | Unknown | Delayed |
| 27 | 2 years | Hypochromic spots, three café-au-lait macules (R) | Unknown | Delayed |
| 28 | 5 years | None | No seizures reported | Normal |
| 29 | 9 years | None | Headache, autonomic seizures with sudden pallor, dizziness, hypotonia (~10 min), weakness | Normal |
| 30 | 4 months | Microcephaly | Generalized hypertonia, upwards gaze deviation | Normal |
| 31 | 7 months | Microcephaly, bilateral hyperreflexia, hypertonia (four limbs) | Multifocal seizures with impaired awareness | Delayed |
| 32 | 3 years | Hyposthenic right upper limb | Head and eye deviation (R), hypertonicity (R upper limb), LoC, eyelid myoclonus | Normal |
| 33 | 9 years | None | Lip deviation (L), drooling, VF, eyelid myoclonus, LoC, postictal weakness and paraesthesia (R) | Normal |
| 34 | 2 years | Multiple café-au-lait macules | VF, sialorrhea, hypertonia, subsequent tonic–clonic contractions, opisthotonus, ocular version, bruxism | Delayed |
| 35 | 8 years | None | Sialorrhea, chewing movements, generalized tremors, buccal rim and gaze deviation (R), eyelid blinking, impaired awareness | Normal |
| 36 | 4 years | Single café-au-lait macule, hyperexcitable patellar reflex | Cyanosis, loss of balance, GTCS, sialorrhea, LoC, profound drowsiness, fear-induced seizures, mastication, bruxism | Normal |
| 37 | 12 years | None | VF, sialorrhea, hypertonia, subsequent tonic–clonic contractions, opisthotonus, ocular version, bruxism | Normal |
| 38 | 13 years | None | VF | Normal |
| 39 | 11 years | Hypochromic macule | Right focal seizures with impaired awareness | Normal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauci, M.C.; Gauci, R.; Ruggieri, M.; Polizzi, A.; Praticò, A.D. Clinical, Genetic, EEG, Neuroimaging Insights and Conservative Treatment in Pediatric Focal Epilepsy: A Retrospective Observational Study. J. Clin. Med. 2025, 14, 2234. https://doi.org/10.3390/jcm14072234
Gauci MC, Gauci R, Ruggieri M, Polizzi A, Praticò AD. Clinical, Genetic, EEG, Neuroimaging Insights and Conservative Treatment in Pediatric Focal Epilepsy: A Retrospective Observational Study. Journal of Clinical Medicine. 2025; 14(7):2234. https://doi.org/10.3390/jcm14072234
Chicago/Turabian StyleGauci, Maria Cristina, Rosaria Gauci, Martino Ruggieri, Agata Polizzi, and Andrea D. Praticò. 2025. "Clinical, Genetic, EEG, Neuroimaging Insights and Conservative Treatment in Pediatric Focal Epilepsy: A Retrospective Observational Study" Journal of Clinical Medicine 14, no. 7: 2234. https://doi.org/10.3390/jcm14072234
APA StyleGauci, M. C., Gauci, R., Ruggieri, M., Polizzi, A., & Praticò, A. D. (2025). Clinical, Genetic, EEG, Neuroimaging Insights and Conservative Treatment in Pediatric Focal Epilepsy: A Retrospective Observational Study. Journal of Clinical Medicine, 14(7), 2234. https://doi.org/10.3390/jcm14072234