Abstract
Islet transplantation has been demonstrated to provide superior glycemic control with reduced glucose lability and hypoglycemic events compared with standard insulin therapy. However, the insulin independence rate after islet transplantation from one donor pancreas has remained low. The low frequency of islet grafting is dependent on poor islet recovery from donors and early islet loss during the first hours following grafting. The reduction in islet mass during pancreas preservation, islet isolation, and islet transplantation leads to β-cell death by apoptosis and the prerecruitment of intracellular death signaling pathways, such as c-Jun NH2-terminal kinase (JNK), which is one of the stress groups of mitogen-activated protein kinases (MAPKs). In this review, we show some of the most recent contributions to the advancement of knowledge of the JNK pathway and several possibilities for the treatment of diabetes using JNK inhibitors.
1. Introduction
Type 1 diabetes mellitus (T1DM) is an autoimmune disease and usually diagnosed at a young age with insulin deficiency. T1DM is characterized by progressive β-cell failure and gradual destruction of β-cells [1]. According to the International Diabetes Federation, approximately 542,000 children 0–14 years of age have T1DM, with 86,000 new cases diagnosed worldwide each year [1]. In the insulitis lesion in T1DM, invading immune cells produce cytokines, such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ [2]. IL-1β, TNF-α, and IFN-γ induce β-cell apoptosis via the activation of β-cell gene networks under the control of the transcription factors nuclear factor-κB (NF-κB) and STAT-1. NF-κB activation leads to the production of nitric oxide (NO) and chemokines and the depletion of endoplasmic reticulum (ER) calcium [3,4,5]. The execution of β-cell death occurs through the activation of mitogen-activated protein kinases (MAPKs), via the triggering of ER stress and the release of mitochondrial death signals.
Pancreatic islet transplantation has recently emerged as one of the most promising therapeutic approaches to improving glycometabolic control in T1DM patients and, in many cases, achieving insulin independence. Application of the Edmonton protocol has markedly improved the outcome [6,7] and the rate of insulin independence after islet transplantation has significantly improved in recent years [8]. However, multiple islet infusions from two or more donors are often required to achieve and maintain insulin independence. Contributions to graft loss include the instant blood-mediated inflammatory reaction (IBMIR), potent host auto- and allo-immune responses, and β-cell toxicity from immunosuppressive agents [9,10,11,12]. Moreover, the isolation procedure of pancreatic islets itself destroys cellular and noncellular components of the pancreatic tissue, which presumably play a role in supporting the survival of islet cells [13,14]. Pancreatic islets are exposed to mechanical and warm/cold ischemic stresses during pancreas procurement; to osmotic and cold ischemic stresses during pancreas preservation; to mechanical, enzymatic, and warm ischemic stresses during pancreas digestion; and to mechanical, osmotic, and cold ischemic stresses during islet purification. These stresses induce β-cell death (Figure 1).
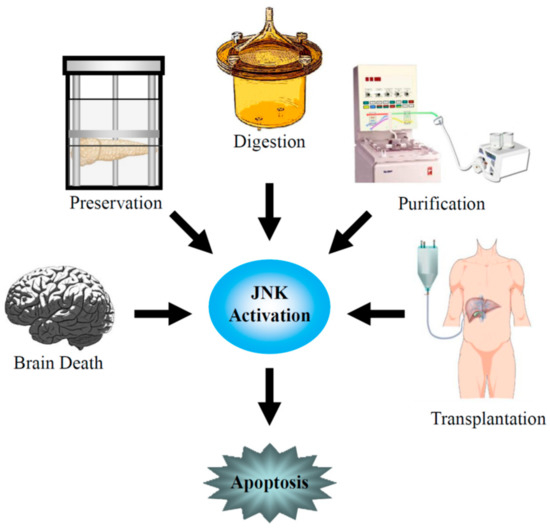
Figure 1.
c-Jun NH2-terminal kinase (JNK) activation during brain death and pancreas procurement, pancreas preservation, islet isolation, and islet transplantation.
β-cell death by apoptosis and the prerecruitment of intracellular death signaling pathways immediately after isolation and transplantation contributes to a reduction of the islet mass [15,16,17]. NF-κB [16] and the stress-associated MAPKs [18,19] mainly act as death-signaling pathways and these factors have been shown to contribute to the apoptosis of pancreatic β-cells. Inhibition of the death-signaling pathways has proven to be beneficial in several models of insulin-producing cell apoptosis in vitro [20,21]. Three major conserved groups of stress-associated MAPKs have been described: p38 kinases (p38 α/β/γ/δ) [22], c-Jun NH2-terminal kinases (JNKs; JNK1/2/3) [23], and extracellular signal-regulated kinases (ERKs; ERK1/2/3) [24]. JNK and p38 are similarly activated by several stresses, such as cold and heat shock, hypo- and hyperosmolarity, shearing stresses, proinflammatory cytokines, cytotoxic drugs, ultraviolet and γ-irradiation, the loss of survival factors, and reactive oxygen species [25,26]. Both p38 and JNK activate downstream nuclear transcription factors, which participate in the cellular response [27,28] by, for example, activating transcription factor-2 (ATF-2) and the activator protein-1 (AP-1), which is formed of heteromers of c-fos and c-Jun [27,28,29,30].
Here, we review the advancement of knowledge on the death-signaling pathways, especially the JNK pathway, during pancreas preservation, islet isolation, and islet transplantation, and the effect of JNK inhibitors for islet transplantation.
2. Donor Organ
The cadaver donor is the principal source of organs for transplantation. However, the successful rate of transplantations, such as those of kidneys, both over the short- and long-term, remains significantly inferior to those from living donors [31]. The differences between cadaveric and living donors are brain death, an optimal health condition of the living donor, a marginal condition of a substantial number of cadaveric donors, and an optimal timing of surgery in the case of living donors in comparison to a long cold ischemia time in cadaveric donors. It is certified that brain death affects the hemodynamic status, inflammatory reactivity, and hormone regulation. The effect of massive acute cerebral injury, as well as hypotension and circulating factors, results in the deterioration of organs following brain death [32,33]. Brain death is characterized by extensive cortical necrosis, which stimulates multiple cell types to produce proinflammatory cytokines, including IL-1β, TNF-α, IFN-γ, and IL-6 [32,33,34,35,36,37]. In the pancreas, donor characteristics such as age, cause of death, length of hospitalization, and medical history have a remarkable impact on islet recovery after isolation [38]. It has been demonstrated that the release of these proinflammatory cytokines, associated with brain death, significantly reduces the islet yield, functionality, viability, and engraftment after transplantation [36]. In this context, islets recovered from brain death donors presented higher nuclear activity of inflammation-related transcription factors, including ATF-2, c-Jun, and NF-κB. Furthermore, it has been demonstrated that macrophages infiltrate islets during brain death and that macrophage-associated inflammatory molecules, such as IL-1β, TNF-α, and IL-6, in islets are induced by brain death [39]. Therefore, the establishment of therapeutic strategies to prevent the deterioration of pancreatic islets during brain death could improve the islet transplant outcome. In addition, the strategies could improve the quality of organs from marginal donors, thus broadening the criteria for donor acceptance for isolation and transplantation.
It has been reported that males are more susceptible to the life-threatening effects of sepsis, hemorrhage, and trauma, compared to females in the proestrus cycle [40,41]. Female sex steroids, such as 17β-estradiol and estrogen, are likely to exhibit protective properties of immune and cardiovascular function after trauma, severe blood loss, and various adverse conditions [40,42]. Estradiol administration reversed the spontaneous increase of proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6 [43]. Moreover, estrogen possesses significant antiapoptotic and antioxidant activities [42,43]. Eckhoff et al. reported that 17β-estradiol treatment significantly decreased proinflammatory cytokine and structural and physiologic derangements in pancreatic islets subsequent to brain death induction [44]. In addition, it was demonstrated that estradiol improves the survival and functionality of human islets after proinflammatory cytokine exposure in vitro and in vivo. The molecular mechanisms involved included the inhibition of JNK activation, NF-κB nuclear translocation, caspase-9 activation, and mitochondrial cytochrome c release [45,46]. The inhibition of JNK activation induced the reduction of JNK targets, including the nuclear activities of transcription factors ATF-2, AP-1, c-Fos, c-Jun, and Jun-D, involved in apoptosis in pancreatic β-cells [46].
Our group investigated whether the administration of JNK inhibitors in human and porcine pancreata immediately after the procurements improves islet isolation results by preventing the apoptosis of islet cells [47]. A low molecular weight JNK inhibitor (SP600125) and a cell-permeable JNK inhibitor were used in porcine and human studies, respectively. The administration of JNK inhibitors in both porcine and human pancreata prevented JNK activation during the isolation procedure and prevented islet apoptosis immediately after isolation. Our data demonstrated that the JNK pathway is the major mediator of islet deterioration during/immediately after isolation and that JNK inhibition before islet isolation could improve the outcomes after pancreatic islet transplantation. The treatment of multiorgan donors with JNK inhibitors or 17β-estradiol could improve the quality of organs from marginal donors and increase human islet yields and functionality, and therefore broaden the criteria for donor acceptance for islet isolation and transplantation.
3. Pancreas Preservation
During pancreas preservation, islets are exposed to serious damaging conditions, resulting in a reduction of islet survival and ultimately graft failure after transplantation. The University of Wisconsin (UW) solution has been recognized as the gold standard in pancreas preservation before islet isolation. We, and other groups, have reported the superiority of the two-layer preservation method (TLM), which employs oxygenated perfluorochemical (PFC) and UW solution, compared with simple cold storage in UW for not only the whole pancreas, but also pancreatic islet transplantation in humans [48,49,50,51]. When TLM is used for pancreas preservation, PFC directly oxygenates the pancreas and results in a high level of adenosine triphosphate (ATP) in pancreatic tissues, which maintains parenchymal and nonparenchymal viability and retains cellular integrity [52,53,54,55]. Matsuda et al. reported the apoptosis pathways of caspase 3, 8, 9, JNK, and p38 in isolated islets after the cold storage of UW solution or TLM [56]. Islet apoptosis in the UW group was significantly increased compared with the fresh (no preservation) and TLM groups. Both caspase 3 and 9 activities in the UW group were higher than in the fresh and TLM groups, with an approximate increase of 2- to 3-fold. On the other hand, there was no significant difference in caspase 8 activity among these three groups. These data suggest that the mitochondrial pathway is largely engaged in islet apoptosis induced by the simple preservation of UW solution, and that TLM blocks to a great extent. On the other hand, JNKs were strongly activated in both the TLM and UW groups, while they were not activated in the fresh group. In contrast, p38 was activated to almost the same levels in these three groups. These findings suggest that pancreas preservation with UW solution or TLM before islet isolation cannot protect against JNK activation.
Our group showed that an intraductal injection of JNK inhibitors before pancreas storage prevented JNK activation during the isolation procedure and improved islet graft survival in humans. [47]. Another group also reported that an intraductal injection of JNK inhibitor in porcine pancreata significantly suppressed mRNA expression levels of IL-1β, TNF-α, IFN-γ, IL-6, IL-8, and macrophage chemoattractant protein-1, as well as the concentration of IL-1β and IL-8, in the culture supernatant [57]. These data suggest that the inhibition of JNK activation during pancreas preservation improves the islet transplant outcome through the reduction of the inflammatory response.
We recently developed a novel preservation solution, the extracellular-type/JNK inhibitor-containing (EJ) solution, for porcine pancreas preservation [58]. After pancreas preservation in EJ solution, JNK activity was maintained at a relatively low level during islet isolation. The islet yield before and after purification was significantly higher in the EJ group than in the UW group or EJ-J (EJ solution without the JNK inhibitor) group. After islet transplantation into streptozotocin-induced diabetic mice, the attainability of post-transplantation normoglycemia was higher in the EJ group than the UW group or EJ-J group. These data suggest that the inhibition of JNK activity for pancreas storage could be useful for preventing islet apoptosis and improving islet transplant outcomes.
4. Islet Isolation and Culture
Pancreatic islets are exposed to mechanical, enzymatic, osmotic, and ischemic stresses during pancreas digestion and islet purification. Our group reported that JNK activity progressively increased during the isolation procedure [47]. Abdelli et al. mapped the major intracellular stress-signaling pathways activated during human islet isolation and following acute cytokine exposure [17]. For the islet isolation procedure, two pathways are involved in islet survival: NF-κB→iNOS and MAPK kinase 7 (MKK7)→JNK/p38→c-fos. Proinflammatory cytokines activate the NF-κB→iNOS and MKK4/MKK3/6→JNK/p38 pathways without the involvement of c-fos. It is also likely that the procedure of islet isolation, together with proinflammatory cytokine production immediately after transplantation, may further synergize to enhance the apoptosis of islets [59]. In the case of other cell types, MKK7 also transduces cytokine signaling [26]. Therefore, the activation of MKK7 after islet purification may sensitize islets to cytokine exposure [60]. The activated pathways return to background levels after the 48 h culture of isolated islets and the expression of MKK7 becomes undetectable [17]. Inhibition of the JNK, p38, and NF-κB pathways throughout the procedures of pancreas preservation, islet isolation, and islet transplantation might result in the reduction of primary nonfunction and the improvement of islet graft survival [20,21,61]. Our group also reported that the treatment of JNK inhibitors before islet isolation prevented JNK activation during the isolation procedure and prevented islet apoptosis immediately after isolation [47].
Three JNK isoforms (JNK1, 2, and 3) have been identified. JNK1 and JNK2 are ubiquitously expressed, while JNK3 expression is restricted to pancreatic islets and the brain [62,63]. In contrast to JNK1 and JNK2, JNK3 exhibits anti-apoptotic activity in insulin-producing cells [63]. Varona-Santos et al. investigated the role of JNK isoforms in pancreatic islets using Jnk1−/− and Jnk2−/− mice [64]. Islets derived from Jnk1−/− mice secreted more insulin and significantly protected cytokine-induced cell death compared with islets derived from wild-type and Jnk2−/− mice. These data suggest that specific JNK1 blockades in islets may be important for islet transplantation [64].
5. Islet Transplantation
The transplantation of isolated islets into the liver through the portal vein is the preferred site for clinical islet transplantation. An early innate inflammatory reaction after intrahepatic islet transplantation strongly affects islet engraftment and survival. This early immune response is triggered by ischemia-reperfusion injury and IBMIR occurring hours and days after islet infusion [65,66,67,68,69,70,71]. IBMIR involves activation of the complement and coagulation cascades, ultimately resulting in clot formation and infiltration of leukocytes into the islets, which leads to disruption of islet integrity and islet destruction [12]. Moreover, the nonspecific activation and dysfunction of intrahepatic endothelial cells after islet transplantation, which are characterized by the production of proinflammatory cytokines such as TNF-α, IL-1β, and IFN-γ, as well as the upregulation of the intracellular adhesion molecule (ICAM)-1, P-selectin, and NO, have been demonstrated [69,70,71,72,73,74,75]. These effects finally induce early graft loss. It has been reported that 25% of the transplanted islets were lost within the first few minutes after intraportal transplantation [76] and that the islet loss after transplantation into the portal vein is widely estimated to be higher (50%–60%) [77,78,79]. To prevent early graft loss, candidate drugs have been reported in clinical and experimental animal studies. Heparin is commonly used for clinical islet transplantation to reduce the impact of coagulation. Low molecular weight dextran sulfate (LMW-DS, MM 5000) is an alternative inhibitor of IBMIR [80,81,82,83,84]. An open randomized multicenter study showed that LMW-DS has a similar efficacy in inhibiting IBMIR to promote islet engraftment when compared with heparin [84]. Activated protein C (APC) is another potent inhibitor which exerts anticoagulant, anti-inflammatory, and antiapoptotic activities by acting directly on cells. It has been reported that the exogenous administration of APC significantly reduced the loss of functional islet mass after intraportal transplantation in diabetic mice [85]. APC is an important physiological anticoagulant generated from protein C by the action of thrombin-thrombomodulin on endothelial cells [86]. APC appears to regulate the inflammatory process in part by blocking the activity of the transcription factor NF-κB by preventing the generation of thrombin and by inhibiting the production of proinflammatory cytokines [86,87,88,89,90]. Our group showed that the double blockage of proinflammatory cytokines, IL-1β and TNF-α, improved the efficacy of clinical islet transplantation [91]. The blockage of TNF-α, eternacept, IL-1β, and anakinra was administered in three patients with type 1 diabetes before and during islet transplantation and all patients achieved insulin independence with normal HbA1c levels by a single infusion from one donor. Although this study used not only the antibody, but also thymoglobulin induction and sirolimus-free immunosuppression, the double blockage of IL-1β and TNF-α could contribute to the prevention of early graft loss.
To evaluate the intracellular stress-signaling pathways of JNK during the islet transplant process, our group measured JNK activity in the liver 1, 3, 6, and 24 h after mouse islet transplantation [92]. The JNK was activated until 1 h after islet transplantation and the activity became gradually higher until 24 h. The evidence has profound implications for IBMIR, the production of proinflammatory cytokine, and subsequent islet apoptosis. Our group also investigated the effect of an intraportal injection of pancreatic islets with JNK inhibitor. Isolated islets with JNK inhibitor were transplanted into diabetic mice through the portal vein and liver samples were collected before transplantation and 1, 3, 6, and 24 h after transplantation. The JNK activity in the liver was suppressed at a low level until 24 h after transplantation. Moreover, the intraportal injection of isolated islets with the JNK inhibitor improved islet graft survival [92]. These data suggest that control of the JNK pathway is extremely important in islet transplantation and that an intraportal injection of isolated islets with JNK inhibitor prevents the activation of JNK in the liver immediately after islet transplantation and improves the outcome for islet transplantation.
Varona-Santos et al. investigated the role of JNK isoforms in transplant recipients using Jnk1−/− and Jnk2−/− mice [64]. When islets derived from wild-type mice were transplanted into diabetic Jnk1−/−recipients, the median time to diabetes reversal was shorter than that for wild-type diabetic recipients. On the other hand, the median time to diabetes reversal in diabetic Jnk2−/− recipients was longer than that for wild-type diabetic recipients when islets derived from wild-type mice were transplanted into diabetic Jnk2−/− recipients. These data suggest that specific JNK1 blockades in recipients may be important for islet transplantation [64].
6. JNK Inhibitors
JNK inhibitors have been expected as drugs to improve islet transplant outcomes (Table 1). The widely used inhibitor of JNKs for research is SP600125 [93]. SP600125 is an ATP-competitive inhibitor and the IC50 values for JNK1 and JNK2 are both 40 nM, while that for JNK3 is 90 nM [94]. On the other hand, SP600125 is >300-fold selective over the related MAPKs, ERK1, and p38-2, and between 10-fold and 100-fold selective over another 14 protein kinases tested [94]. We and another group showed the efficacy of SP600125 during pancreas preservation for islet transplantation [47,57]. SP600125 prevented JNK activation during islet isolation and improved isle viability and the islet transplant outcome. However, the ATP-competitive inhibitor has several degrees of toxicity and lacks the required specificity because it inhibits the phosphorylation of all JNK substrates [95].

Table 1.
c-Jun NH2-terminal kinase (JNK) inhibitors used for islet transplantation.
Peptide inhibitors of JNK have also been developed, which are ATP-noncompetitive [93]. The peptide inhibitors of JNK are based on JNK-interacting protein-1 (JIP1), also known as islet-brain-1 (IB1). JIP1 has been discovered to have a JNK inhibitory property and its minimum inhibitory sequence has also been identified [20,96]. For efficient delivery of the JNK inhibitory peptide (JNKI) (Figure 2) into pancreatic islets, our group synthesized JNKI as a C-terminal fusion protein with 11-arginine (11R). Poly-arginine facilitates the uptake of peptides into mammalian cells more efficiently than TAT or other cell-penetrating peptides [97,98,99,100]. 11R-JNKI prevented JNK activation during pancreas preservation and islet isolation [47], islet culture [101], and immediately after islet transplantation [92], resulting in an improvement of islet graft survival. Another group also reported that TAT-JNKI reduced the islet loss in culture and protected against cell death through the regulation of AKT/GSK3B activity [102]. Our group recently developed a more efficient JNK inhibitory peptide [58,103]. The N-terminal amino acids of JNKI include two arginine and one lysin (RPKR) (Figure 2). Since poly-arginine/lysin facilitates the uptake of peptides and proteins into mammalian cells, our group hypothesized that the transduction efficacy of 11R-JNKI may not be reduced after the deletion of three arginine and two glycine-linkers. Moreover, we investigated whether C-terminal deletion peptides of JNKI can inhibit JNK activity (Figure 2). One of the peptides, 8R-sJNKI(-9), efficiently prevented JNK activation at one tenth of the concentration of 11R-JNKI, suggesting that 8R-sJNKI(-9) inhibits islet apoptosis and improves islet function more efficiently than 11R-JNKI. It has been reported that, when the specificity of sJNKI was investigated in assays of 40 different protein kinases, only the JNKs and their upstream activators MKK7 and MKK4 were affected, emphasizing the specificity of inhibition [104].
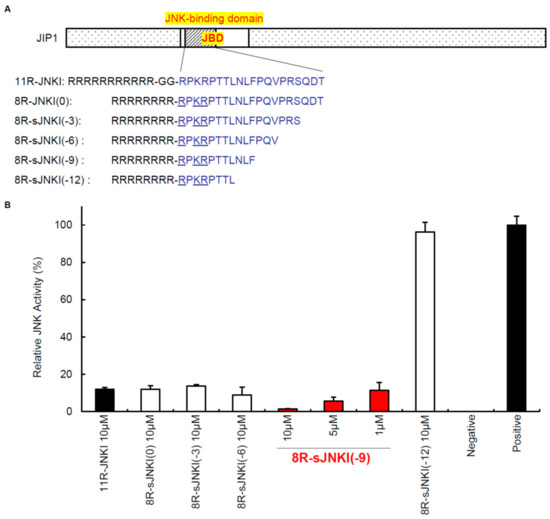
Figure 2.
Cell-permeable JNK inhibitors. (A) The sequences of JNK inhibitory peptides. The peptide inhibitors of JNK are based on JNK-interacting protein-1 (JIP1), which was discovered to have a JNK inhibitory property. (B) Inhibition of JNK activation. MIN6 cells, which are part of the pancreatic β-cell line, were cultured with 1–10 μM of 11R-JNKI, 8R-JNKI(0), 8R-sJNKI(-3), 8R-sJNKI(-6), 8R-sJNKI (-9), or 8R-sJNKI(-12) for 23 h. The cells were then treated with 1 μg/mL of anisomycin for 1 h to stimulate the activation of JNK, after which the JNK activity was examined by western blotting. The cell lysates from MIN6 cells cultured with and without 1 μg/mL of anisomycin for 1 h were used as positive and negative controls, respectively. The data are expressed with the JNK activity of the positive and negative controls, which were arbitrarily set at 100 and 0, respectively.
It has been reported that exenatide, a glucagon-like peptide-1 receptor agonist, inhibited JNK activation and caspase-3 activation, resulting in the inhibition of β-cell apoptosis [105] and that 17β-estoradiol reduced JNK activation, nuclear AP-1, c-fos, Jun-D, and ATF-2 activities and enhanced islet viability and islet mass [46]. Prolactin and α-1 antitrypsin also inhibited JNK activation [106,107,108].
7. Future Perspective
The activation of JNK is induced during pancreas preservation and JNK activity is progressively increased during the isolation procedure. In addition, JNK is activated in the transplanted liver immediately after islet transplantation. In diabetes, JNK plays an important role in various tissues due to the phenomenon known as “glucose toxicity” and activation of the JNK pathway interferes with insulin biosynthesis [109], β-cell function [109,110], and insulin action [111,112,113]. The JNK inhibition from pancreas preservation and isolation, and throughout the transplantation procedure might prove critical for the maintenance of islet cell mass and improve isle graft function. The current challenge in finding new successful anti-JNK therapies is to design isoform-selective inhibitors of the JNKs. The regulation of intracellular signaling pathways, including JNK, may become a new therapeutic strategy to improve graft survival in clinical islet transplantation.
Both JNK and p38 are preferentially activated in response to the processing of islets for transplantation and by the inflammation associated with islet transplantation. Some small molecules that inhibit p38 activity suppress the production of proinflammatory cytokines and improve islet engraftment [114,115,116]. Since p38 participates in another signaling cascade controlling cellular responses to cytokines and stress, the inhibition of both JNK and p38 may enhance the inhibition of isle apoptosis. Some groups indicate cytokine-mediated β-cell necrosis as an additional possibility [117,118]. Collier et al. showed that proinflammatory cytokines cause β-cell cytotoxicity primarily through a nonapoptotic mechanism linked to a decline in ATP levels [117]. Steer et al. showed that IL-1 induces β-cell necrosis [118]. The inhibition of not only apoptosis but also necrosis may become a new therapeutic strategy to improve the outcome of islet transplantation.
Author Contributions
Investigation, supervision, writing—original draft and Writing—review and editing: H.N.
Funding
This work was supported in part by JSPS KAKENHI Grant Numbers 19K09051, 17H00769, 19H01064, and 17H04412, and the Okinawa Science and Technology Innovation System Construction Project.
Acknowledgments
We thank Chika Miyagi-Shiohira, Naomi Kakazu, Maki Higa, Yuki Kawahira, and Ikue Honda (University of the Ryukyus) for office processing and technical support.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| JNK | c-Jun NH2-terminal kinase |
| MAPKs | mitogen-activated protein kinases |
| T1DM | type 1 diabetes mellitus |
| IL | interleukin |
| TNF | tumor necrosis factor |
| IFN | interferon |
| NF-κB | nuclear factor-κB |
| NO | nitric oxide |
| ER | endoplasmic reticulum |
| IBMIR | instant blood-mediated inflammatory reaction |
| ERKs | extracellular signal–regulated kinases |
| ATF-2 | activating transcription factor-2 |
| AP-1 | activator protein-1 |
| UW | University of Wisconsin solution |
| PFC | perfluorochemical |
| TLM | two-layer preservation method |
| ATP | adenosine triphosphate |
| EJ | extracellular-type/JNK inhibitor-containing solution |
| MKK | MAPK kinase |
| ICAM | intracellular adhesion molecule |
| APC | activated protein C |
| JIP1 | JNK-interacting protein-1 |
| IB1 | islet-brain-1 |
| JNKI | JNK inhibitory peptide |
| 11R | 11-arginine |
References
- International Diabetes Federation. IDF Diabetes Atlas, 7th ed. 2015. Available online: www.diabetesatlas.org (accessed on 1 June 2016).
- Mandrup-Poulsen, T.; Bendtzen, K.; Nerup, J.; Dinarello, C.A.; Svenson, M.; Nielsen, J.H. Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia 1986, 29, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.A.; Kwon, G.; Marino, M.H.; Rodi, C.P.; Sullivan, P.M.; Turk, J.; McDaniel, M.L. Tyrosine kinase inhibitors prevent cytokine-induced expression of iNOS and COX-2 by human islets. Am. J. Physiol. 1996, 270, C1581–C1587. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Dypbukt, J.M.; Brüne, B.; Nicotera, P. Interleukin-1 beta-induced nitric oxide production activates apoptosis in pancreatic RINm5F cells. Exp. Cell Res. 1994, 213, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Fujii, J.; Seo, H.G.; Suzuki, K.; Matsuoka, T.; Nakamura, M.; Tatsumi, H.; Yamasaki, Y.; Kamada, T.; Taniguchi, N. Apoptotic cell death triggered by nitric oxide in pancreatic beta-cells. Diabetes 1995, 44, 733–738. [Google Scholar] [CrossRef]
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef]
- Matsumoto, S.; Okitsu, T.; Iwanaga, Y.; Noguchi, H.; Nagata, H.; Yonekawa, Y.; Yamada, Y.; Fukuda, K.; Tsukiyama, K.; Suzuki, H.; et al. Insulin independence after living-donor distal pancreatectomy and islet allotransplantation. Lancet 2005, 365, 1642–1644. [Google Scholar] [CrossRef]
- Fiorina, P.; Shapiro, A.M.; Ricordi, C.; Secchi, A. The clinical impact of islet transplantation. Am. J. Transplant. 2008, 8, 1990–1997. [Google Scholar] [CrossRef]
- Shapiro, A.M.; Gallant, H.L.; Hao, E.G.; Lakey, J.R.; McCready, T.; Rajotte, R.V.; Yatscoff, R.W.; Kneteman, N.M. The portal immunosuppressive storm: Relevance to islet transplantation? Ther. Drug Monit. 2005, 27, 35–37. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef]
- Kourtzelis, I.; Kotlabova, K.; Lim, J.H.; Mitroulis, I.; Ferreira, A.; Chen, L.S.; Gercken, B.; Steffen, A.; Kemter, E.; Klotzsche-von Ameln, A.; et al. Developmental endothelial locus-1 modulates platelet-monocyte interactions and instant blood-mediated inflammatory reaction in islet transplantation. Thromb. Haemost. 2016, 115, 781–788. [Google Scholar] [CrossRef]
- Nilsson, B.; Ekdahl, K.N.; Korsgren, O. Control of instant blood-mediated inflammatory reaction to improve islets of Langerhans engraftment. Curr. Opin. Organ. Transplant. 2011, 16, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Davalli, A.M.; Scaglia, L.; Zangen, D.H.; Hollister, J.; Bonner-Weir, S.; Weir, G.C. Vulnerability of islets in the immediate posttransplantation period. Dynamic changes in structure and function. Diabetes 1996, 45, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L.; Wang, R.; Paraskevas, S.; Maysinger, D. Structural and functional changes resulting from islet isolation lead to islet cell death. Surgery 1999, 126, 393–398. [Google Scholar] [CrossRef]
- Thomas, D.; Yang, H.; Boffa, D.J.; Ding, R.; Sharma, V.K.; Lagman, M.; Li, B.; Hering, B.; Mohanakumar, T.; Lakey, J.; et al. Proapoptotic Bax is hyperexpressed in isolated human islets compared with antiapoptotic Bcl-2. Transplantation 2002, 74, 1489–1496. [Google Scholar] [CrossRef]
- Rehman, K.K.; Bertera, S.; Bottino, R.; Balamurugan, A.N.; Mai, J.C.; Mi, Z.; Trucco, M.; Robbins, P.D. Protection of islets by in situ peptide-mediated transduction of the Ikappa B kinase inhibitor Nemo-binding domain peptide. J. Biol. Chem. 2003, 278, 9862–9868. [Google Scholar] [CrossRef]
- Abdelli, S.; Ansite, J.; Roduit, R.; Borsello, T.; Matsumoto, I.; Sawada, T.; Allaman-Pillet, N.; Henry, H.; Beckmann, J.S.; Hering, B.J.; et al. Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes 2004, 53, 2815–2823. [Google Scholar] [CrossRef][Green Version]
- Ammendrup, A.; Maillard, A.; Nielsen, K.; Aabenhus Andersen, N.; Serup, P.; Dragsbaek Madsen, O.; Mandrup-Poulsen, T.; Bonny, C. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 2000, 49, 1468–1476. [Google Scholar] [CrossRef]
- Saldeen, J.; Lee, J.C.; Welsh, N. Role of p38 mitogen-activated protein kinase (p38 MAPK) in cytokine-induced rat islet cell apoptosis. Biochem. Pharmacol. 2001, 61, 1561–1569. [Google Scholar] [CrossRef]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-permeable peptide inhibitors of JNK: Novel blockers of beta-cell death. Diabetes 2001, 50, 77–82. [Google Scholar] [CrossRef]
- Baker, M.S.; Chen, X.; Cao, X.C.; Kaufman, D.B. Expression of a dominant negative inhibitor of NF-kappaB protects MIN6 beta-cells from cytokine-induced apoptosis. J. Surg. Res. 2001, 97, 117–122. [Google Scholar] [CrossRef]
- Keesler, G.A.; Bray, J.; Hunt, J.; Johnson, D.A.; Gleason, T.; Yao, Z.; Wang, S.W.; Parker, C.; Yamane, H.; Cole, C.; et al. Purification and activation of recombinant p38 isoforms alpha, beta, gamma, and delta. Protein Expr. Purif. 1998, 14, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the c-Jun N-terminal kinase. Biochem. Soc. Symp. 1999, 64, 1–12. [Google Scholar] [PubMed]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radziejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [CrossRef]
- Clerk, A.; Fuller, S.J.; Michael, A.; Sugden, P.H. Stimulation of “stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J. Biol. Chem. 1998, 273, 7228–7234. [Google Scholar] [CrossRef]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. Philos. Trans. R Soc. Lond. B Biol. Sci. 1996, 351, 127–134. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, J.; MacGibbon, G.; Dragunow, M.; Cooper, G.J. Increased expression and activation of c-Jun contributes to human amylin-induced apoptosis in pancreatic islet beta-cells. J. Mol. Biol. 2002, 324, 271–285. [Google Scholar] [CrossRef]
- Wang, J.; Van De Water, T.R.; Bonny, C.; de Ribaupierre, F.; Puel, J.L.; Zine, A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J. Neurosci. 2003, 23, 8596–8607. [Google Scholar] [CrossRef]
- Terasaki, P.I.; Cecka, J.M.; Gjertson, D.W.; Takemoto, S. High survival rates of kidney transplants from spousal and living unrelated donors. N. Engl. J. Med. 1995, 333, 333–336. [Google Scholar] [CrossRef]
- Pratschke, J.; Wilhelm, M.J.; Kusaka, M.; Laskowski, I.; Tilney, N.L. A model of gradual onset brain death for transplant-associated studies in rats. Transplantation 2000, 69, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Nadeau, K.C.; Hancock, W.W.; Mackenzie, H.S.; Shaw, G.D.; Waaga, A.M.; Chandraker, A.; Sayegh, M.H.; Tilney, N.L. Effects of explosive brain death on cytokine activation of peripheral organs in the rat. Transplantation 1998, 65, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Kusaka, M.; Pratschke, J.; Wilhelm, M.J.; Ziai, F.; Zandi-Nejad, K.; Mackenzie, H.S.; Hancock, W.W.; Tilney, N.L. Activation of inflammatory mediators in rat renal isografts by donor brain death. Transplantation 2000, 69, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.J.; Pratschke, J.; Beato, F.; Taal, M.; Kusaka, M.; Hancock, W.W.; Tilney, N.L. Activation of the heart by donor brain death accelerates acute rejection after transplantation. Circulation 2000, 102, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.L.; Eckstein, C.; Smyth, C.A.; Sellers, M.T.; Vilatoba, M.; Bilbao, G.; Rahemtulla, F.G.; Young, C.J.; Thompson, J.A.; Chaudry, I.H.; et al. Brain death significantly reduces isolated pancreatic islet yields and functionality in vitro and in vivo after transplantation in rats. Diabetes 2003, 52, 2935–2942. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pratschke, J.; Wilhelm, M.J.; Kusaka, M.; Hancock, W.W.; Tilney, N.L. Activation of proinflammatory genes in somatic organs as a consequence of brain death. Transplant. Proc. 1999, 31, 1003–1005. [Google Scholar] [CrossRef]
- Lakey, J.R.; Burridge, P.W.; Shapiro, A.M. Technical aspects of islet preparation and transplantation. Transpl. Int. 2003, 16, 613–632. [Google Scholar] [CrossRef]
- Toyama, H.; Takada, M.; Suzuki, Y.; Kuroda, Y. Activation of macrophage-associated molecules after brain death in islets. Cell Transplant. 2003, 12, 27–32. [Google Scholar] [CrossRef]
- Angele, M.K.; Schwacha, M.G.; Ayala, A.; Chaudry, I.H. Effect of gender and sex hormones on immune responses following shock. Shock 2000, 14, 81–90. [Google Scholar] [CrossRef]
- Jarrar, D.; Wang, P.; Knöferl, M.W.; Kuebler, J.F.; Cioffi, W.G.; Bland, K.I.; Chaudry, I.H. Insight into the mechanism by which estradiol improves organ functions after trauma-hemorrhage. Surgery 2000, 128, 246–252. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Pfeilschifter, J.; Köditz, R.; Pfohl, M.; Schatz, H. Changes in proinflammatory cytokine activity after menopause. Endocr. Rev. 2002, 23, 90–119. [Google Scholar] [CrossRef] [PubMed]
- Eckhoff, D.E.; Eckstein, C.; Smyth, C.A.; Vilatoba, M.; Bilbao, G.; Rahemtulla, F.G.; Young, C.J.; Anthony Thompson, J.; Chaudry, I.H.; Contreras, J.L. Enhanced isolated pancreatic islet recovery and functionality in rats by 17beta-estradiol treatment of brain death donors. Surgery 2004, 136, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.L.; Smyth, C.A.; Bilbao, G.; Young, C.J.; Thompson, J.A.; Eckhoff, D.E. 17beta-Estradiol protects isolated human pancreatic islets against proinflammatory cytokine-induced cell death: Molecular mechanisms and islet functionality. Transplantation 2002, 74, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Eckhoff, D.E.; Smyth, C.A.; Eckstein, C.; Bilbao, G.; Young, C.J.; Thompson, J.A.; Contreras, J.L. Suppression of the c-Jun N-terminal kinase pathway by 17beta-estradiol can preserve human islet functional mass from proinflammatory cytokine-induced destruction. Surgery 2003, 134, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Matsumoto, S.; Onaca, N.; Naziruddin, B.; Jackson, A.; Ikemoto, T.; Shimoda, M.; Fujita, Y.; Chujo, D.; Iwanaga, Y.; et al. Ductal injection of JNK inhibitors before pancreas preservation prevents islet apoptosis and improves islet graft function. Hum. Gene Ther. 2009, 20, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Levy, M.F.; Kobayashi, N.; Matsumoto, S. Pancreas preservation by the two-layer method: Does it have a beneficial effect compared with simple preservation in University of Wisconsin solution? Cell Transplant. 2009, 18, 497–503. [Google Scholar] [CrossRef]
- Hiraoka, K.; Trexler, A.; Eckman, E.; Stage, A.; Nevile, S.; Sageshima, J.; Shibata, S.; Sutherland, D.E.; Hering, B.J. Successful pancreas preservation before islet isolation by the simplified two-layer cold storage method. Transplant. Proc. 2001, 33, 952–953. [Google Scholar] [CrossRef]
- Tanioka, Y.; Sutherland, D.E.; Kuroda, Y.; Gilmore, T.R.; Asaheim, T.C.; Kronson, J.W.; Leone, J.P. Excellence of the two-layer method (University of Wisconsin solution/perfluorochemical) in pancreas preservation before islet isolation. Surgery 1997, 122, 435–441. [Google Scholar] [CrossRef]
- Noguchi, H.; Ueda, M.; Nakai, Y.; Iwanaga, Y.; Okitsu, T.; Nagata, H.; Yonekawa, Y.; Kobayashi, N.; Nakamura, T.; Wada, H.; et al. Modified two-layer preservation method (M-Kyoto/PFC) improves islet yields in islet isolation. Am. J. Transplant. 2006, 6, 496–504. [Google Scholar] [CrossRef]
- Tanioka, Y.; Kuroda, Y.; Kim, Y.; Matsumoto, S.; Suzuki, Y.; Ku, Y.; Fujita, H.; Saitoh, Y. The effect of ouabain (inhibitor of an ATP-dependent Na+/K+ pump) on the pancreas graft during preservation by the two-layer method. Transplantation 1996, 62, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Fujino, Y.; Kawamura, T.; Suzuki, Y.; Fujiwara, H.; Saitoh, Y. Mechanism of oxygenation of pancreas during preservation by a two-layer (Euro-Collins’ solution/perfluorochemical) cold-storage method. Transplantation 1990, 49, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Fujino, Y.; Morita, A.; Ku, Y.; Saitoh, Y. Correlation between high adenosine triphosphate tissue concentration and good posttransplant outcome for the canine pancreas graft after preservation by the two-layer cold storage method. Transplantation 1991, 52, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Fujita, H.; Matsumoto, S.; Suzuki, Y.; Kim, Y.; Tanioka, Y.; Ku, Y. Protection of canine pancreatic microvascular endothelium against cold ischemic injury during preservation by the two-layer method. Transplantation 1997, 64, 948–953. [Google Scholar] [CrossRef]
- Matsuda, T.; Suzuki, Y.; Tanioka, Y.; Toyama, H.; Kakinoki, K.; Hiraoka, K.; Fujino, Y.; Kuroda, Y. Pancreas preservation by the 2-layer cold storage method before islet isolation protects isolated islets against apoptosis through the mitochondrial pathway. Surgery 2003, 134, 437–445. [Google Scholar] [CrossRef]
- Jin, S.M.; Kim, K.S.; Lee, S.Y.; Gong, C.H.; Park, S.K.; Shin, J.S.; Park, C.G.; Kim, S.J. The sequential combination of a JNK inhibitor and simvastatin protects porcine islets from peritransplant apoptosis and inflammation. Cell Transplant. 2011, 20, 1139–1151. [Google Scholar] [CrossRef]
- Noguchi, H.; Miyagi-Shiohira, C.; Nakashima, Y.; Ebi, N.; Hamada, E.; Tamaki, Y.; Kuwae, K.; Kitamura, S.; Kobayashi, N.; Saitoh, I.; et al. A Novel Preservation Solution Containing a JNK Inhibitory Peptide Efficiently Improves Islet Yield for Porcine Islet Isolation. Transplantation 2019, 103, 344–352. [Google Scholar] [CrossRef]
- Davalli, A.M.; Ogawa, Y.; Ricordi, C.; Scharp, D.W.; Bonner-Weir, S.; Weir, G.C. A selective decrease in the beta cell mass of human islets transplanted into diabetic nude mice. Transplantation 1995, 59, 817–820. [Google Scholar] [CrossRef]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): Differences revealed by gene targeting. Bioessays 2006, 28, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Abdelli, S.; Puyal, J.; Bielmann, C.; Buchillier, V.; Abderrahmani, A.; Clarke, P.G.; Beckmann, J.S.; Bonny, C. JNK3 is abundant in insulin-secreting cells and protects against cytokine-induced apoptosis. Diabetologia 2009, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Varona-Santos, J.L.; Pileggi, A.; Molano, R.D.; Sanabria, N.Y.; Ijaz, A.; Atsushi, M.; Ichii, H.; Pastori, R.L.; Inverardi, L.; Ricordi, C.; et al. c-Jun N-terminal kinase 1 is deleterious to the function and survival of murine pancreatic islets. Diabetologia 2008, 51, 2271–2280. [Google Scholar] [CrossRef][Green Version]
- Moberg, L.; Johansson, H.; Lukinius, A.; Berne, C.; Foss, A.; Källen, R.; Østraat, Ø.; Salmela, K.; Tibell, A.; Tufveson, G.; et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet 2002, 360, 2039–2045. [Google Scholar] [CrossRef]
- Bennet, W.; Sundberg, B.; Growth, C.G.; Brendel, M.D.; Brandhorst, D.; Brandhorst, H.; Bretzel, R.G.; Elgue, G.; Larsson, R.; Nilsson, B.; et al. Incompatibility between human blood and isolated islets of Langerhans: A finding with implications for clinical intraportal islet transplantation? Diabetes 1999, 48, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Moberg, L.; Olsson, A.; Berne, C.; Felldin, M.; Foss, A.; Källen, R.; Salmela, K.; Tibell, A.; Tufveson, G.; Nilsson, B.; et al. Nicotinamide inhibits tissue factor expression in isolated human pancreatic islets: Implications for clinical islet transplantation. Transplantation 2003, 76, 1285–1288. [Google Scholar] [CrossRef]
- Badet, L.; Titus, T.; Metzen, E.; Handa, A.; McShane, P.; Chang, L.W.; Giangrande, P.; Gray, D.W. The interaction between primate blood and mouse islets induces accelerated clotting with islet destruction. Xenotransplantation 2002, 9, 91–96. [Google Scholar] [CrossRef]
- Bottino, R.; Fernandez, L.A.; Ricordi, C.; Lehmann, R.; Tsan, M.F.; Oliver, R.; Inverardi, L. Transplantation of allogeneic islets of Langerhans in the rat liver: Effects of macrophage depletion on graft survival and microenvironment activation. Diabetes 1998, 47, 316–323. [Google Scholar] [CrossRef]
- Fernandez, L.; Bottino, R.; Oliver, R.; Lehmann, R.; Ricordi, C.; Fu Tsan, M.; Inverardi, L. Endothelial cell dysfunction after intraportal islet cell transplant in rats. Transplant. Proc. 1997, 29, 2064–2065. [Google Scholar] [CrossRef]
- Fontaine, M.J.; Blanchard, J.; Rastellini, C.; Lazda, V.; Herold, K.C.; Pollak, R. Pancreatic islets activate portal vein endothelial cells in vitro. Ann. Clin. Lab. Sci. 2002, 32, 352–361. [Google Scholar]
- Arita, S.; Une, S.; Ohtsuka, S.; Atiya, A.; Kasraie, A.; Shevlin, L.; Mullen, Y. Prevention of primary islet isograft nonfunction in mice with pravastatin. Transplantation 1998, 65, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.B.; Gores, P.F.; Field, M.J.; Farney, A.C.; Gruber, S.A.; Stephanian, E.; Sutherland, D.E. Effect of 15-deoxyspergualin on immediate function and long-term survival of transplanted islets in murine recipients of a marginal islet mass. Diabetes 1994, 43, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Kenmochi, T.; Miyamoto, M.; Mullen, Y. Protection of mouse islet isografts from nonspecific inflammatory damage by recipient treatment with nicotinamide and 15-deoxyspergualin. Cell Transplant. 1996, 5, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Xenos, E.S.; Stevens, R.B.; Sutherland, D.E.; Lokeh, A.; Ansite, J.D.; Casanova, D.; Gores, P.F.; Platt, J.L. The role of nitric oxide in IL-1 beta-mediated dysfunction of rodent islets of Langerhans. Implications for the function of intrahepatic islet grafts. Transplantation 1994, 57, 1208–1212. [Google Scholar] [CrossRef]
- Eriksson, O.; Eich, T.; Sundin, A.; Tibell, A.; Tufveson, G.; Andersson, H.; Felldin, M.; Foss, A.; Kyllönen, L.; Langstrom, B.; et al. Positron emission tomography in clinical islet transplantation. Am. J. Transplant. 2009, 9, 2816–2824. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Larson, C.S.; Baker, M.S.; Kaufman, D.B. In vivo bioluminescence imaging of transplanted islets and early detection of graft rejection. Transplantation 2006, 81, 1421–1427. [Google Scholar] [CrossRef]
- Saudek, F.; Jirák, D.; Girman, P.; Herynek, V.; Dezortová, M.; Kríz, J.; Peregrin, J.; Berková, Z.; Zacharovová, K.; Hájek, M. Magnetic resonance imaging of pancreatic islets transplanted into the liver in humans. Transplantation 2010, 90, 1602–1606. [Google Scholar] [CrossRef]
- Eich, T.; Eriksson, O.; Lundgren, T.; Nordic Network for Clinical Islet Transplantation. Visualization of early engraftment in clinical islet transplantation by positron-emission tomography. N. Engl. J. Med. 2007, 356, 2754–2755. [Google Scholar] [CrossRef]
- Wuillemin, W.A.; te Velthuis, H.; Lubbers, Y.T.; de Ruig, C.P.; Eldering, E.; Hack, C.E. Potentiation of C1 inhibitor by glycosaminoglycans: Dextran sulfate species are effective inhibitors of in vitro complement activation in plasma. J. Immunol. 1997, 159, 1953–1960. [Google Scholar]
- Fiorante, P.; Banz, Y.; Mohacsi, P.J.; Kappeler, A.; Wuillemin, W.A.; Macchiarini, P.; Roos, A.; Daha, M.R.; Schaffner, T.; Haeberli, A.; et al. Low molecular weight dextran sulfate prevents complement activation and delays hyperacute rejection in pig-to-human xenotransplantation models. Xenotransplantation 2001, 8, 24–35. [Google Scholar] [CrossRef]
- Johansson, H.; Goto, M.; Dufrane, D.; Siegbahn, A.; Elgue, G.; Gianello, P.; Korsgren, O.; Nilsson, B. Low molecular weight dextran sulfate: A strong candidate drug to block IBMIR in clinical islet transplantation. Am. J. Transplant. 2006, 6, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.; Magnusson, C.; Lundgren, T.; Korsgren, O.; Nilsson, B. Low molecular weight dextran sulfate is well tolerated in humans and increases endogenous expression of islet protective hepatocyte growth factor. Transplantation 2008, 86, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Von Zur-Mühlen, B.; Lundgren, T.; Bayman, L.; Berne, C.; Bridges, N.; Eggerman, T.; Foss, A.; Goldstein, J.; Jenssen, T.; Jorns, C.; et al. Open Randomized Multicenter Study to Evaluate Safety and Efficacy of Low Molecular Weight Sulfated Dextran in Islet Transplantation. Transplantation 2019, 103, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.L.; Eckstein, C.; Smyth, C.A.; Bilbao, G.; Vilatoba, M.; Ringland, S.E.; Young, C.; Thompson, J.A.; Fernández, J.A.; Griffin, J.H.; et al. Activated protein C preserves functional islet mass after intraportal transplantation: A novel link between endothelial cell activation, thrombosis, inflammation, and islet cell death. Diabetes 2004, 53, 2804–2814. [Google Scholar] [CrossRef]
- Griffin, J.H.; Zlokovic, B.; Fernández, J.A. Activated protein C: Potential therapy for severe sepsis, thrombosis, and stroke. Semin. Hematol. 2002, 39, 197–205. [Google Scholar] [CrossRef]
- Joyce, D.E.; Gelbert, L.; Ciaccia, A.; DeHoff, B.; Grinnell, B.W. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J. Biol Chem. 2001, 276, 11199–11203. [Google Scholar] [CrossRef]
- White, B.; Schmidt, M.; Murphy, C.; Livingstone, W.; O’Toole, D.; Lawler, M.; O’Neill, L.; Kelleher, D.; Schwarz, H.P.; Smith, O.P. Activated protein C inhibits lipopolysaccharide-induced nuclear translocation of nuclear factor kappaB (NF-kappaB) and tumour necrosis factor alpha (TNF-alpha) production in the THP-1 monocytic cell line. Br. J. Haematol. 2000, 110, 130–134. [Google Scholar] [CrossRef]
- Grey, S.T.; Tsuchida, A.; Hau, H.; Orthner, C.L.; Salem, H.H.; Hancock, W.W. Selective inhibitory effects of the anticoagulant activated protein C on the responses of human mononuclear phagocytes to LPS, IFN-gamma, or phorbol ester. J. Immunol. 1994, 153, 3664–3672. [Google Scholar]
- Shibata, M.; Kumar, S.R.; Amar, A.; Fernandez, J.A.; Hofman, F.; Griffin, J.H.; Zlokovic, B.V. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation 2001, 103, 1799–1805. [Google Scholar] [CrossRef]
- Matsumoto, S.; Takita, M.; Chaussabel, D.; Noguchi, H.; Shimoda, M.; Sugimoto, K.; Itoh, T.; Chujo, D.; SoRelle, J.; Onaca, N.; et al. Improving efficacy of clinical islet transplantation with iodixanol-based islet purification, thymoglobulin induction, and blockage of IL-1β and TNF-α. Cell Transplant. 2011, 20, 1641–1647. [Google Scholar] [CrossRef]
- Noguchi, H.; Nakai, Y.; Ueda, M.; Masui, Y.; Futaki, S.; Kobayashi, N.; Hayashi, S.; Matsumoto, S. Activation of c-Jun NH2-terminal kinase (JNK) pathway during islet transplantation and prevention of islet graft loss by intraportal injection of JNK inhibitor. Diabetologia 2007, 50, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A. Therapeutic promise of JNK ATP-noncompetitive inhibitors. Trends Mol. Med. 2005, 11, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Messoussi, A.; Feneyrolles, C.; Bros, A.; Deroide, A.; Daydé-Cazals, B.; Chevé, G.; Van Hijfte, N.; Fauvel, B.; Bougrin, K.; Yasri, A. Recent progress in the design, study, and development of c-Jun N-terminal kinase inhibitors as anticancer agents. Chem. Biol. 2014, 21, 1433–1443. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Ngoei, K.R.; Zhao, T.T.; Yeap, Y.Y.; Ng, D.C. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta 2010, 1804, 463–475. [Google Scholar] [CrossRef]
- Noguchi, H.; Matsushita, M.; Okitsu, T.; Moriwaki, A.; Tomizawa, K.; Kang, S.; Li, S.T.; Kobayashi, N.; Matsumoto, S.; Tanaka, K.; et al. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat. Med. 2004, 10, 305–309. [Google Scholar] [CrossRef]
- Noguchi, H.; Sugimoto, K.; Miyagi-Shiohira, C.; Nakashima, Y.; Kobayashi, N.; Saitoh, I.; Watanabe, M.; Noguchi, Y. RCAN-11R peptide provides immunosuppression for fully mismatched islet allografts in mice. Sci. Rep. 2017, 7, 3043. [Google Scholar] [CrossRef]
- Noguchi, H.; Matsushita, M.; Kobayashi, N.; Levy, M.F.; Matsumoto, S. Recent advances in protein transduction technology. Cell Transplant. 2010, 19, 649–654. [Google Scholar] [CrossRef]
- Matsushita, M.; Tomizawa, K.; Moriwaki, A.; Li, S.T.; Terada, H.; Matsui, H. A high-efficiency protein transduction system demonstrating the role of PKA in long-lasting long-term potentiation. J. Neurosci. 2001, 21, 6000–6007. [Google Scholar] [CrossRef]
- Noguchi, H.; Nakai, Y.; Matsumoto, S.; Kawaguchi, M.; Ueda, M.; Okitsu, T.; Iwanaga, Y.; Yonekawa, Y.; Nagata, H.; Minami, K.; et al. Cell permeable peptide of JNK inhibitor prevents islet apoptosis immediately after isolation and improves islet graft function. Am. J. Transplant. 2005, 5, 1848–1855. [Google Scholar] [CrossRef]
- Fornoni, A.; Pileggi, A.; Molano, R.D.; Sanabria, N.Y.; Tejada, T.; Gonzalez-Quintana, J.; Ichii, H.; Inverardi, L.; Ricordi, C.; Pastori, R.L. Inhibition of c-jun N terminal kinase (JNK) improves functional beta cell mass in human islets and leads to AKT and glycogen synthase kinase-3 (GSK-3) phosphorylation. Diabetologia 2008, 51, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Miyagi-Shiohira, C.; Nakashima, Y.; Ebi, N.; Hamada, E.; Tamaki, Y.; Kuwae, K.; Kobayashi, N.; Saitoh, I.; Watanabe, M. Modified cell-permeable JNK inhibitors efficiently prevents islet apoptosis and improves the outcome of islet transplantation. Sci. Rep. 2018, 8, 11082. [Google Scholar] [CrossRef] [PubMed]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Ao, Z.; Kieffer, T.J.; Chen, H.; Safikhan, N.; Thompson, D.M.; Meloche, M.; Warnock, G.L.; Marzban, L. The glucagon-like peptide-1 receptor agonist exenatide restores impaired pro-islet amyloid polypeptide processing in cultured human islets: Implications in type 2 diabetes and islet transplantation. Diabetologia 2013, 56, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Z.; Gou, W.; Adams, D.B.; Cui, W.; Morgan, K.A.; Strange, C.; Wang, H. α-1 Antitrypsin Enhances Islet Engraftment by Suppression of Instant Blood-Mediated Inflammatory Reaction. Diabetes 2017, 66, 970–980. [Google Scholar] [CrossRef]
- Nardelli, T.R.; Vanzela, E.C.; Benedicto, K.C.; Brozzi, F.; Fujita, A.; Cardozo, A.K.; Eizirik, D.L.; Boschero, A.C.; Ortis, F. Prolactin protects against cytokine-induced beta-cell death by NFκB and JNK inhibition. J. Mol. Endocrinol. 2018, 61, 25–36. [Google Scholar] [CrossRef]
- Wang, J.; Gou, W.; Kim, D.S.; Strange, C.; Wang, H. Clathrin-mediated Endocytosis of Alpha-1 Antitrypsin is Essential for its Protective Function in Islet Cell Survival. Theranostics 2019, 9, 3940–3951. [Google Scholar] [CrossRef]
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018. [Google Scholar] [CrossRef]
- Kawamori, D.; Kajimoto, Y.; Kaneto, H.; Umayahara, Y.; Fujitani, Y.; Miyatsuka, T.; Watada, H.; Leibiger, I.B.; Yamasaki, Y.; Hori, M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes 2003, 52, 2896–2904. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.M.; Davis, R.J. Targeting JNK for therapeutic benefit: From junk to gold? Nat. Rev. Drug Discov. 2003, 2, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Omori, K.; Vuong, T.; Pascual, M.; Valiente, L.; Ferreri, K.; Todorov, I.; Kuroda, Y.; Smith, C.V.; Kandeel, F.; et al. Inhibition of p38 pathway suppresses human islet production of pro-inflammatory cytokines and improves islet graft function. Am. J. Transplant. 2005, 5, 484–493. [Google Scholar] [CrossRef]
- Ito, T.; Omori, K.; Rawson, J.; Todorov, I.; Asari, S.; Kuroda, A.; Shintaku, J.; Itakura, S.; Ferreri, K.; Kandeel, F.; et al. Improvement of canine islet yield by donor pancreas infusion with a p38MAPK inhibitor. Transplantation 2008, 86, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Todorov, I.; Shintaku, J.; Rawson, J.; Al-Abdullah, I.H.; Higgins, L.S.; Medicherla, S.; Kandeel, F.; Mullen, Y. P38alpha-selective mitogen-activated protein kinase inhibitor for improvement of cultured human islet recovery. Pancreas 2010, 39, 436–443. [Google Scholar] [CrossRef]
- Collier, J.J.; Fueger, P.T.; Hohmeier, H.E.; Newgard, C.B. Pro- and antiapoptotic proteins regulate apoptosis but do not protect against cytokine-mediated cytotoxicity in rat islets and beta-cell lines. Diabetes 2006, 55, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Steer, S.A.; Scarim, A.L.; Chambers, K.T.; Corbett, J.A. Interleukin-1 stimulates beta-cell necrosis and release of the immunological adjuvant HMGB1. PLoS Med. 2006, 3, e17. [Google Scholar]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).