IL-1β and Statin Treatment in Patients with Myocardial Infarction and Diabetic Cardiomyopathy




Abstract
1. Introduction
2. Statins in Myocardial Infarction and Diabetic Cardiomyopathy
2.1. Myocardial Infarction
2.2. Diabetic Cardiomyopathy
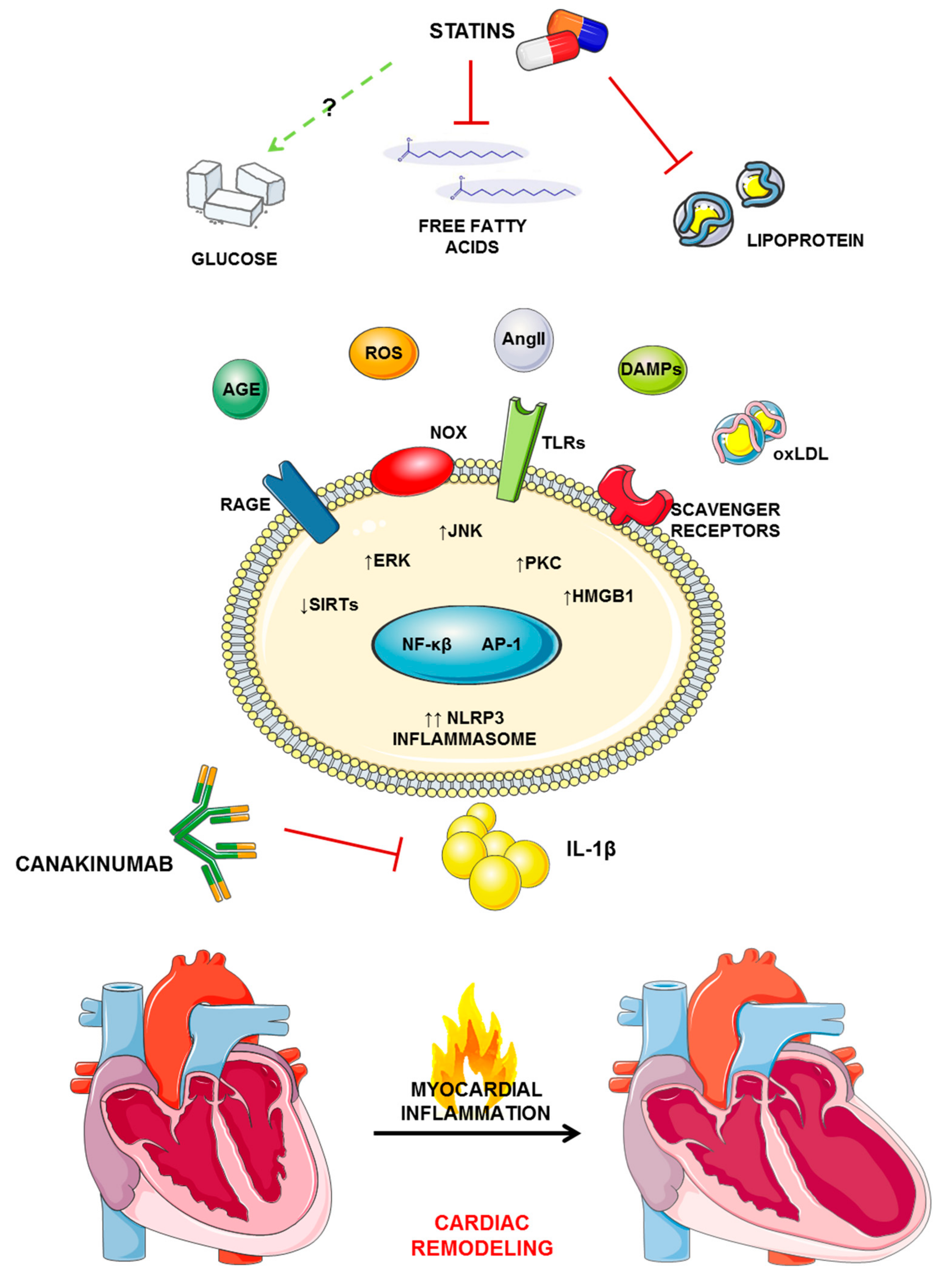
3. Statins, Inflammation, and IL-1β
4. Perspective
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Endo, A.; Kuroda, M. Citrinin, an inhibitor of cholesterol synthesis. J. Antibiot. (Tokyo) 1976, 29, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of hmg-coa reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.; Wun, C.C.; et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Long-Term Intervention with Pravastatin in Ischaemic Disease Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N. Engl. J. Med. 1998, 339, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Schonbeck, U.; Libby, P. Inflammation, immunity, and hmg-coa reductase inhibitors: Statins as antiinflammatory agents? Circulation 2004, 109, 18–26. [Google Scholar] [CrossRef]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef]
- Liberale, L.; Montecucco, F.; Camici, G.G.; Dallegri, F.; Vecchie, A.; Carbone, F.; Bonaventura, A. Treatment with proprotein convertase subtilisin/kexin type 9 (pcsk9) inhibitors to reduce cardiovascular inflammation and outcomes. Curr. Med. Chem. 2017, 24, 1403–1416. [Google Scholar] [CrossRef]
- Carbone, F.; Liberale, L.; Bonaventura, A.; Cea, M.; Montecucco, F. Targeting inflammation in primary cardiovascular prevention. Curr. Pharm. Des. 2016, 22, 5662–5675. [Google Scholar] [CrossRef]
- Montecucco, F.; Liberale, L.; Bonaventura, A.; Vecchie, A.; Dallegri, F.; Carbone, F. The role of inflammation in cardiovascular outcome. Curr. Atheroscler. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Bonaventura, A.; Montecucco, F.; Dallegri, F.; Carbone, F.; Luscher, T.F.; Camici, G.G.; Liberale, L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 2019, 115, 1266–1285. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Camici, G.G. The role of vascular aging in atherosclerotic plaque development and vulnerability. Curr. Pharm. Des. 2019. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Bonaventura, A.; Liberale, L.; Paolino, S.; Torre, F.; Dallegri, F.; Montecucco, F.; Cutolo, M. Atherosclerosis in rheumatoid arthritis: Promoters and opponents. Clin. Rev. Allergy Immunol. 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-dose methotrexate for the prevention of atherosclerotic events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Dendukuri, N.; Rich, B.; Nadeau, L.; Helin-Salmivaara, A.; Garbe, E.; Brophy, J.M. Risk of acute myocardial infarction with nsaids in real world use: Bayesian meta-analysis of individual patient data. BMJ 2017, 357, 1909. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Baylis, R.A.; Gomez, D.; Mallat, Z.; Pasterkamp, G.; Owens, G.K. The cantos trial: One important step for clinical cardiology but a giant leap for vascular biology. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 174–177. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D.; Group E.S.C.S.D. Fourth universal definition of myocardial infarction (2018). Eur. Heart J. 2019, 40, 237–269. [Google Scholar] [CrossRef]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 esc/eas guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2019. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 european guidelines on cardiovascular disease prevention in clinical practice: The sixth joint task force of the european society of cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of 10 societies and by invited experts)developed with the special contribution of the european association for cardiovascular prevention & rehabilitation (eacpr). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 aha/acc/aacvpr/aapa/abc/acpm/ada/ags/apha/aspc/nla/pcna guideline on the management of blood cholesterol: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 2019, 73, 285–350. [Google Scholar] [CrossRef] [PubMed]
- Byrne, P.; Cullinan, J.; Murphy, C.; Smith, S.M. Cross-sectional analysis of the prevalence and predictors of statin utilisation in ireland with a focus on primary prevention of cardiovascular disease. BMJ Open 2018, 8, 18524. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.L.; Bekelman, J.E.; Liu, Y.; Bach, P.B.; Basch, E.M.; Elkin, E.B.; Zelefsky, M.J.; Scardino, P.T.; Begg, C.B.; Schrag, D. Physician visits prior to treatment for clinically localized prostate cancer. Arch. Intern. Med. 2010, 170, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Petretta, M.; Costanzo, P.; Perrone-Filardi, P.; Chiariello, M. Impact of gender in primary prevention of coronary heart disease with statin therapy: A meta-analysis. Int. J. Cardiol. 2010, 138, 25–31. [Google Scholar] [CrossRef]
- Brugts, J.J.; Yetgin, T.; Hoeks, S.E.; Gotto, A.M.; Shepherd, J.; Westendorp, R.G.; de Craen, A.J.; Knopp, R.H.; Nakamura, H.; Ridker, P.; et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: Meta-analysis of randomised controlled trials. BMJ 2009, 338, 2376. [Google Scholar] [CrossRef]
- de Vries, F.M.; Denig, P.; Pouwels, K.B.; Postma, M.J.; Hak, E. Primary prevention of major cardiovascular and cerebrovascular events with statins in diabetic patients: A meta-analysis. Drugs 2012, 72, 2365–2373. [Google Scholar] [CrossRef]
- Byrne, P.; Cullinan, J.; Smith, A.; Smith, S.M. Statins for the primary prevention of cardiovascular disease: An overview of systematic reviews. BMJ Open 2019, 9, 23085. [Google Scholar] [CrossRef]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The scandinavian simvastatin survival study (4s). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M.; et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Rodriguez, F.; Maron, D.J.; Knowles, J.W.; Virani, S.S.; Lin, S.; Heidenreich, P.A. Association between intensity of statin therapy and mortality in patients with atherosclerotic cardiovascular disease. JAMA Cardiol. 2017, 2, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.; Bowman, L.; Wallendszus, K.; Bulbulia, R.; Rahimi, K.; Haynes, R.; Parish, S.; Peto, R.; Collins, R. Intensive lowering of ldl cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: A double-blind randomised trial. Lancet 2010, 376, 1658–1669. [Google Scholar] [PubMed]
- Ference, B.A.; Majeed, F.; Penumetcha, R.; Flack, J.M.; Brook, R.D. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in npc1l1, hmgcr, or both: A 2 × 2 factorial mendelian randomization study. J. Am. Coll. Cardiol. 2015, 65, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Holscher, M.E.; Bode, C.; Bugger, H. Diabetic cardiomyopathy: Does the type of diabetes matter? Int. J. Mol. Sci. 2016, 17, 2136. [Google Scholar] [CrossRef]
- Zarich, S.W.; Arbuckle, B.E.; Cohen, L.R.; Roberts, M.; Nesto, R.W. Diastolic abnormalities in young asymptomatic diabetic patients assessed by pulsed doppler echocardiography. J. Am. Coll. Cardiol. 1988, 12, 114–120. [Google Scholar] [CrossRef]
- Palmieri, V.; Bella, J.N.; Arnett, D.K.; Liu, J.E.; Oberman, A.; Schuck, M.Y.; Kitzman, D.W.; Hopkins, P.N.; Morgan, D.; Rao, D.C.; et al. Effect of type 2 diabetes mellitus on left ventricular geometry and systolic function in hypertensive subjects: Hypertension genetic epidemiology network (hypergen) study. Circulation 2001, 103, 102–107. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- Frati, G.; Schirone, L.; Chimenti, I.; Yee, D.; Biondi-Zoccai, G.; Volpe, M.; Sciarretta, S. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc. Res. 2017, 113, 378–388. [Google Scholar] [CrossRef]
- Thomas, C.M.; Yong, Q.C.; Rosa, R.M.; Seqqat, R.; Gopal, S.; Casarini, D.E.; Jones, W.K.; Gupta, S.; Baker, K.M.; Kumar, R. Cardiac-specific suppression of nf-kappab signaling prevents diabetic cardiomyopathy via inhibition of the renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. Nf-kappab-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type ii diabetes. Cardiovasc. Res. 2010, 85, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Antras, J.; Picatoste, B.; Ramirez, E.; Egido, J.; Tunon, J.; Lorenzo, O. Targeting metabolic disturbance in the diabetic heart. Cardiovasc. Diabetol. 2015, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Akhmedov, A.; Melina, G.; Mohammed, S.A.; Othman, A.; Ambrosini, S.; Wijnen, W.J.; Sada, L.; Ciavarella, G.M.; Liberale, L.; et al. Obesity-induced activation of jund promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur. Heart J. 2019, 40, 997–1008. [Google Scholar] [CrossRef]
- Van Linthout, S.; Riad, A.; Dhayat, N.; Spillmann, F.; Du, J.; Dhayat, S.; Westermann, D.; Hilfiker-Kleiner, D.; Noutsias, M.; Laufs, U.; et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia 2007, 50, 1977–1986. [Google Scholar] [CrossRef]
- Carillion, A.; Feldman, S.; Na, N.; Biais, M.; Carpentier, W.; Birenbaum, A.; Cagnard, N.; Loyer, X.; Bonnefont-Rousselot, D.; Hatem, S.; et al. Atorvastatin reduces beta-adrenergic dysfunction in rats with diabetic cardiomyopathy. PLoS ONE 2017, 12, 180103. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, Y.; Zhang, M.; An, F. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting nlrp3 inflammasome and mapk pathways in a type 2 diabetes rat model. Cardiovasc. Drugs Ther. 2014, 28, 33–43. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. Mrc/bhf heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.; Livingstone, S.J.; Thomason, M.J.; Mackness, M.I.; Charlton-Menys, V.; Fuller, J.H.; et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the collaborative atorvastatin diabetes study (cards): Multicentre randomised placebo-controlled trial. Lancet 2004, 364, 685–696. [Google Scholar] [CrossRef]
- Gaede, P.; Lund-Andersen, H.; Parving, H.H.; Pedersen, O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 2008, 358, 580–591. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 esc guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the easd. Eur. Heart J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Paseban, M.; Butler, A.E.; Sahebkar, A. Mechanisms of statin-induced new-onset diabetes. J. Cell. Physiol. 2019, 234, 12551–12561. [Google Scholar] [CrossRef] [PubMed]
- Barylski, M.; Nikolic, D.; Banach, M.; Toth, P.P.; Montalto, G.; Rizzo, M. Statins and new-onset diabetes. Curr. Pharm. Des. 2014, 20, 3657–3664. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- de Lemos, J.A.; Blazing, M.A.; Wiviott, S.D.; Lewis, E.F.; Fox, K.A.; White, H.D.; Rouleau, J.L.; Pedersen, T.R.; Gardner, L.H.; Mukherjee, R.; et al. Early intensive vs. a delayed conservative simvastatin strategy in patients with acute coronary syndromes: Phase z of the a to z trial. JAMA 2004, 292, 1307–1316. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- Liu, L.; Moesner, P.; Kovach, N.L.; Bailey, R.; Hamilton, A.D.; Sebti, S.M.; Harlan, J.M. Integrin-dependent leukocyte adhesion involves geranylgeranylated protein(s). J. Biol. Chem. 1999, 274, 33334–33340. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Tupper, J.C.; Bannerman, D.D.; Winn, R.K.; Sebti, S.M.; Hamilton, A.D.; Harlan, J.M. Inhibition of protein geranylgeranylation and rhoa/rhoa kinase pathway induces apoptosis in human endothelial cells. J. Biol. Chem. 2002, 277, 15309–15316. [Google Scholar] [CrossRef]
- Rasmussen, L.M.; Hansen, P.R.; Nabipour, M.T.; Olesen, P.; Kristiansen, M.T.; Ledet, T. Diverse effects of inhibition of 3-hydroxy-3-methylglutaryl-coa reductase on the expression of vcam-1 and e-selectin in endothelial cells. Biochem. J. 2001, 360, 363–370. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating rho gtpases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. Ras proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Nimnual, A.S.; Taylor, L.J.; Bar-Sagi, D. Redox-dependent downregulation of rho by rac. Nat. Cell Biol. 2003, 5, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 7432–7437. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.C.; Asiimwe, E.; Birkenstamm, J.R.; Kim, S.Y.; Campbell, P.M. Analyzing ras-associated cell proliferation signaling. Methods Mol. Biol. 2014, 1170, 393–409. [Google Scholar] [PubMed]
- Treasure, C.B.; Klein, J.L.; Weintraub, W.S.; Talley, J.D.; Stillabower, M.E.; Kosinski, A.S.; Zhang, J.; Boccuzzi, S.J.; Cedarholm, J.C.; Alexander, R.W. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N. Engl. J. Med. 1995, 332, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J.; Meredith, I.T.; Yeung, A.C.; Frei, B.; Selwyn, A.P.; Ganz, P. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N. Engl. J. Med. 1995, 332, 488–493. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Greer, J.J.; Cameron, S.J.; Matsushita, K.; Morrell, C.N.; Talbot-Fox, K.; Baldwin, W.M., 3rd; Lefer, D.J.; Lowenstein, C.J. Hmg-coa reductase inhibitors inhibit endothelial exocytosis and decrease myocardial infarct size. Circ. Res. 2005, 96, 1185–1192. [Google Scholar] [CrossRef]
- Meda, C.; Plank, C.; Mykhaylyk, O.; Schmidt, K.; Mayer, B. Effects of statins on nitric oxide/cgmp signaling in human umbilical vein endothelial cells. Pharmacol. Rep. 2010, 62, 100–112. [Google Scholar] [CrossRef]
- Dichtl, W.; Dulak, J.; Frick, M.; Alber, H.F.; Schwarzacher, S.P.; Ares, M.P.; Nilsson, J.; Pachinger, O.; Weidinger, F. Hmg-coa reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 58–63. [Google Scholar] [CrossRef]
- Crisby, M.; Nordin-Fredriksson, G.; Shah, P.K.; Yano, J.; Zhu, J.; Nilsson, J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: Implications for plaque stabilization. Circulation 2001, 103, 926–933. [Google Scholar] [CrossRef]
- Aikawa, M.; Rabkin, E.; Sugiyama, S.; Voglic, S.J.; Fukumoto, Y.; Furukawa, Y.; Shiomi, M.; Schoen, F.J.; Libby, P. An hmg-coa reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001, 103, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Xenos, E.S.; Stevens, S.L.; Freeman, M.B.; Cassada, D.C.; Goldman, M.H. Nitric oxide mediates the effect of fluvastatin on intercellular adhesion molecule-1 and platelet endothelial cell adhesion molecule-1 expression on human endothelial cells. Ann. Vasc. Surg. 2005, 19, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ikeda, U.; Yamamoto, K.; Shimada, K. Regulation of interleukin-8 expression by hmg-coa reductase inhibitors in human vascular smooth muscle cells. Atherosclerosis 2002, 165, 51–55. [Google Scholar] [CrossRef]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat. Med. 2001, 7, 687–692. [Google Scholar] [CrossRef]
- Simon, D.I.; Dhen, Z.; Seifert, P.; Edelman, E.R.; Ballantyne, C.M.; Rogers, C. Decreased neointimal formation in mac-1(-/-) mice reveals a role for inflammation in vascular repair after angioplasty. J. Clin. Investig. 2000, 105, 293–300. [Google Scholar] [CrossRef]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [CrossRef]
- Hillyard, D.Z.; Cameron, A.J.; McDonald, K.J.; Thomson, J.; MacIntyre, A.; Shiels, P.G.; Panarelli, M.; Jardine, A.G. Simvastatin inhibits lymphocyte function in normal subjects and patients with cardiovascular disease. Atherosclerosis 2004, 175, 305–313. [Google Scholar] [CrossRef]
- Henriksbo, B.D.; Schertzer, J.D. Is immunity a mechanism contributing to statin-induced diabetes? Adipocyte 2015, 4, 232–238. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of cantos and beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The nlrp3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: Nf-kappab activating pattern recognition and cytokine receptors license nlrp3 inflammasome activation by regulating nlrp3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Nunez, G. Cutting edge: Tnf-alpha mediates sensitization to atp and silica via the nlrp3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Yao, X.; Li, H.; Xue, G.; Guo, Q.; Yang, G.; An, L.; Zhang, Y.; Meng, G. Cutting edge: Traf6 mediates tlr/il-1r signaling-induced nontranscriptional priming of the nlrp3 inflammasome. J. Immunol. 2017, 199, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. Nlrp3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate nlrp3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. Clics-dependent chloride efflux is an essential and proximal upstream event for nlrp3 inflammasome activation. Nat. Commun. 2017, 8, 202. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the nalp3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of nlrp3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, W.; Lu, C.; Ma, Z.; Jiang, S.; Gu, C.; Acuna-Castroviejo, D.; Yang, Y. Targeting nlrp3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome in cardiovascular disorders. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2765–2779. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of nlrp3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.G.; Bonaventura, A.; Abbate, A. Drugs to inhibit the nlrp3 inflammasome: Not always on target. J. Cardiovasc. Pharmacol. 2019, 74, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Marette, A. Statin-induced insulin resistance through inflammasome activation: Sailing between scylla and charybdis. Diabetes 2014, 63, 3569–3571. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Banach, M.; Malodobra-Mazur, M.; Gluba, A.; Katsiki, N.; Rysz, J.; Dobrzyn, A. Statin therapy and new-onset diabetes: Molecular mechanisms and clinical relevance. Curr. Pharm. Des. 2013, 19, 4904–4912. [Google Scholar] [CrossRef] [PubMed]
- Henriksbo, B.D.; Lau, T.C.; Cavallari, J.F.; Denou, E.; Chi, W.; Lally, J.S.; Crane, J.D.; Duggan, B.M.; Foley, K.P.; Fullerton, M.D.; et al. Fluvastatin causes nlrp3 inflammasome-mediated adipose insulin resistance. Diabetes 2014, 63, 3742–3747. [Google Scholar] [CrossRef]
- Massonnet, B.; Normand, S.; Moschitz, R.; Delwail, A.; Favot, L.; Garcia, M.; Bourmeyster, N.; Cuisset, L.; Grateau, G.; Morel, F.; et al. Pharmacological inhibitors of the mevalonate pathway activate pro-il-1 processing and il-1 release by human monocytes. Eur. Cytokine Netw. 2009, 20, 112–120. [Google Scholar] [CrossRef]
- Montero, M.T.; Hernandez, O.; Suarez, Y.; Matilla, J.; Ferruelo, A.J.; Martinez-Botas, J.; Gomez-Coronado, D.; Lasuncion, M.A. Hydroxymethylglutaryl-coenzyme a reductase inhibition stimulates caspase-1 activity and th1-cytokine release in peripheral blood mononuclear cells. Atherosclerosis 2000, 153, 303–313. [Google Scholar] [CrossRef]
- Liao, Y.H.; Lin, Y.C.; Tsao, S.T.; Lin, Y.C.; Yang, A.J.; Huang, C.T.; Huang, K.C.; Lin, W.W. Hmg-coa reductase inhibitors activate caspase-1 in human monocytes depending on atp release and p2 × 7 activation. J. Leukoc. Biol. 2013, 93, 289–299. [Google Scholar] [CrossRef]
- Frenkel, J.; Rijkers, G.T.; Mandey, S.H.; Buurman, S.W.; Houten, S.M.; Wanders, R.J.; Waterham, H.R.; Kuis, W. Lack of isoprenoid products raises ex vivo interleukin-1beta secretion in hyperimmunoglobulinemia d and periodic fever syndrome. Arthritis Rheum. 2002, 46, 2794–2803. [Google Scholar] [CrossRef]
- Henriksbo, B.D.; Tamrakar, A.K.; Xu, J.; Duggan, B.M.; Cavallari, J.F.; Phulka, J.; Stampfli, M.R.; Ashkar, A.A.; Schertzer, J.D. Statins promote interleukin-1beta-dependent adipocyte insulin resistance through lower prenylation, not cholesterol. Diabetes 2019, 68, 1441–1448. [Google Scholar] [CrossRef]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchie, A.; Diaz-Canestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The pathophysiological role of neutrophil extracellular traps in inflammatory diseases. Thromb. Haemost. 2018, 118, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Dallegri, F.; Montecucco, F.; Carbone, F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb. Haemost. 2017, 117, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Bertolotto, M.; Carbone, F.; Contini, P.; Wust, P.; Spinella, G.; Pane, B.; Palombo, D.; Bonaventura, A.; Pende, A.; et al. Resistin exerts a beneficial role in atherosclerotic plaque inflammation by inhibiting neutrophil migration. Int. J. Cardiol. 2018, 272, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Rigamonti, F.; Burger, F.; Roth, A.; Bertolotto, M.; Spinella, G.; Pane, B.; Palombo, D.; Pende, A.; Bonaventura, A.; et al. Serum levels of osteopontin predict major adverse cardiovascular events in patients with severe carotid artery stenosis. Int. J. Cardiol. 2018, 255, 195–199. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Group, C.T. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the cantos randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the nlrp3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, 11765. [Google Scholar] [CrossRef]
- Warner, S.J.; Auger, K.R.; Libby, P. Interleukin 1 induces interleukin 1. Ii. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J. Immunol. 1987, 139, 1911–1917. [Google Scholar]
- Liberale, L.; Bonaventura, A.; Vecchie, A.; Casula, M.; Dallegri, F.; Montecucco, F.; Carbone, F. The role of adipocytokines in coronary atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 10. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Hartman, M.H.T.; Groot, H.E.; Leach, I.M.; Karper, J.C.; van der Harst, P. Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure. Trends Cardiovasc. Med. 2018, 28, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Salloum, F.N.; Vecile, E.; Das, A.; Hoke, N.N.; Straino, S.; Biondi-Zoccai, G.G.; Houser, J.E.; Qureshi, I.Z.; Ownby, E.D.; et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 2008, 117, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Varma, A.; Salloum, F.N.; Das, A.; Seropian, I.M.; Toldo, S.; Smithson, L.; Hoke, N.N.; Chau, V.Q.; Robati, R.; et al. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J. Cardiovasc. Pharmacol. 2010, 55, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Schatz, A.M.; Mezzaroma, E.; Chawla, R.; Stallard, T.W.; Stallard, W.C.; Jahangiri, A.; Van Tassell, B.W.; Abbate, A. Recombinant human interleukin-1 receptor antagonist provides cardioprotection during myocardial ischemia reperfusion in the mouse. Cardiovasc. Drugs Ther. 2012, 26, 273–276. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Van Tassell, B.W.; Farkas, D.; Marchetti, C.; Voelkel, N.F.; Abbate, A. Interleukin-1beta blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp. Physiol. 2013, 98, 734–745. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Bressi, E.; Marchetti, C.; Carbone, S.; Sonnino, C.; Van Tassell, B.W.; Abbate, A. Interleukin-1beta blockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J. Cardiovasc. Pharmacol. 2014, 64, 1–6. [Google Scholar] [CrossRef]
- De Jesus, N.M.; Wang, L.; Lai, J.; Rigor, R.R.; Francis Stuart, S.D.; Bers, D.M.; Lindsey, M.L.; Ripplinger, C.M. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm 2017, 14, 727–736. [Google Scholar] [CrossRef]
- Mauro, A.G.; Mezzaroma, E.; Torrado, J.; Kundur, P.; Joshi, P.; Stroud, K.; Quaini, F.; Lagrasta, C.A.; Abbate, A.; Toldo, S. Reduction of myocardial ischemia-reperfusion injury by inhibiting interleukin-1 alpha. J. Cardiovasc. Pharmacol. 2017, 69, 156–160. [Google Scholar] [CrossRef]
- Harouki, N.; Nicol, L.; Remy-Jouet, I.; Henry, J.P.; Dumesnil, A.; Lejeune, A.; Renet, S.; Golding, F.; Djerada, Z.; Wecker, D.; et al. The il-1beta antibody gevokizumab limits cardiac remodeling and coronary dysfunction in rats with heart failure. JACC Basic Transl. Sci. 2017, 2, 418–430. [Google Scholar] [CrossRef]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the virginia commonwealth university-anakinra remodeling trial (2) (vcu-art2) pilot study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Oddi Erdle, C.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 blockade in recently decompensated systolic heart failure: Results from redhart (recently decompensated heart failure anakinra response trial). Circ. Heart Fail. 2017, 10, 4373. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Arena, R.; Biondi-Zoccai, G.; Canada, J.M.; Oddi, C.; Abouzaki, N.A.; Jahangiri, A.; Falcao, R.A.; Kontos, M.C.; Shah, K.B.; et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the d-hart pilot study). Am. J. Cardiol. 2014, 113, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Kontos, M.C.; Abouzaki, N.A.; Melchior, R.D.; Thomas, C.; Van Tassell, B.W.; Oddi, C.; Carbone, S.; Trankle, C.R.; Roberts, C.S.; et al. Comparative safety of interleukin-1 blockade with anakinra in patients with st-segment elevation acute myocardial infarction (from the vcu-art and vcu-art2 pilot studies). Am. J. Cardiol. 2015, 115, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Pedersen, T.R.; Park, J.G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; Toth, K.; Gouni-Berthold, I.; Lopez-Miranda, J.; Schiele, F.; et al. Clinical efficacy and safety of achieving very low ldl-cholesterol concentrations with the pcsk9 inhibitor evolocumab: A prespecified secondary analysis of the fourier trial. Lancet 2017, 390, 1962–1971. [Google Scholar] [CrossRef]
- Peiro, C.; Lorenzo, O.; Carraro, R.; Sanchez-Ferrer, C.F. Il-1beta inhibition in cardiovascular complications associated to diabetes mellitus. Front. Pharmacol. 2017, 8, 363. [Google Scholar] [CrossRef]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (virginia commonwealth university anakinra remodeling trial [vcu-art] pilot study). Am. J. Cardiol. 2010, 105, 1371–1377. [Google Scholar] [CrossRef]
- Morton, A.C.; Rothman, A.M.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-st elevation acute coronary syndromes: The mrc-ila heart study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Lipinski, M.J.; Appleton, D.; Roberts, C.S.; Kontos, M.C.; Abouzaki, N.; Melchior, R.; Mueller, G.; Garnett, J.; Canada, J.; et al. Rationale and design of the virginia commonwealth university-anakinra remodeling trial-3 (vcu-art3): A randomized, placebo-controlled, double-blinded, multicenter study. Clin. Cardiol. 2018, 41, 1004–1008. [Google Scholar] [CrossRef]


{kind=link}
| Author | Year | Drug(dose) | Schedule | Results |
|---|---|---|---|---|
| Abbate A. et al. [113] | 2008 | Anakinra (1 mg/kg) | Immediate or delayed (24 h after ischemia) and then daily for 6 days. | Anakinra-treated mice showed signs of more favorable ventricular remodeling. |
| Van Tassell et al. [114] | 2010 | IL-1 Trap (1, 5 or 30 mg/kg) | Every 48 h after surgery. | Mice treated with 5 or 30 mg/kg of IL-1 Trap had more favorable cardiac remodeling and echocardiographic assessment of infarct size at 7 days. |
| Toldo et al. [115] | 2012 | rhIL-1Ra | 10 mg/kg given either 30 min or 4 h prior to surgery | Irrespective of dose, treated mice showed marked cardio-protection in terms of LVEF and the reduction of the infarct size. |
| Toldo et al. [116] | 2013 | Anti-IL-1β Ab | 10 mg/kg immediately after surgery and then 1 week later. | When compared with control vehicle, anti-IL-1β Ab limit left ventricular enlargement and improve systolic dysfunction by inhibiting cardiomyocyte apoptosis. |
| Toldo et al. [117] | 2014 | Anti-IL-1β Ab | 10 mg/kg 1 week after surgery and then weekly for 9 weeks. | After 10 weeks, anti-IL-1β Ab prevents reduction of LVEF, impairment in the myocardial performance index. and contractile reserve. |
| De Jesus et al. [118] | 2017 | Anakinra (10 mg/kg) | Daily, starting 24 h after surgery | Anakinra improved conduction velocity and reduced action potential duration dispersion, thus determining a reduction of spontaneous and inducible ventricular arrhythmias. |
| Mauro et al. [119] | 2017 | IL-1α-blocking antibody (15 μg/kg) | Single dose after reperfusion | At 24 h, IL-1α blockade significantly reduced inflammasome formation and infarct size, thus preserving LVFS. |
| Herouki et al. [120] | 2017 | Anti-IL-1β Ab | Single dose after reperfusion or 7 days after reperfusion | Immediate, but not delayed, administration of anti-IL-1β Ab reduces ischemia/reperfusion-related infarct size, left ventricular remodeling, and heart-failure-related coronary dysfunction. |
| Author | Year | Drug | Treatment | Disease (cohort) | Results |
|---|---|---|---|---|---|
| Abbate et al. VCU-ART [128] | 2010 | Anakinra | 100 mg/daily sc for 14 days | STEMI (n = 10) | In this pilot double blind RCT, treatment with anakinra showed to be safe and to reduce left ventricular remodeling (assessed by both echocardiography and cardiac magnetic resonance) after STEMI as compared to placebo. |
| Morton et al. MRC-ILA-HEART [129] | 2015 | Anakinra | 100 mg/daily sc for 14 days | NSTEMI (n = 182) | In this proof-of-principle double blind RCT, patients treated with anakinra showed reduced levels of hsCRP and IL-6 as compared to those receiving a placebo. |
| Abbate et al. VCU-ART2 [121] | 2013 | Anakinra | 100 mg/daily sc for 14 days | STEMI (n = 30) | In this pilot double blind RCT, treatment with anakinra could reduce hsCRP levels as compared to a placebo. Anakinra-treated patients also showed a numerically lower incidence of heart failure, although this was not statistically significant. |
| Ridker et al. CANTOS [125] | 2019 | Canakinumab | 50, 100 or 150 mg/daily sc every 3 months | STEMI (n = 10’061) | In this double blind RCT, treatment with canakinumab after STEMI was shown to dose-dependently reduce hospitalization for heart failure and the composite of hospitalization for heart failure or heart-failure-related mortality as compared to a placebo. |
| Van Tassell et al. VCU-ART3 [130] | 2019 | Anakinra | 100 mg once or twice/daily for 14 days | STEMI (n = 99) | Preliminary results of this double blind RCT were presented at the 2019 Congress of the European Society of Cardiology. Patients treated with anakinra showed significant improvement in cardiac systolic function after STEMI, as compared to a placebo. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liberale, L.; Carbone, F.; Camici, G.G.; Montecucco, F. IL-1β and Statin Treatment in Patients with Myocardial Infarction and Diabetic Cardiomyopathy. J. Clin. Med. 2019, 8, 1764. https://doi.org/10.3390/jcm8111764
Liberale L, Carbone F, Camici GG, Montecucco F. IL-1β and Statin Treatment in Patients with Myocardial Infarction and Diabetic Cardiomyopathy. Journal of Clinical Medicine. 2019; 8(11):1764. https://doi.org/10.3390/jcm8111764
Chicago/Turabian StyleLiberale, Luca, Federico Carbone, Giovanni G. Camici, and Fabrizio Montecucco. 2019. "IL-1β and Statin Treatment in Patients with Myocardial Infarction and Diabetic Cardiomyopathy" Journal of Clinical Medicine 8, no. 11: 1764. https://doi.org/10.3390/jcm8111764
APA StyleLiberale, L., Carbone, F., Camici, G. G., & Montecucco, F. (2019). IL-1β and Statin Treatment in Patients with Myocardial Infarction and Diabetic Cardiomyopathy. Journal of Clinical Medicine, 8(11), 1764. https://doi.org/10.3390/jcm8111764