Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression


Abstract
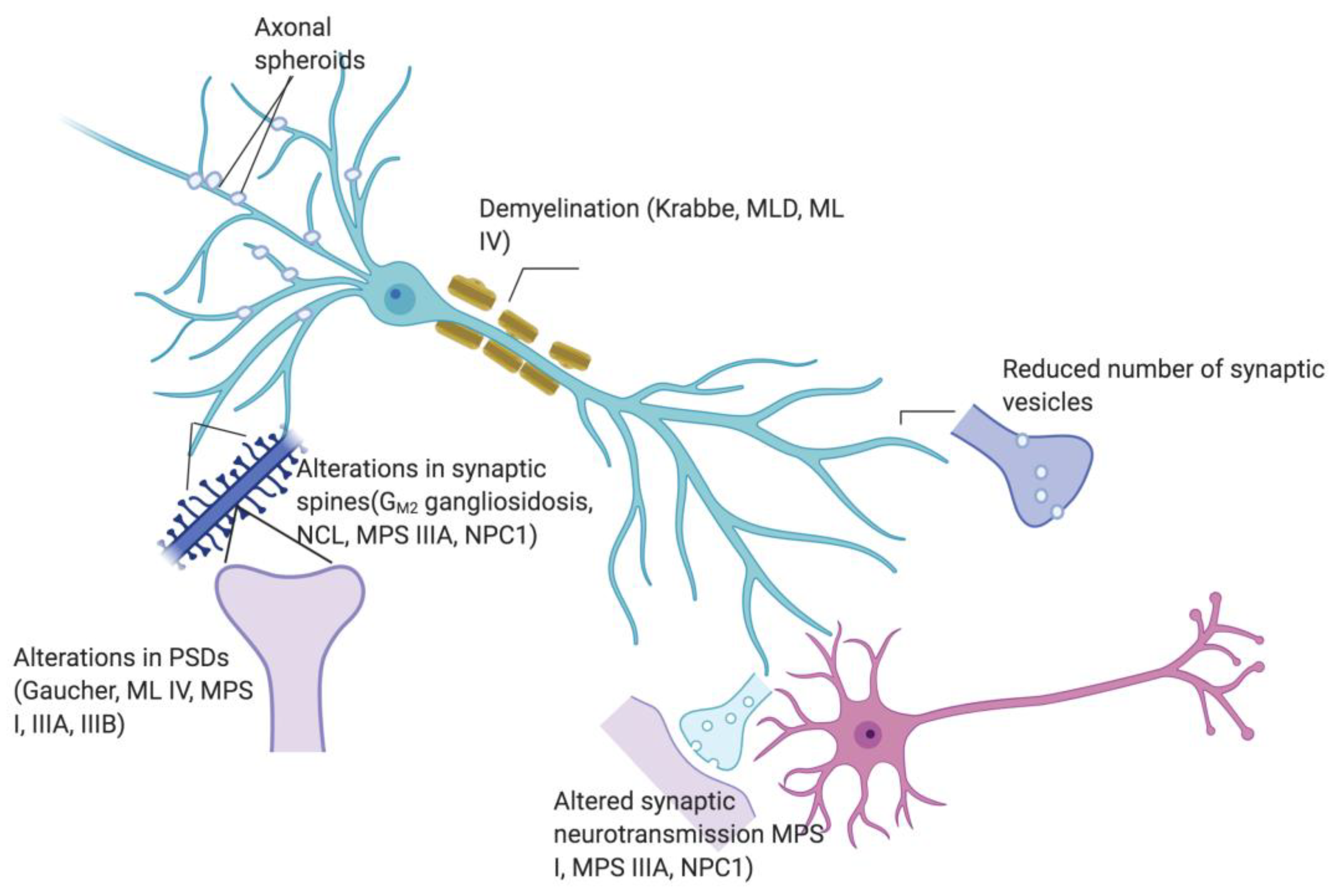
1. Introduction
2. Main Aspects of Central Nervous System (CNS) Pathology in Neurological LSD
2.1. Impairment of Endo- and Exocytosis
2.2. Axonopathy
2.3. Changes in Synaptic Proteins

{kind=link}
| LSD | Protein | Function | Change | Sample/Region | Reference |
|---|---|---|---|---|---|
| MPS I | Syntaxin-1A | SNARE complex | Reduction | Hippocampus | Baldo et al. (2015) [59] |
| Amphiphysin | Exocytosis of synaptic vesicles | Reduction | Hippocampus | ||
| Complexin-1 | Exocytosis of synaptic vesicles | Reduction | Hippocampus | ||
| Synaptophysin | Synaptic vesicle membrane protein involved in endocytosis | Reduction | Hippocampus | ||
| MAP1A | Microtubule cross-linking protein | Reduction | Hippocampus | ||
| MAP1B | Microtubule cross-linking protein | Reduction | Hippocampus | ||
| PSD-95 | Postsynaptic density protein | Increased | Hippocampus | ||
| VAMP2 | Vesicle-associated membrane protein 2 in the synaptic vesicles | Reduction | Primary motor, somatosensory and parietal areas of cerebral cortex | Wilkinson et al. (2012) [34] | |
| Homer-1 | Protein in the postsynaptic density of excitatory synapses | Reduction | Primary motor, somatosensory and parietal areas of cerebral cortex | Wilkinson et al. (2012) [34] | |
| MPS IIIA | SNAP25 | t-SNARE | Reduction | Synaptosomes | Sambri et al. (2017) [57] |
| VAMP2 | Vesicle-associated membrane protein 2 in the synaptic vesicles | Reduction | Primary motor, somatosensory and parietal areas of cerebral cortex; synaptosomes | Wilkinson et al. (2012) [34]; Sambri et al. (2017) [57] | |
| Homer-1 | Protein in the postsynaptic density of excitatory synapses | Reduction | Primary motor, somatosensory and parietal areas of cerebral cortex | Wilkinson et al. (2012) [34] | |
| PSD-95 | Postsynaptic density protein | Increased | Cortical layers I, II/III and V | Dwyer et al. (2017) [64] | |
| α-Synuclein | Presynaptic chaperone | Decreased | Cultured neurons and brain homogenates | Sambri et al. (2017) [57] | |
| CSPα | Presynaptic chaperone | Decreased | Cultured neurons and brain homogenates | ||
| MPS IIIB | Synaptophysin | Synaptic vesicle membrane protein involved in endocytosis | Reduction | Rostral cortex | Vitry et al. (2009) [84] |
| VAMP2 | Vesicle-associated membrane protein 2 in the synaptic vesicles | Reduction | Suprachiasmatic Nucleus; Primary motor, somatosensory and parietal areas of cerebral cortex | Canals et al (2010) [65]; Wilkinson et al. (2012) [34] | |
| Homer-1 | Protein in the postsynaptic density of excitatory synapses | Reduction | Primary motor, somatosensory and parietal areas of cerebral cortex | Wilkinson et al. (2012) [34] | |
| MPS VII | Synaptophysin | Synaptic vesicle membrane protein involved in endocytosis | Reduction | iPSC-derived neurospheroids | Bayo-Puxan et al. (2018) [85] |
| GAD67 | Enzyme that catalyzes the production of GABA | Reduction | iPSC-derived neurospheroids | ||
| Niemann-Pick Type C | Synaptophysin | Synaptic vesicle membrane protein involved in endocytosis | Aggregation | Striatum, substantia nigra, white matter tracts and thalamus | Pressey et al. (2012) [38] |
| VAMP2 | Vesicle-associated membrane protein 2 in the synaptic vesicles | Aggregation | Striatum, substantia nigra, white matter tracts and thalamus | ||
| Krabbe | Dynein | Retrograde transport of synaptic vesicles | Reduction | Dorsal Root Ganglia Neurons | Teixeira at al. (2014) [62] |
| Gaucher | α-synuclein | regulation of synaptic vesicle trafficking and neurotransmitter release | Accumulation | Striatum | Ginns et al. (2013) [37] |
| CLN3 | Synaptophysin | Synaptic vesicle membrane protein involved in endocytosis | Reduction | Thalamic nuclei | Hurtado et al., (2017) [83] |
| CLN5 | α-synuclein | regulation of synaptic vesicle trafficking and neurotransmitter release | Reduction | Synaptosomes | Amorim et al., (2015) [87] |
| CSP-α | Presynaptic chaperone | Reduction | Synaptosomes | ||
| α-neurofascin | Cell adhesion molecule | Reduction | Synaptosomes | ||
| CLN6 | Synaptophysin | Synaptic vesicle membrane protein involved in endocytosis | Reduction | Occipital and parietal lobes | Kanninen et al. (2013) [86] |
| Syntaxin-6 | Intracellular vesicle trafficking | Reduction | Occipital lobe |
2.4. Alterations in Generation and Recycling of Synaptic Vesicles
2.5. Defects in Synaptic Spines
2.6. Changes in Postsynaptic Density
3. Functional Synaptic Defects
4. iPSCs as An Emerging Model to Study Human Neuronal Dysfunction in LSD
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hers, H.G. alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem. J. 1963, 86, 11–16. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Kingma, S.D.; Bodamer, O.A.; Wijburg, F.A. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef]
- Filocamo, M.; Morrone, A. Lysosomal storage disorders: Molecular basis and laboratory testing. Hum. Genomics 2011, 5, 156–169. [Google Scholar] [CrossRef]
- Di Ronza, A.; Bajaj, L.; Sharma, J.; Sanagasetti, D.; Lotfi, P.; Adamski, C.J.; Collette, J.; Palmieri, M.; Amawi, A.; Popp, L.; et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. Vol. 2018, 20, 1370–1377. [Google Scholar] [CrossRef]
- Moammar, H.; Cheriyan, G.; Mathew, R.; Al-Sannaa, N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983-2008. Ann. Saudi Med. 2010, 30, 271–277. [Google Scholar] [CrossRef]
- Pastores, G.M.; Maegawa, G.H. Clinical neurogenetics: Neuropathic lysosomal storage disorders. Neurol. Clin. 2013, 31, 1051–1071. [Google Scholar] [CrossRef]
- Boustany, R.M. Lysosomal storage diseases--the horizon expands. Nat. Rev. Neurol. 2013, 9, 583–598. [Google Scholar] [CrossRef]
- Fraldi, A.; Klein, A.D.; Medina, D.L.; Settembre, C. Brain Disorders Due to Lysosomal Dysfunction. Annu. Rev. Neurosci. 2016, 39, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Jardim, L.B.; Villanueva, M.M.; de Souza, C.F.; Netto, C.B. Clinical aspects of neuropathic lysosomal storage disorders. J. Inherit Metab. Dis. 2010, 33, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Barkhuizen, M.; Anderson, D.G.; Grobler, A.F. Advances in GBA-associated Parkinson’s disease--Pathology, presentation and therapies. Neurochem. Int. 2016, 93, 6–25. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; International Parkinson’s Disease Genomics Consortium; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Li, G.; Cui, S.; Du, J.; Liu, J.; Zhang, P.; Fu, Y.; He, Y.; Zhou, H.; Ma, J.; Chen, S. Association of GALC, ZNF184, IL1R2 and ELOVL7 With Parkinson’s Disease in Southern Chinese. Front. Aging. Neurosci. 2018, 10, 402. [Google Scholar] [CrossRef]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin Med. 2020, 9. [Google Scholar] [CrossRef]
- Oberstadt, M.; Classen, J.; Arendt, T.; Holzer, M. TDP-43 and Cytoskeletal Proteins in ALS. Mol. Neurobiol. 2018, 55, 3143–3151. [Google Scholar] [CrossRef] [PubMed]
- Dardis, A.; Zampieri, S.; Canterini, S.; Newell, K.L.; Stuani, C.; Murrell, J.R.; Ghetti, B.; Fiorenza, M.T.; Bembi, B.; Buratti, E. Altered localization and functionality of TAR DNA Binding Protein 43 (TDP-43) in niemann- pick disease type C. Acta Neuropathol. Commun. 2016, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Tropea, T.F.; Mak, J.; Guo, M.H.; Xie, S.X.; Suh, E.; Rick, J.; Siderowf, A.; Weintraub, D.; Grossman, M.; Irwin, D.; et al. TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann. Neurol. 2019, 85, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.A.; Zheng, Y.; Murphy, K.; Huang, M.; Hu, F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum. Mol. Genet. 2013, 22, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Klein, Z.A.; Takahashi, H.; Ma, M.; Stagi, M.; Zhou, M.; Lam, T.T.; Strittmatter, S.M. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron 2017, 95, 281–296.e286. [Google Scholar] [CrossRef]
- Schwenk, B.M.; Lang, C.M.; Hogl, S.; Tahirovic, S.; Orozco, D.; Rentzsch, K.; Lichtenthaler, S.F.; Hoogenraad, C.C.; Capell, A.; Haass, C.; et al. The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 2014, 33, 450–467. [Google Scholar] [CrossRef]
- Tetreault, M.; Gonzalez, M.; Dicaire, M.J.; Allard, P.; Gehring, K.; Leblanc, D.; Leclerc, N.; Schondorf, R.; Mathieu, J.; Zuchner, S.; et al. Adult-onset painful axonal polyneuropathy caused by a dominant NAGLU mutation. Brain 2015, 138, 1477–1483. [Google Scholar] [CrossRef][Green Version]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44, 120. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Quan, S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst. Rev. 2017, 11, CD011539. [Google Scholar] [CrossRef]
- Li, M. Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases. Pediatr. Ann. 2018, 47, e191–e197. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Hoogerbrugge, P.M.; Brouwer, O.F.; Bordigoni, P.; Ringden, O.; Kapaun, P.; Ortega, J.J.; O’Meara, A.; Cornu, G.; Souillet, G.; Frappaz, D.; et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow Transplantation. Lancet 1995, 345, 1398–1402. [Google Scholar] [CrossRef]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Martins, C.; Hulkova, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef]
- Grishchuk, Y.; Sri, S.; Rudinskiy, N.; Ma, W.; Stember, K.G.; Cottle, M.W.; Sapp, E.; Difiglia, M.; Muzikansky, A.; Betensky, R.A.; et al. Behavioral deficits, early gliosis, dysmyelination and synaptic dysfunction in a mouse model of mucolipidosis IV. Acta Neuropathol. Commun. 2014, 2, 133. [Google Scholar] [CrossRef]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef]
- Pressey, S.N.; Smith, D.A.; Wong, A.M.; Platt, F.M.; Cooper, J.D. Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol. Dis. 2012, 45, 1086–1100. [Google Scholar] [CrossRef]
- Baudry, M.; Yao, Y.; Simmons, D.; Liu, J.; Bi, X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: Immunohistochemical observations of microglia and astroglia. Exp. Neurol. 2003, 184, 887–903. [Google Scholar] [CrossRef]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef]
- Pshezhetsky, A.V. Crosstalk between 2 organelles: Lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C. Rare Dis. 2015, 3, e1049793. [Google Scholar] [CrossRef]
- Bruyere, J.; Roy, E.; Ausseil, J.; Lemonnier, T.; Teyre, G.; Bohl, D.; Etienne-Manneville, S.; Lortat-Jacob, H.; Heard, J.M.; Vitry, S. Heparan sulfate saccharides modify focal adhesions: Implication in mucopolysaccharidosis neuropathophysiology. J. Mol. Biol. 2015, 427, 775–791. [Google Scholar] [CrossRef]
- Kavetsky, L.; Green, K.K.; Boyle, B.R.; Yousufzai, F.A.K.; Padron, Z.M.; Melli, S.E.; Kuhnel, V.L.; Jackson, H.M.; Blanco, R.E.; Howell, G.R.; et al. Increased interactions and engulfment of dendrites by microglia precede Purkinje cell degeneration in a mouse model of Niemann Pick Type-C. Sci. Rep. 2019, 9, 14722. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin Invest 2017, 127, 3201–3209. [Google Scholar] [CrossRef]
- Vitner, E.B.; Dekel, H.; Zigdon, H.; Shachar, T.; Farfel-Becker, T.; Eilam, R.; Karlsson, S.; Futerman, A.H. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum. Mol. Genet. 2010, 19, 3583–3590. [Google Scholar] [CrossRef]
- Lange, J.; Haslett, L.J.; Lloyd-Evans, E.; Pocock, J.M.; Sands, M.S.; Williams, B.P.; Cooper, J.D. Compromised astrocyte function and survival negatively impact neurons in infantile neuronal ceroid lipofuscinosis. Acta Neuropathol. Commun. 2018, 6, 74. [Google Scholar] [CrossRef]
- Parviainen, L.; Dihanich, S.; Anderson, G.W.; Wong, A.M.; Brooks, H.R.; Abeti, R.; Rezaie, P.; Lalli, G.; Pope, S.; Heales, S.J.; et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol. Commun. 2017, 5, 74. [Google Scholar] [CrossRef]
- Pontikis, C.C.; Cotman, S.L.; MacDonald, M.E.; Cooper, J.D. Thalamocortical neuron loss and localized astrocytosis in the Cln3Deltaex7/8 knock-in mouse model of Batten disease. Neurobiol. Dis. 2005, 20, 823–836. [Google Scholar] [CrossRef]
- Thelen, M.; Damme, M.; Schweizer, M.; Hagel, C.; Wong, A.M.; Cooper, J.D.; Braulke, T.; Galliciotti, G. Disruption of the autophagy-lysosome pathway is involved in neuropathology of the nclf mouse model of neuronal ceroid lipofuscinosis. PLoS ONE 2012, 7, e35493. [Google Scholar] [CrossRef]
- Oswald, M.J.; Palmer, D.N.; Kay, G.W.; Shemilt, S.J.; Rezaie, P.; Cooper, J.D. Glial activation spreads from specific cerebral foci and precedes neurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6). Neurobiol. Dis. 2005, 20, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Dwek, R.A.; Butters, T.D.; Platt, F.M. Storage solutions: Treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 2005, 6, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S.; Lee, J.H. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 2008, 4, 590–599. [Google Scholar] [CrossRef]
- Cao, Y.; Espinola, J.A.; Fossale, E.; Massey, A.C.; Cuervo, A.M.; MacDonald, M.E.; Cotman, S.L. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2006, 281, 20483–20493. [Google Scholar] [CrossRef]
- Koike, M.; Shibata, M.; Waguri, S.; Yoshimura, K.; Tanida, I.; Kominami, E.; Gotow, T.; Peters, C.; von Figura, K.; Mizushima, N.; et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am. J. Pathol. 2005, 167, 1713–1728. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef]
- Sambri, I.; D’Alessio, R.; Ezhova, Y.; Giuliano, T.; Sorrentino, N.C.; Cacace, V.; De Risi, M.; Cataldi, M.; Annunziato, L.; De Leonibus, E.; et al. Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol. Med. 2017, 9, 112–132. [Google Scholar] [CrossRef]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem. Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef]
- Baldo, G.; Lorenzini, D.M.; Santos, D.S.; Mayer, F.Q.; Vitry, S.; Bigou, S.; Heard, J.M.; Matte, U.; Giugliani, R. Shotgun proteomics reveals possible mechanisms for cognitive impairment in Mucopolysaccharidosis I mice. Mol. Genet. Metab. 2015, 114, 138–145. [Google Scholar] [CrossRef]
- Karabelas, A.B.; Walkley, S.U. Altered patterns of evoked synaptic activity in cortical pyramidal neurons in feline ganglioside storage disease. Brain Res. 1985, 339, 329–336. [Google Scholar] [CrossRef]
- Purpura, D.P.; Highstein, S.M.; Karabelas, A.B.; Walkley, S.U. Intracellular recording and HRP-staining of cortical neurons in feline ganglioside storage disease. Brain Res. 1980, 181, 446–449. [Google Scholar] [CrossRef]
- Teixeira, C.A.; Miranda, C.O.; Sousa, V.F.; Santos, T.E.; Malheiro, A.R.; Solomon, M.; Maegawa, G.H.; Brites, P.; Sousa, M.M. Early axonal loss accompanied by impaired endocytosis, abnormal axonal transport, and decreased microtubule stability occur in the model of Krabbe’s disease. Neurobiol. Dis. 2014, 66, 92–103. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, S.; Xia, D.; Xia, J.; Chen, G.; Duan, S.; Luo, J. Defects of synaptic vesicle turnover at excitatory and inhibitory synapses in Niemann-Pick C1-deficient neurons. Neuroscience 2010, 167, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, C.A.; Scudder, S.L.; Lin, Y.; Dozier, L.E.; Phan, D.; Allen, N.J.; Patrick, G.N.; Esko, J.D. Neurodevelopmental Changes in Excitatory Synaptic Structure and Function in the Cerebral Cortex of Sanfilippo Syndrome IIIA Mice. Sci. Rep. 2017, 7, 46576. [Google Scholar] [CrossRef] [PubMed]
- Canals, I.; Soriano, J.; Orlandi, J.G.; Torrent, R.; Richaud-Patin, Y.; Jimenez-Delgado, S.; Merlin, S.; Follenzi, A.; Consiglio, A.; Vilageliu, L.; et al. Activity and High-Order Effective Connectivity Alterations in Sanfilippo C Patient-Specific Neuronal Networks. Stem. Cell Rep. 2015, 5, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Zweifel, L.S.; Kuruvilla, R.; Ginty, D.D. Functions and mechanisms of retrograde neurotrophin signalling. Nat. Rev. Neurosci. 2005, 6, 615–625. [Google Scholar] [CrossRef]
- Button, R.W.; Roberts, S.L.; Willis, T.L.; Hanemann, C.O.; Luo, S. Accumulation of autophagosomes confers cytotoxicity. J. Biol. Chem. 2017, 292, 13599–13614. [Google Scholar] [CrossRef]
- Sharma, S.; Lindau, M. t-SNARE Transmembrane Domain Clustering Modulates Lipid Organization and Membrane Curvature. J. Am. Chem. Soc. 2017, 139, 18440–18443. [Google Scholar] [CrossRef]
- Button, R.W.; Luo, S. The formation of autophagosomes during lysosomal defect: A new source of cytotoxicity. Autophagy 2017, 13, 1797–1798. [Google Scholar] [CrossRef]
- March, P.A.; Thrall, M.A.; Brown, D.E.; Mitchell, T.W.; Lowenthal, A.C.; Walkley, S.U. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol. 1997, 94, 164–172. [Google Scholar] [CrossRef]
- Walkley, S.U.; Baker, H.J.; Rattazzi, M.C.; Haskins, M.E.; Wu, J.Y. Neuroaxonal dystrophy in neuronal storage disorders: Evidence for major GABAergic neuron involvement. J. Neurol. Sci. 1991, 104, 1–8. [Google Scholar] [CrossRef]
- Oya, Y.; Nakayasu, H.; Fujita, N.; Suzuki, K.; Suzuki, K. Pathological study of mice with total deficiency of sphingolipid activator proteins (SAP knockout mice). Acta Neuropathol. 1998, 96, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bergner, C.G.; van der Meer, F.; Winkler, A.; Wrzos, C.; Turkmen, M.; Valizada, E.; Fitzner, D.; Hametner, S.; Hartmann, C.; Pfeifenbring, S.; et al. Microglia damage precedes major myelin breakdown in X-linked adrenoleukodystrophy and metachromatic leukodystrophy. Glia 2019, 67, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nagara, H.; Kobayashi, T.; Goto, I. The twitcher mouse: Accumulation of galactosylsphingosine and pathology of the sciatic nerve. Brain Res. 1988, 454, 340–346. [Google Scholar] [CrossRef]
- Smith, B.; Galbiati, F.; Castelvetri, L.C.; Givogri, M.I.; Lopez-Rosas, A.; Bongarzone, E.R. Peripheral neuropathy in the Twitcher mouse involves the activation of axonal caspase 3. ASN Neuro 2011, 3. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Morgan, A. Chaperoning the SNAREs: A role in preventing neurodegeneration? Nat. Cell Biol. 2011, 13, 8–9. [Google Scholar] [CrossRef]
- Sharma, M.; Burre, J.; Sudhof, T.C. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39. [Google Scholar] [CrossRef]
- Von Schantz, C.; Kielar, C.; Hansen, S.N.; Pontikis, C.C.; Alexander, N.A.; Kopra, O.; Jalanko, A.; Cooper, J.D. Progressive thalamocortical neuron loss in Cln5 deficient mice: Distinct effects in Finnish variant late infantile NCL. Neurobiol. Dis. 2009, 34, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Kielar, C.; Wishart, T.M.; Palmer, A.; Dihanich, S.; Wong, A.M.; Macauley, S.L.; Chan, C.H.; Sands, M.S.; Pearce, D.A.; Cooper, J.D.; et al. Molecular correlates of axonal and synaptic pathology in mouse models of Batten disease. Hum. Mol. Genet. 2009, 18, 4066–4080. [Google Scholar] [CrossRef] [PubMed]
- Llavero Hurtado, M.; Fuller, H.R.; Wong, A.M.S.; Eaton, S.L.; Gillingwater, T.H.; Pennetta, G.; Cooper, J.D.; Wishart, T.M. Proteomic mapping of differentially vulnerable pre-synaptic populations identifies regulators of neuronal stability in vivo. Sci. Rep. 2017, 7, 12412. [Google Scholar] [CrossRef]
- Vitry, S.; Ausseil, J.; Hocquemiller, M.; Bigou, S.; Dos Santos Coura, R.; Heard, J.M. Enhanced degradation of synaptophysin by the proteasome in mucopolysaccharidosis type IIIB. Mol. Cell Neurosci. 2009, 41, 8–18. [Google Scholar] [CrossRef]
- Bayo-Puxan, N.; Terrasso, A.P.; Creyssels, S.; Simao, D.; Begon-Pescia, C.; Lavigne, M.; Salinas, S.; Bernex, F.; Bosch, A.; Kalatzis, V.; et al. Lysosomal and network alterations in human mucopolysaccharidosis type VII iPSC-derived neurons. Sci. Rep. 2018, 8, 16644. [Google Scholar] [CrossRef]
- Kanninen, K.M.; Grubman, A.; Meyerowitz, J.; Duncan, C.; Tan, J.L.; Parker, S.J.; Crouch, P.J.; Paterson, B.M.; Hickey, J.L.; Donnelly, P.S.; et al. Increased zinc and manganese in parallel with neurodegeneration, synaptic protein changes and activation of Akt/GSK3 signaling in ovine CLN6 neuronal ceroid lipofuscinosis. PLoS ONE 2013, 8, e58644. [Google Scholar] [CrossRef]
- Amorim, I.S.; Mitchell, N.L.; Palmer, D.N.; Sawiak, S.J.; Mason, R.; Wishart, T.M.; Gillingwater, T.H. Molecular neuropathology of the synapse in sheep with CLN5 Batten disease. Brain Behav. 2015, 5, e00401. [Google Scholar] [CrossRef]
- Kwon, S.E.; Chapman, E.R. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 2011, 70, 847–854. [Google Scholar] [CrossRef]
- Conde, C.; Caceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Alberts, B. Molecular Biology of the Cell; Garland Science Taylor & Francis: New York, NY, USA, 2008. [Google Scholar]
- Kreutzberg, G.W. Neuronal dynamics and axonal flow. IV. Blockage of intra-axonal enzyme transport by colchicine. Proc. Natl. Acad. Sci. USA 1969, 62, 722–728. [Google Scholar] [CrossRef]
- Vale, R.D.; Reese, T.S.; Sheetz, M.P. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 1985, 42, 39–50. [Google Scholar] [CrossRef]
- Paschal, B.M.; Shpetner, H.S.; Vallee, R.B. MAP 1C is a microtubule-activated ATPase which translocates microtubules in vitro and has dynein-like properties. J. Cell Biol. 1987, 105, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, S.O.; Betz, W.J. Synaptic vesicle pools. Nat. Rev. Neurosci. 2005, 6, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Virmani, T.; Gupta, P.; Liu, X.; Kavalali, E.T.; Hofmann, S.L. Progressively reduced synaptic vesicle pool size in cultured neurons derived from neuronal ceroid lipofuscinosis-1 knockout mice. Neurobiol. Dis. 2005, 20, 314–323. [Google Scholar] [CrossRef]
- Spacek, J.; Harris, K.M. Three-dimensional organization of cell adhesion junctions at synapses and dendritic spines in area CA1 of the rat hippocampus. J. Comp. Neurol. 1998, 393, 58–68. [Google Scholar] [CrossRef]
- Arellano, J.I.; Espinosa, A.; Fairen, A.; Yuste, R.; DeFelipe, J. Non-synaptic dendritic spines in neocortex. Neuroscience 2007, 145, 464–469. [Google Scholar] [CrossRef]
- Chen, J.L.; Villa, K.L.; Cha, J.W.; So, P.T.; Kubota, Y.; Nedivi, E. Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron 2012, 74, 361–373. [Google Scholar] [CrossRef]
- Sorra, K.E.; Harris, K.M. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus 2000, 10, 501–511. [Google Scholar] [CrossRef]
- Lang, C.; Barco, A.; Zablow, L.; Kandel, E.R.; Siegelbaum, S.A.; Zakharenko, S.S. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 16665–16670. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef]
- Padamsey, Z.; McGuinness, L.; Bardo, S.J.; Reinhart, M.; Tong, R.; Hedegaard, A.; Hart, M.L.; Emptage, N.J. Activity-Dependent Exocytosis of Lysosomes Regulates the Structural Plasticity of Dendritic Spines. Neuron 2017, 93, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Padamsey, Z.; McGuinness, L.; Emptage, N.J. Inhibition of lysosomal Ca(2+) signalling disrupts dendritic spine structure and impairs wound healing in neurons. Commun. Integr. Biol. 2017, 10, e1344802. [Google Scholar] [CrossRef] [PubMed]
- Purpura, D.P.; Suzuki, K. Distortion of neuronal geometry and formation of aberrant synapses in neuronal storage disease. Brain Res. 1976, 116, 1–21. [Google Scholar] [CrossRef]
- Bible, E.; Gupta, P.; Hofmann, S.L.; Cooper, J.D. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol. Dis. 2004, 16, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Tiscione, S.A.; Vivas, O.; Ginsburg, K.S.; Bers, D.M.; Ory, D.S.; Santana, L.F.; Dixon, R.E.; Dickson, E.J. Disease-associated mutations in Niemann-Pick type C1 alter ER calcium signaling and neuronal plasticity. J. Cell Biol. 2019, 218, 4141–4156. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Hoogenraad, C.C. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef]
- Gulley, R.L.; Reese, T.S. Cytoskeletal organization at the postsynaptic complex. J. Cell Biol. 1981, 91, 298–302. [Google Scholar] [CrossRef]
- Landis, D.M.; Reese, T.S. Cytoplasmic organization in cerebellar dendritic spines. J. Cell Biol. 1983, 97, 1169–1178. [Google Scholar] [CrossRef]
- Harris, K.M.; Weinberg, R.J. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect Biol. 2012, 4. [Google Scholar] [CrossRef]
- Lisman, J.E.; Harris, K.M. Quantal analysis and synaptic anatomy--integrating two views of hippocampal plasticity. Trends Neurosci. 1993, 16, 141–147. [Google Scholar] [CrossRef]
- Harris, K.M.; Sultan, P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Neuropharmacology 1995, 34, 1387–1395. [Google Scholar] [CrossRef]
- Schikorski, T.; Stevens, C.F. Morphological correlates of functionally defined synaptic vesicle populations. Nat. Neurosci. 2001, 4, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Sans, N.; Petralia, R.S.; Wang, Y.X.; Blahos, J., 2nd; Hell, J.W.; Wenthold, R.J. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J. Neurosci. 2000, 20, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Sans, N.; Wang, Y.X.; Wenthold, R.J. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol. Cell Neurosci. 2005, 29, 436–452. [Google Scholar] [CrossRef][Green Version]
- Sheng, M.; Kim, E. The postsynaptic organization of synapses. Cold Spring Harb. Perspect Biol. 2011, 3. [Google Scholar] [CrossRef]
- Goo, M.S.; Sancho, L.; Slepak, N.; Boassa, D.; Deerinck, T.J.; Ellisman, M.H.; Bloodgood, B.L.; Patrick, G.N. Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Biol. 2017, 216, 2499–2513. [Google Scholar] [CrossRef]
- Koster, K.P.; Francesconi, W.; Berton, F.; Alahmadi, S.; Srinivas, R.; Yoshii, A. Developmental NMDA receptor dysregulation in the infantile neuronal ceroid lipofuscinosis mouse model. eLife 2019, 8. [Google Scholar] [CrossRef]
- Imrie, J.; Dasgupta, S.; Besley, G.T.; Harris, C.; Heptinstall, L.; Knight, S.; Vanier, M.T.; Fensom, A.H.; Ward, C.; Jacklin, E.; et al. The natural history of Niemann-Pick disease type C in the UK. J. Inherit Metab. Dis. 2007, 30, 51–59. [Google Scholar] [CrossRef]
- Voikar, V.; Rauvala, H.; Ikonen, E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav. Brain Res. 2002, 132, 1–10. [Google Scholar] [CrossRef]
- Sevin, M.; Lesca, G.; Baumann, N.; Millat, G.; Lyon-Caen, O.; Vanier, M.T.; Sedel, F. The adult form of Niemann-Pick disease type C. Brain 2007, 130, 120–133. [Google Scholar] [CrossRef]
- Amritraj, A.; Peake, K.; Kodam, A.; Salio, C.; Merighi, A.; Vance, J.E.; Kar, S. Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am. J. Pathol. 2009, 175, 2540–2556. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.L.; Su, L.D.; Li, Q.; Wang, X.X.; Shen, Y. Cerebellar long-term depression is deficient in Niemann-Pick type C disease mice. Cerebellum 2011, 10, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qin, L.; Li, Y.; Yu, H.; Zhang, Z.; Tao, C.; Liu, Y.; Xue, Y.; Zhang, X.; Xu, Z.; et al. Presynaptic Endosomal Cathepsin D Regulates the Biogenesis of GABAergic Synaptic Vesicles. Cell Rep. 2019, 28, 1015–1028 e1015. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, B.; Lange, M.D.; Werner, C.; O’Leary, A.; Weishaupt, A.; Popp, S.; Pearce, D.A.; Wiendl, H.; Reif, A.; Pape, H.C.; et al. Defective synaptic transmission causes disease signs in a mouse model of juvenile neuronal ceroid lipofuscinosis. eLife 2017, 6. [Google Scholar] [CrossRef]
- Ahrens-Nicklas, R.C.; Tecedor, L.; Hall, A.F.; Lysenko, E.; Cohen, A.S.; Davidson, B.L.; Marsh, E.D. Neuronal network dysfunction precedes storage and neurodegeneration in a lysosomal storage disorder. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Stroobants, S.; Damme, M.; Van der Jeugd, A.; Vermaercke, B.; Andersson, C.; Fogh, J.; Saftig, P.; Blanz, J.; D’Hooge, R. Long-term enzyme replacement therapy improves neurocognitive functioning and hippocampal synaptic plasticity in immune-tolerant alpha-mannosidosis mice. Neurobiol. Dis. 2017, 106, 255–268. [Google Scholar] [CrossRef]
- Gografe, S.I.; Sanberg, P.R.; Chamizo, W.; Monforte, H.; Garbuzova-Davis, S. Novel pathologic findings associated with urinary retention in a mouse model of mucopolysaccharidosis type IIIB. Comp. Med. 2009, 59, 139–146. [Google Scholar]
- Valenzano, K.J.; Khanna, R.; Powe, A.C.; Boyd, R.; Lee, G.; Flanagan, J.J.; Benjamin, E.R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev. Technol. 2011, 9, 213–235. [Google Scholar] [CrossRef]
- Cho, M.S.; Lee, Y.E.; Kim, J.Y.; Chung, S.; Cho, Y.H.; Kim, D.S.; Kang, S.M.; Lee, H.; Kim, M.H.; Kim, J.H.; et al. Highly efficient and large-scale generation of functional dopamine neurons from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3392–3397. [Google Scholar] [CrossRef]
- Sundberg, M.; Tochitsky, I.; Buchholz, D.E.; Winden, K.; Kujala, V.; Kapur, K.; Cataltepe, D.; Turner, D.; Han, M.J.; Woolf, C.J.; et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 2018, 23, 2167–2183. [Google Scholar] [CrossRef]
- Muguruma, K.; Nishiyama, A.; Ono, Y.; Miyawaki, H.; Mizuhara, E.; Hori, S.; Kakizuka, A.; Obata, K.; Yanagawa, Y.; Hirano, T.; et al. Ontogeny-recapitulating generation and tissue integration of ES cell-derived Purkinje cells. Nat. Neurosci. 2010, 13, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Stanslowsky, N.; Reinhardt, P.; Glass, H.; Kalmbach, N.; Naujock, M.; Hensel, N.; Lubben, V.; Pal, A.; Venneri, A.; Lupo, F.; et al. Neuronal Dysfunction in iPSC-Derived Medium Spiny Neurons from Chorea-Acanthocytosis Patients Is Reversed by Src Kinase Inhibition and F-Actin Stabilization. J. Neurosci. 2016, 36, 12027–12043. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, M.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Zheng, W. Patient iPSC-derived neural stem cells exhibit phenotypes in concordance with the clinical severity of mucopolysaccharidosis I. Hum. Mol. Genet. 2018, 27, 3612–3626. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Mei, A.; Paquola, A.C.M.; Stern, S.; Bardy, C.; Klug, J.R.; Kim, S.; Neshat, N.; Kim, H.J.; Ku, M.; et al. Efficient Generation of CA3 Neurons from Human Pluripotent Stem Cells Enables Modeling of Hippocampal Connectivity In Vitro. Cell Stem Cell 2018, 22, 684–697 e689. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Florer, J.; Mayhew, C.N.; Jia, Z.; Zhao, Z.; Xu, K.; Ran, H.; Liou, B.; Zhang, W.; Setchell, K.D.; et al. Properties of neurons derived from induced pluripotent stem cells of Gaucher disease type 2 patient fibroblasts: Potential role in neuropathology. PLoS ONE 2015, 10, e0118771. [Google Scholar] [CrossRef]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet 2015, 24, 5775–5788. [Google Scholar] [CrossRef]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis. Model Mech. 2019, 12. [Google Scholar] [CrossRef]
- Duarte, A.J.; Ribeiro, D.; Santos, R.; Moreira, L.; Braganca, J.; Amaral, O. Induced pluripotent stem cell line (INSAi001-A) from a Gaucher disease type 3 patient compound heterozygote for mutations in the GBA1 gene. Stem Cell Res. 2019, 41, 101595. [Google Scholar] [CrossRef]
- Tofoli, F.A.; Chien, H.F.; Barbosa, E.R.; Pereira, L.V. Generation of three human induced pluripotent stem cell (hiPSC) lines derived from one Gaucher disease patient with Parkinson’s disease and two unrelated Parkinson’s disease patients with GBA mutations. Stem Cell Res. 2019, 39, 101519. [Google Scholar] [CrossRef]
- Nagel, M.; Reichbauer, J.; Bohringer, J.; Schelling, Y.; Krageloh-Mann, I.; Schule, R.; Ulmer, U. Generation of two iPSC lines derived from two unrelated patients with Gaucher disease. Stem Cell Res. 2019, 35, 101336. [Google Scholar] [CrossRef]
- Peng, Y.; Liou, B.; Inskeep, V.; Blackwood, R.; Mayhew, C.N.; Grabowski, G.A.; Sun, Y. Intravenous infusion of iPSC-derived neural precursor cells increases acid beta-glucosidase function in the brain and lessens the neuronopathic phenotype in a mouse model of Gaucher disease. Hum. Mol. Genet. 2019, 28, 3406–3421. [Google Scholar] [CrossRef]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F.; Group, N.-C.G.W. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.K.; Park, M.H.; Hong, Y.R.; Marti, H.H.; Kim, H.; Okada, Y.; Otsu, M.; Seo, E.J.; Park, J.H.; et al. Pathological roles of the VEGF/SphK pathway in Niemann-Pick type C neurons. Nat. Commun. 2014, 5, 5514. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, M.; Peter, F.; Joost, S.; Trilck, M.; Rolfs, A.; Frech, M.J. Decreased calcium flux in Niemann-Pick type C1 patient-specific iPSC-derived neurons due to higher amount of calcium-impermeable AMPA receptors. Mol. Cell Neurosci. 2017, 83, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Rauva, K.; Blom, T.; von Schantz-Fant, C.; Blom, T.; Jalanko, A.; Kyttala, A. Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Gomez-Giro, G.; Arias-Fuenzalida, J.; Jarazo, J.; Zeuschner, D.; Ali, M.; Possemis, N.; Bolognin, S.; Halder, R.; Jager, C.; Kuper, W.F.E.; et al. Synapse alterations precede neuronal damage and storage pathology in a human cerebral organoid model of CLN3-juvenile neuronal ceroid lipofuscinosis. Acta Neuropathol. Commun. 2019, 7, 222. [Google Scholar] [CrossRef]
- Son, M.Y.; Kwak, J.E.; Seol, B.; Lee, D.Y.; Jeon, H.; Cho, Y.S. A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J. Pathol. 2015, 237, 98–110. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsubara, T.; Sato, T.; Yanagisawa, K. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid beta-protein fibrillogenesis. Biochim. Biophys. Acta 2008, 1778, 2717–2726. [Google Scholar] [CrossRef]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef]
- Rybova, J.; Ledvinova, J.; Sikora, J.; Kuchar, L.; Dobrovolny, R. Neural cells generated from human induced pluripotent stem cells as a model of CNS involvement in mucopolysaccharidosis type II. J. Inherit Metab. Dis. 2018, 41, 221–229. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pará, C.; Bose, P.; Pshezhetsky, A.V. Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. J. Clin. Med. 2020, 9, 616. https://doi.org/10.3390/jcm9030616
Pará C, Bose P, Pshezhetsky AV. Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. Journal of Clinical Medicine. 2020; 9(3):616. https://doi.org/10.3390/jcm9030616
Chicago/Turabian StylePará, Camila, Poulomee Bose, and Alexey V. Pshezhetsky. 2020. "Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression" Journal of Clinical Medicine 9, no. 3: 616. https://doi.org/10.3390/jcm9030616
APA StylePará, C., Bose, P., & Pshezhetsky, A. V. (2020). Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. Journal of Clinical Medicine, 9(3), 616. https://doi.org/10.3390/jcm9030616