Mechanotransduction in Wound Healing and Fibrosis

 , ,
, ,  and
and 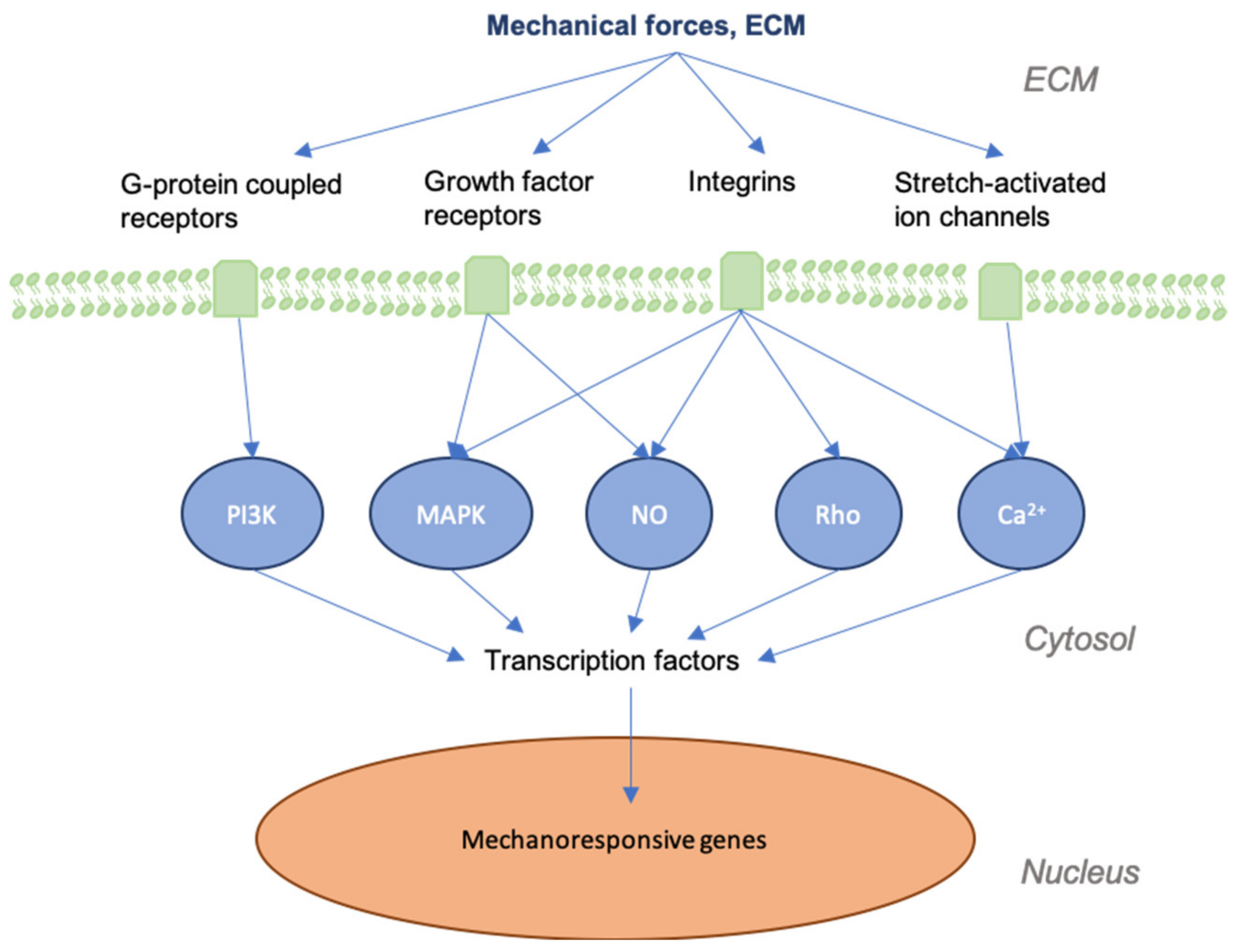{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Wound Healing and Fibrosis in the Skin
3. Mechanotransduction in Skin and Wounds
4. Tensegrity and CTF
5. TGF-β
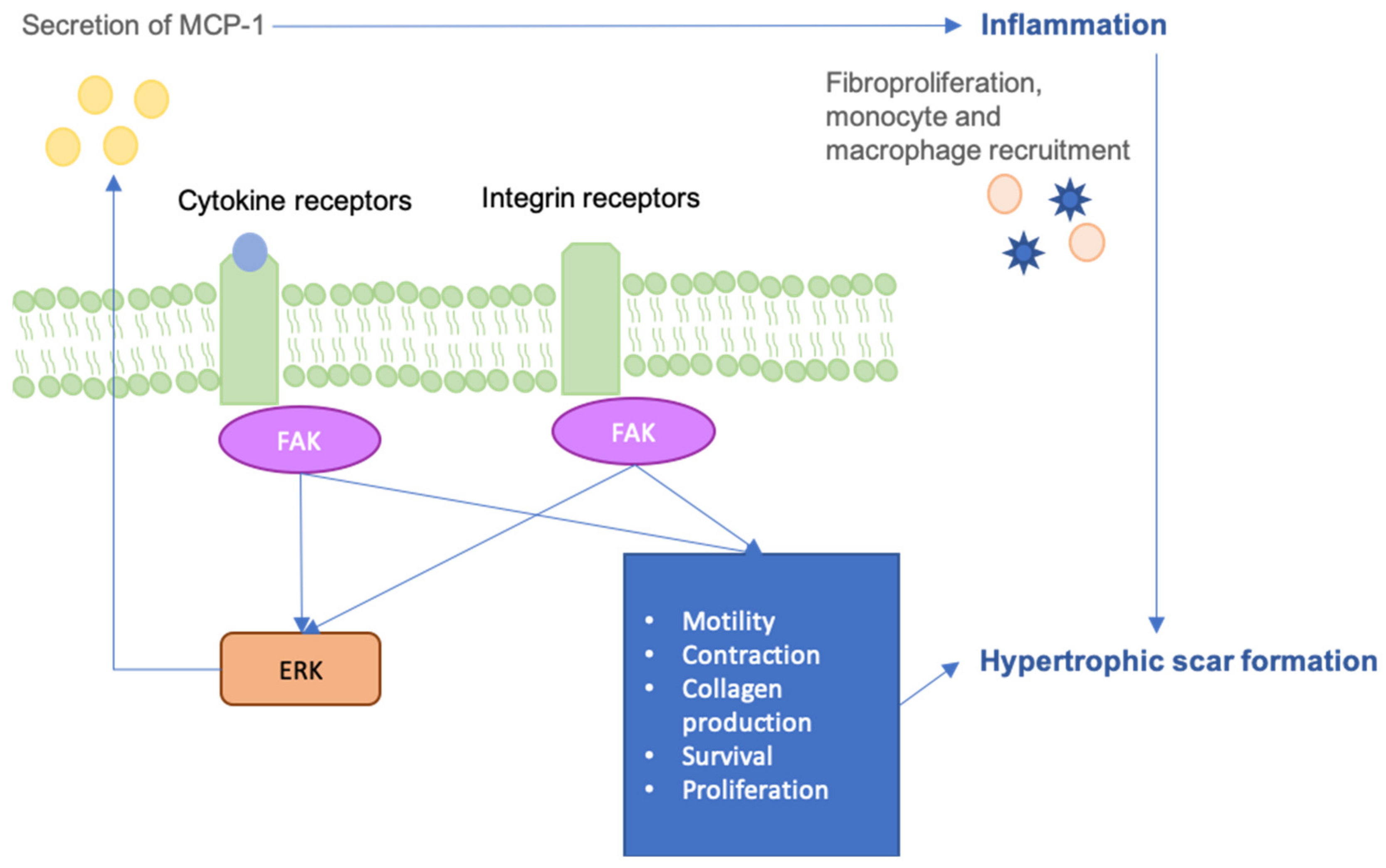
6. FAK-ERK-MCP1 Pathway
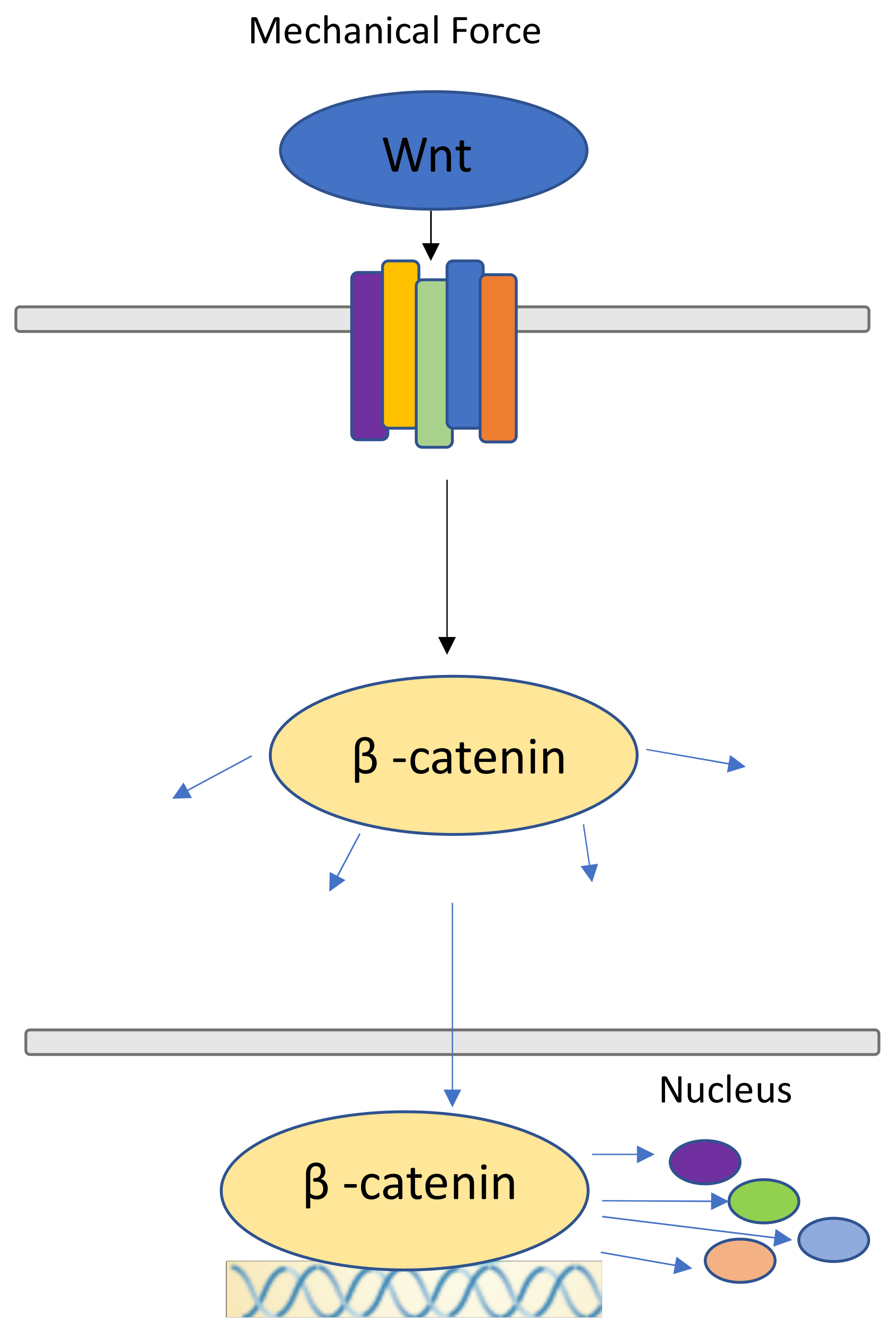
7. Canonical Wnt Pathway and β-catenin Pathway
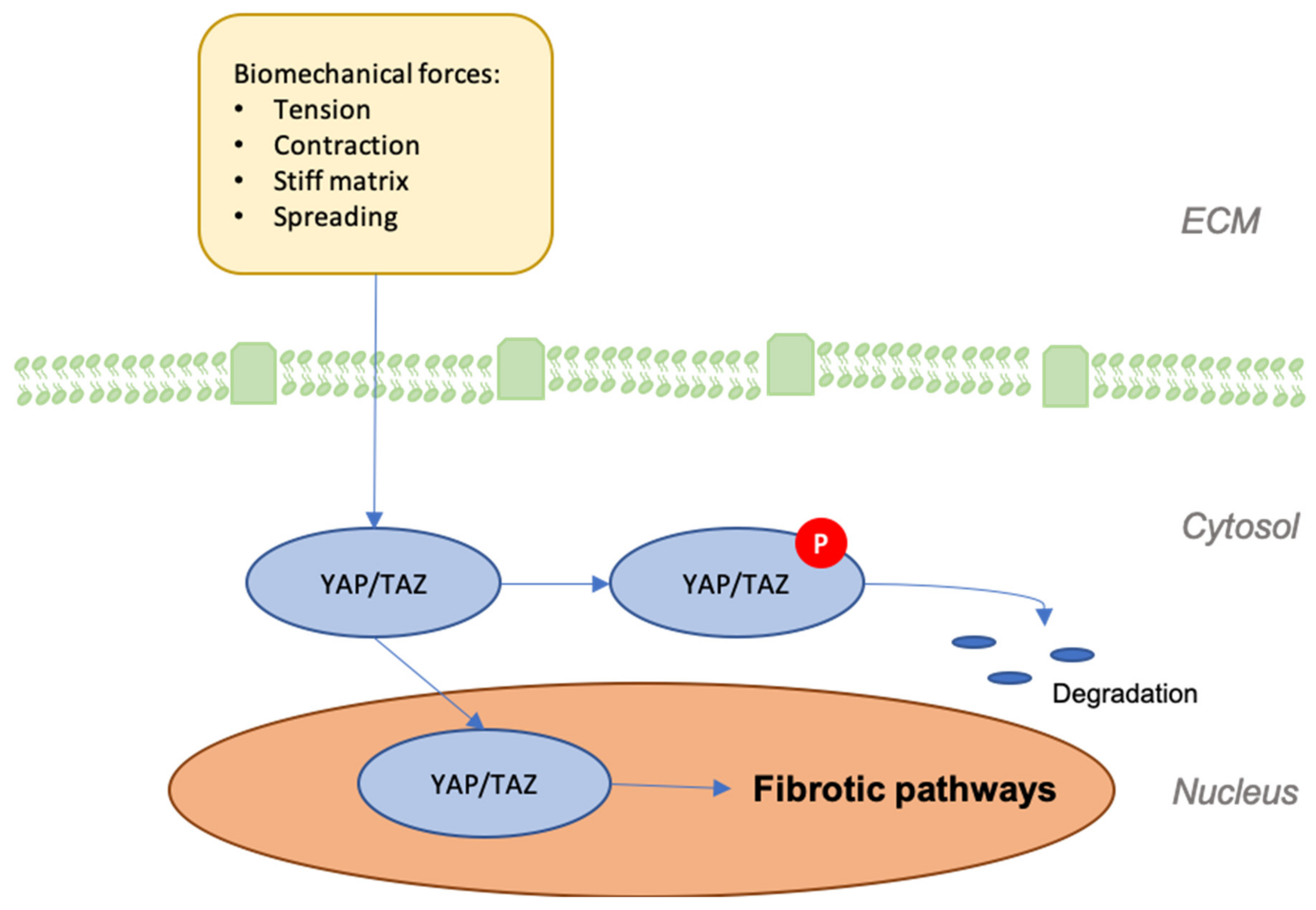
8. YAP/TAZ
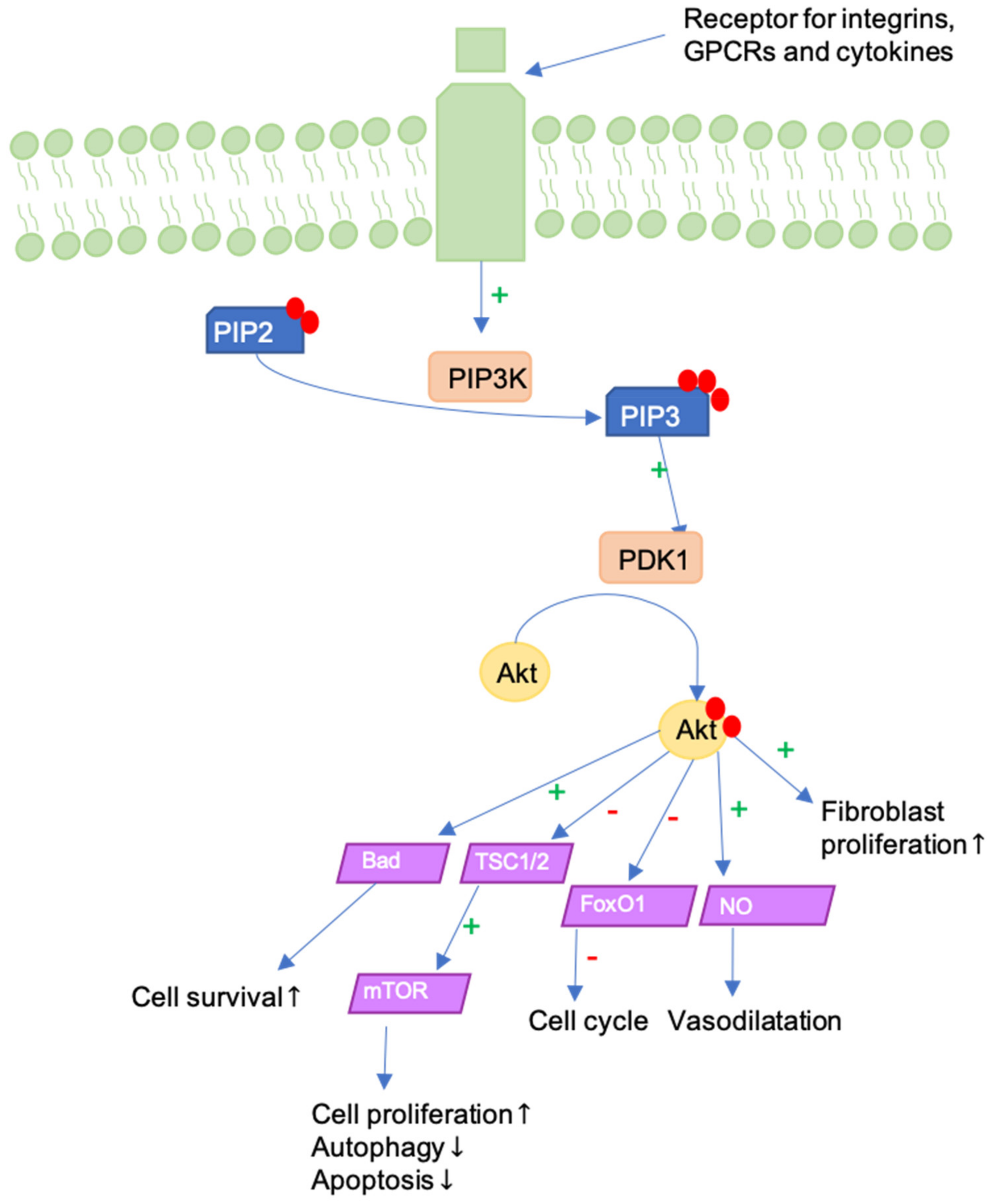
9. ILK-PI3K/Akt Pathway
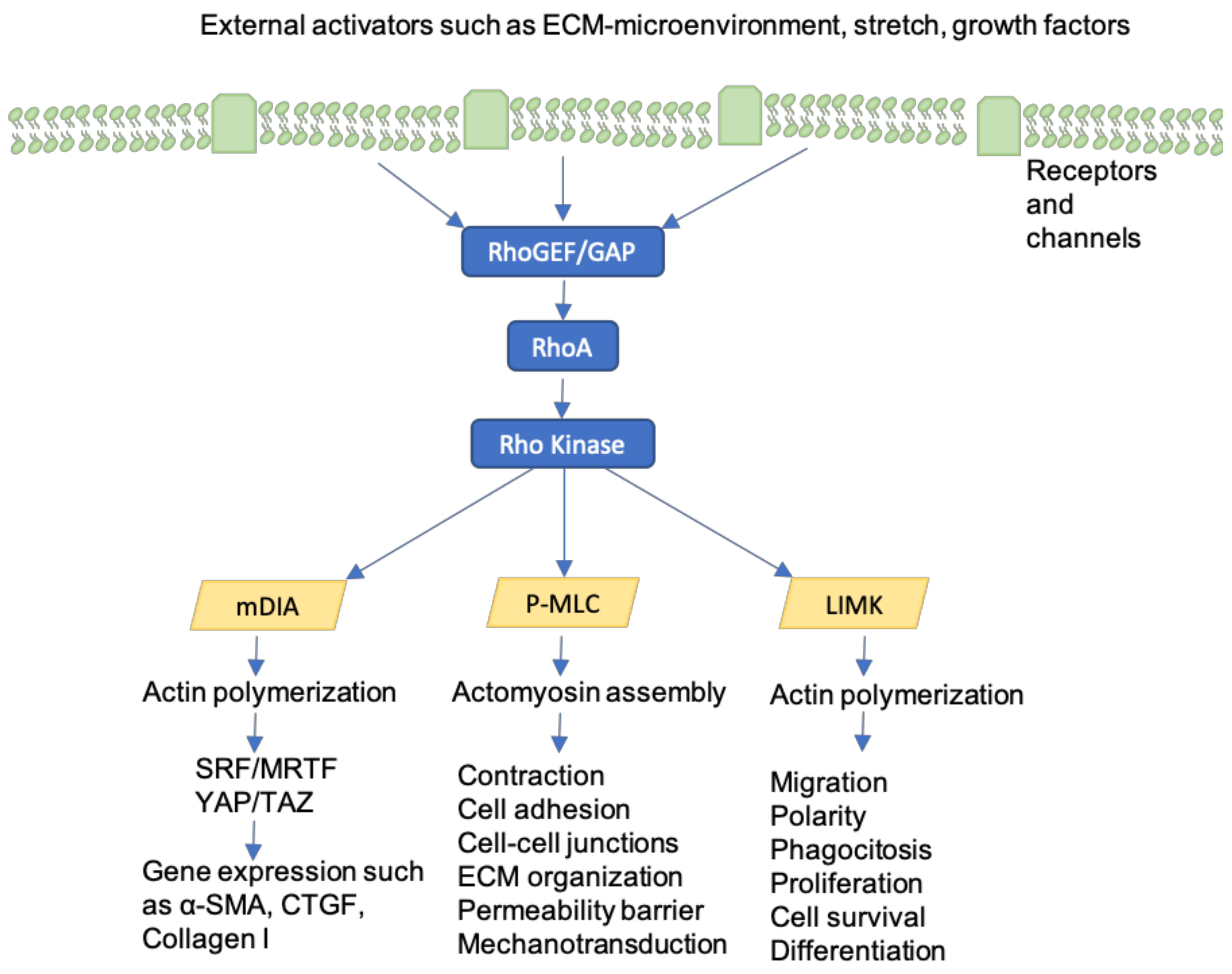
10. Rho-GTPases
11. Calcium-Dependent ion Channels
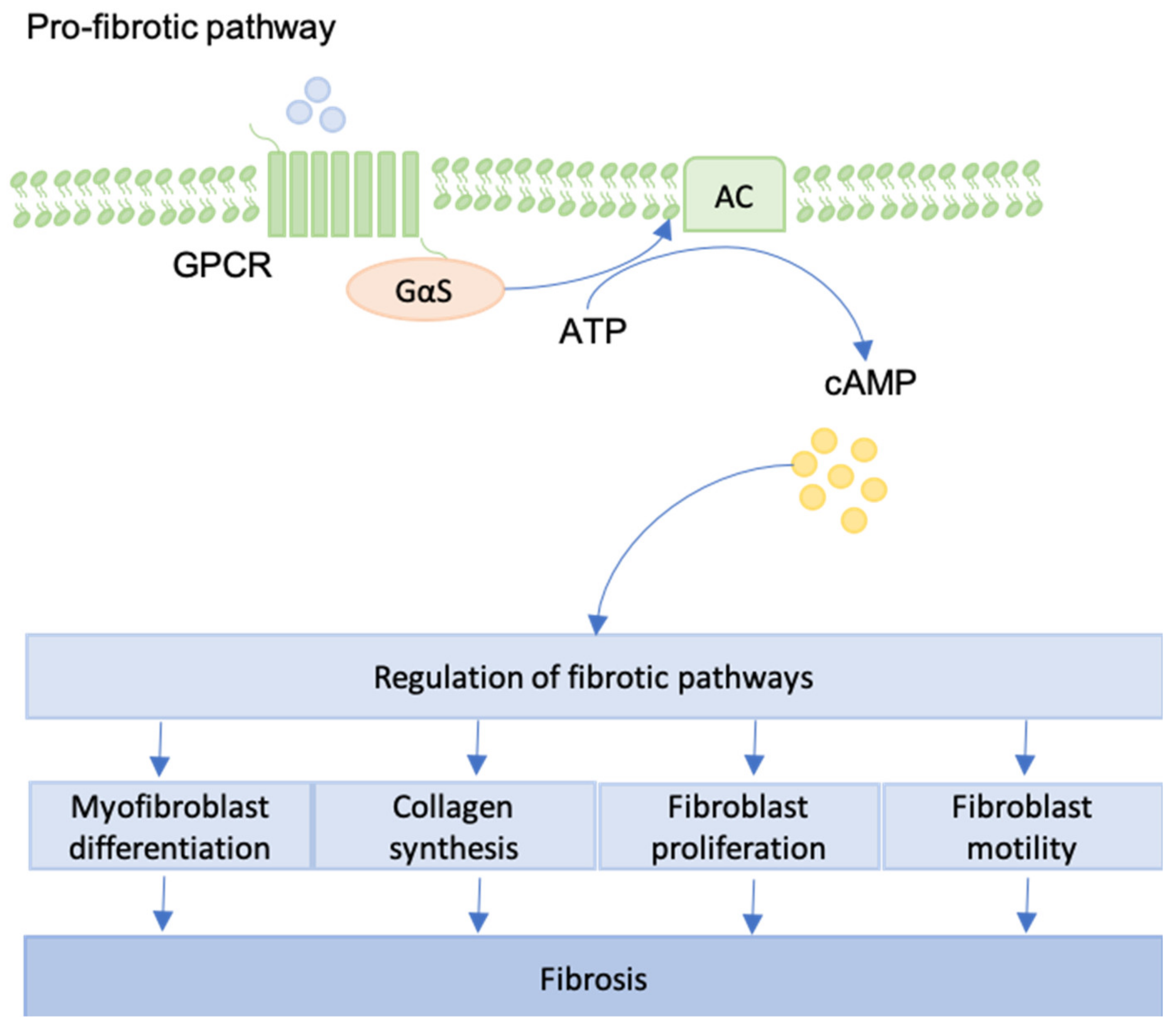
12. GPCRs
13. Future Perspectives
14. Conclusions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | Alpha Smooth Muscle Actin |
| Ca2+ | calcium |
| CaSR | calcium-sensing receptors |
| CCB | calcium channel-blocker |
| CTF | cell traction force |
| DNA | deoxyribonucleic acid |
| ECF | extracellular fluid |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-related kinase |
| FAK | focal adhesion kinase |
| FAKI | focal adhesion kinase inhibitor |
| GPCR | G protein-coupled receptor |
| HCN | hyperpolarization-activated Cyclic Nucleotide-gated |
| HTS | hypertrophic scar |
| ILK | integrin-linked kinase |
| IPF | idiopathic pulmonary fibrosis |
| K+ | kalium |
| LAP | latency-associated peptide |
| LTBP-1 | latent TGF-β-binding-protein-1 |
| miRNA | microRNA |
| MMP | matrix metalloproteinase |
| mRNA | messenger RNAs |
| MS | mechanosensitive |
| MSCs | mechanosensitive ion channels |
| mTOR | mammalian target of rapamycine |
| MYLK | myosin light chain kinase |
| Na+ | natrium |
| NO | nitric oxide |
| PAI-1 | plasminogen activator inhibitor type-1 |
| PI3K | phosphoinositide 3-kinase |
| RAN | ribonucleic acid |
| RTK | receptor tyrosine kinase |
| ROCK | rho-associated coiled-coil kinase |
| ROS | reactive oxygen species |
| siRNA | small interfering RNA |
| TAZ | tafazzin |
| TGFβ | transforming growth factor beta |
| TIMP | tissue inhibitors of metalloproteinase |
| TRP | transient receptor potential |
| TRPV4 | transient receptor potential vanilloid 4 |
| VAS | visual analogue scale |
| YAP | yes-associated protein |
References
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Masur, S.K.; Dewal, H.S.; Dinh, T.T.; Erenburg, I.; Petridou, S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc. Natl. Acad. Sci. USA 1996, 93, 4219–4223. [Google Scholar] [CrossRef] [PubMed]
- Gosain, A.; DiPietro, L.A. Aging and wound healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef]
- Wong, V.W.; Levi, K.; Akaishi, S.; Schultz, G.; Dauskardt, R.H. Scar zones: Region-specific differences in skin tension may determine incisional scar formation. Plast Reconstr. Surg. 2012, 129, 1272–1276. [Google Scholar] [CrossRef]
- Bayat, A.; McGrouther, D.A.; Ferguson, M.W. Skin scarring. BMJ 2003, 326, 88–92. [Google Scholar] [CrossRef]
- Barnes, L.A.; Marshall, C.D.; Leavitt, T.; Hu, M.S.; Moore, A.L.; Gonzalez, J.G.; Longaker, M.T.; Gurtner, G.C. Mechanical Forces in Cutaneous Wound Healing: Emerging Therapies to Minimize Scar Formation. Adv. Wound Care 2018, 7, 47–56. [Google Scholar] [CrossRef]
- Sharma, M.; Wakure, A. Scar revision. Indian J. Plast Surg. 2013, 46, 408–418. [Google Scholar] [CrossRef]
- Block, L.; Gosain, A.; King, T.W. Emerging Therapies for Scar Prevention. Adv. Wound Care (New Rochelle) 2015, 4, 607–614. [Google Scholar] [CrossRef]
- Matsumoto, E.A.; Liang, H.; Mahadevan, L. Topology, Geometry, and Mechanics of Z-Plasty. Phys. Rev. Lett. 2018, 120, 068101. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, C.L.; Homa, K. Evaluation of a new wound closure device for linear surgical incisions: 3M Steri-Strip S Surgical Skin Closure versus subcuticular closure. Plast Reconstr. Surg. 2010, 125, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Nast, A.; Eming, S.; Fluhr, J.; Fritz, K.; Gauglitz, G.; Hohenleutner, S.; Panizzon, R.G.; Sebastian, G.; Sporbeck, B.; Koller, J. German S2k guidelines for the therapy of pathological scars (hypertrophic scars and keloids). J. Dtsch. Dermatol. Ges. 2012, 10, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Control of capillary growth and differentiation by extracellular matrix. Use of a tensegrity (tensional integrity) mechanism for signal processing. Chest 1991, 99, 34–40. [Google Scholar] [CrossRef]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef]
- Ingber, D.E. Tensegrity I. Cell structure and hierarchical systems biology. J. Cell Sci. 2003, 116, 1157–1173. [Google Scholar] [CrossRef]
- Ingber, D.E. Tensegrity II. How structural networks influence cellular information processing networks. J. Cell Sci. 2003, 116, 1397–1408. [Google Scholar] [CrossRef]
- Knapik, D.M.; Perera, P.; Nam, J.; Blazek, A.D.; Rath, B.; Leblebicioglu, B.; Das, H.; ChuWu, L.; Hewett, T.E.; Agarwal, S.K.J.; et al. Mechanosignaling in bone health, trauma and inflammation. Antioxid. Redox Signal. 2014, 20, 970–985. [Google Scholar] [CrossRef]
- Young, S.R.; Gerard-O’Riley, R.; Kim, J.B.; Pavalko, F.M. Focal adhesion kinase is important for fluid shear stress-induced mechanotransduction in osteoblasts. J. Bone Miner. Res. 2009, 24, 411–424. [Google Scholar] [CrossRef]
- Marjoram, R.J.; Lessey, E.C.; Burridge, K. Regulation of RhoA activity by adhesion molecules and mechanotransduction. Curr. Mol. Med. 2014, 14, 199–208. [Google Scholar] [CrossRef]
- Alenghat, F.J.; Ingber, D.E. Mechanotransduction: All signals point to cytoskeleton, matrix, and integrins. Sci. STKE 2002, 119, pe6. [Google Scholar] [CrossRef] [PubMed]
- Sukharev, S.; Betanzos, M.; Chiang, C.S.; Guy, H.R. The gating mechanism of the large mechanosensitive channel MscL. Nature 2001, 409, 720–724. [Google Scholar] [CrossRef]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Dame, M.K.; Rittie, L.; Fligiel, S.E.G.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased collagen production in chronologically aged skin: Roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am. J. Pathol. 2006, 168, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R.; Hsu, C.K. Mechanobiological dysregulation of the epidermis and dermis in skin disorders and in degeneration. J. Cell. Mol. Med. 2013, 17, 817–822. [Google Scholar] [CrossRef]
- Fligiel, S.E.; Varani, J.; Datta, S.C.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Collagen degradation in aged/photodamaged skin in vivo and after exposure to matrix metalloproteinase-1 in vitro. J. Investig. Dermatol. 2003, 120, 842–848. [Google Scholar] [CrossRef]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 2011, 18, 148–152. [Google Scholar] [CrossRef]
- Aarabi, S.; Bhatt, K.A.; Shi, Y.; Paterno, J.; Chang, E.I.; Loh, S.A.; Holmes, J.W.; Longaker, M.T.; Yee, H.; Gurtner, G.C. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 2007, 21, 3250–3261. [Google Scholar] [CrossRef]
- Longaker, M.T.; Rohrich, R.J.; Greenberg, L.; Furnas, H.; Wald, R.; Bansal, V.; Seify, H.; Tran, A.; Weston, J.; Korman, J.M.; et al. A randomized controlled trial of the embrace advanced scar therapy device to reduce incisional scar formation. Plast Reconstr. Surg. 2014, 134, 536–546. [Google Scholar] [CrossRef]
- Lim, A.F.; Weintraub, J.; Kaplan, E.N.; Januszyk, M.; Cowley, C.; McLaughlin, P.; Beasley, B.; Gurtner, G.C.; Longaker, M.T. The embrace device significantly decreases scarring following scar revision surgery in a randomized controlled trial. Plast Reconstr. Surg. 2014, 133, 398–405. [Google Scholar] [CrossRef]
- Orgill, D.P.; Ogawa, R. Discussion: The embrace device significantly decreases scarring following scar revision surgery in a randomized controlled trial. Plast Reconstr. Surg. 2014, 133, 406–407. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Dauskardt, R.H.; Wong, V.W.; Bhatt, K.A.; Wu, K.; Vial, I.N.; Padois, K.; Korman, J.M.; Longaker, M.T. Improving cutaneous scar formation by controlling the mechanical environment: Large animal and phase I studies. Ann. Surg. 2011, 254, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Lionelli, G.T.; Lawrence, W.T. Wound dressings. Surg. Clin. N. Am. 2003, 83, 617–638. [Google Scholar] [CrossRef]
- Reiffel, R.S. Prevention of hypertrophic scars by long-term paper tape application. Plast Reconstr. Surg. 1995, 96, 1715–1718. [Google Scholar] [CrossRef]
- Rosengren, H.; Askew, D.A.; Heal, C.; Buettner, P.G.; Humphreys, W.O.; Semmens, L.A. Does taping torso scars following dermatologic surgery improve scar appearance? Derm. Pr. Concept. 2013, 3, 75–83. [Google Scholar] [CrossRef]
- Atkinson, J.A.; McKenna, K.T.; Barnett, A.G.; McGrath, D.J.; Rudd, M. A randomized, controlled trial to determine the efficacy of paper tape in preventing hypertrophic scar formation in surgical incisions that traverse Langer’s skin tension lines. Plast Reconstr. Surg. 2005, 116, 1648–1656. [Google Scholar] [CrossRef]
- Gieni, R.S.; Hendzel, M.J. Mechanotransduction from the ECM to the genome: Are the pieces now in place? J. Cell. Biochem. 2008, 104, 1964–1987. [Google Scholar] [CrossRef]
- Wang, J.H.; Lin, J.S. Cell traction force and measurement methods. Biomech. Model. Mechanobiol. 2007, 6, 361–371. [Google Scholar] [CrossRef]
- Duscher, D.; Maan, Z.N.; Wong, V.W.; Rennert, R.C.; Januszyk, M.; Rodrigues, M.; Hu, M.; Whitmore, A.J.; Whittam, A.J.; Longaker, M.T.; et al. Mechanotransduction and fibrosis. J. Biomech. 2014, 47, 1997–2005. [Google Scholar] [CrossRef]
- Noguera, R.; Nieto, O.A.; Tadeo, I.; Fariñas, F.; Alvaro, T. Extracellular matrix, biotensegrity and tumor microenvironment. An update and overview. Histol. Histopathol. 2012, 27, 693–705. [Google Scholar]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharm. 2017, 8, 461. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G. Tissue mechanics and fibrosis. Biochim. Biophys. Acta 2013, 1832, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent TGF-β1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Vindevoghel, L.; Lechleider, R.J.; Uitto, J.; Roberts, A.B.; Mauviel, A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene 2001, 20, 3332–3340. [Google Scholar] [CrossRef]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef]
- Milani, S.; Herbst, H.; Schuppan, D.; Stein, H.; Surrenti, C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am. J. Pathol. 1991, 139, 1221–1229. [Google Scholar]
- Occleston, N.L.; Laverty, H.G.; O’Kane, S.; Ferguson, M.W. Prevention and reduction of scarring in the skin by Transforming Growth Factor beta 3 (TGFbeta3): From laboratory discovery to clinical pharmaceutical. J. Biomater. Sci. Polym. Ed. 2008, 19, 1047–1063. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Duncan, J.; Bond, J.; Bush, J.; Durani, P.; So, K.; Taylor, L.; Chantrey, J.; Mason, T.; James, G.; et al. Prophylactic administration of avotermin for improvement of skin scarring: Three double-blind, placebo-controlled, phase I/II studies. Lancet 2009, 373, 1264–1274. [Google Scholar] [CrossRef]
- Voelker, J.; Berg, P.H.; Sheetz, M. Anti-TGF- β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Azuma, A. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2012, 6, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hermida, N.; López, B.; González, A.; Dotor, J.; Lasarte, J.J.; Sarobe, P.; Borrás-Cuesta, F.; Díez, J. A synthetic peptide from transforming growth factor-beta1 type III receptor prevents myocardial fibrosis in spontaneously hypertensive rats. Cardiovasc. Res. 2009, 81, 601–609. [Google Scholar] [CrossRef]
- Santiago, B.; Gutierrez-Cañas, I.; Dotor, J.; Palao, G.; Lasarte, J.J.; Ruiz, J.; Prieto, J.; Borrás-Cuesta, F.; Pablos, J.L. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J. Investig. Dermatol. 2005, 125, 450–455. [Google Scholar] [CrossRef]
- Januszyk, M.; Kwon, S.H.; Wong, V.W.; Padmanabhan, J.; Maan, Z.N.; Whittam, A.J.; Major, M.R.; Gurtner, G.C. The Role of Focal Adhesion Kinase in Keratinocyte Fibrogenic Gene Expression. Int. J. Mol. Sci. 2017, 18, 1915. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef]
- David, F.S.; Zage, P.E.; Marcantonio, E.E. Integrins interact with focal adhesions through multiple distinct pathways. J. Cell. Physiol. 1999, 181, 74–82. [Google Scholar] [CrossRef]
- Liu, W.; Ma, K.; Kwon, S.H.; Garg, R.; Patta, Y.R.; Fujiwara, T.; Gurtner, G.C. The Abnormal Architecture of Healed Diabetic Ulcers Is the Result of FAK Degradation by Calpain 1. J. Investig. Dermatol. 2017, 137, 1155–1165. [Google Scholar] [CrossRef]
- Wong, V.W.; Garg, R.K.; Sorkin, M.; Rustad, K.C.; Akaishi, S.; Levi, K.; Nelson, E.R.; Tran, M.; Rennert, R.; Liu, W.; et al. Loss of keratinocyte focal adhesion kinase stimulates dermal proteolysis through upregulation of MMP9 in wound healing. Ann. Surg. 2014, 260, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Li, X.; Wu, X.; Hui, B.; Han, S.; Gao, J.; Li, Y.; Shi, J.; Zhu, H.; Zhao, B.; et al. Simultaneous deactivation of FAK and Src improves the pathology of hypertrophic scar. Sci. Rep. 2016, 6, 26023. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Sorkin, M.; Glotzbach, J.P.; Longaker, M.T.; Gurtner, G.C. Surgical approaches to create murine models of human wound healing. J. Biomed. Biotechnol. 2011, 2011, 969618. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Kwon, S.H.; Padmanabhan, J.; Duscher, D.; Trotsyuk, A.A.; Dong, Y.; Inayathullah, M.; Rajadas, J.; Gurtner, G.C. Controlled Delivery of a Focal Adhesion Kinase Inhibitor Results in Accelerated Wound Closure with Decreased Scar Formation. J. Investig. Dermatol. 2018, 138, 2452–2460. [Google Scholar] [CrossRef] [PubMed]
- Widelitz, R.B. Wnt signaling in skin organogenesis. Organogels 2008, 4, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lim, X.; Nusse, R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Yonemura, S. A mechanism of mechanotransduction at the cell-cell interface: Emergence of alpha-catenin as the center of a force-balancing mechanism for morphogenesis in multicellular organisms. BioEssays 2011, 33, 732–736. [Google Scholar] [CrossRef]
- Leckband, D.E.; de Rooij, J. Cadherin adhesion and mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef]
- Wu, M.; Fannin, J.; Rice, K.M.; Wang, B.; Blough, E.R. Effect of aging on cellular mechanotransduction. Ageing Res. Rev. 2011, 10, 1–15. [Google Scholar] [CrossRef]
- Cheon, S.S.; Cheah, A.Y.L.; Turley, S.; Nadesan, P.; Poon, R.; Clevers, H.; Alman, B.A. beta-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc. Natl. Acad. Sci. USA 2002, 99, 6973–6978. [Google Scholar] [CrossRef]
- Wei, J.; Melichian, D.; Komura, K.; Hinchcliff, M.E.; Lam, A.P.; Lafyatis, R.; Gottardi, C.; MacDougald, O.A.; Varga, J. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: A novel mouse model for scleroderma? Arthritis Rheum. 2011, 63, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, R.; Farina, G.; Bayle, J.; Dimarzio, M.; Pendergrass, S.A.; Milano, A.; Whitfield, M.L.; Lafyatis, R. Antagonistic effect of the matricellular signaling protein CCN3 on TGF-beta- and Wnt-mediated fibrillinogenesis in systemic sclerosis and Marfan syndrome. J. Investig. Dermatol. 2010, 130, 1514–1523. [Google Scholar] [CrossRef]
- Bastakoty, D.; Young, P.P. Wnt/beta-catenin pathway in tissue injury: Roles in pathology and therapeutic opportunities for regeneration. FASEB J. 2016, 30, 3271–3284. [Google Scholar] [CrossRef]
- Amini-Nik, S.; Cambridge, E.; Yu, W.; Guo, A.; Whetstone, H.; Nadesan, P.; Poon, R.; Hinz, B.; Alman, B.A. beta-Catenin-regulated myeloid cell adhesion and migration determine wound healing. J. Clin. Investig. 2014, 124, 2599–2610. [Google Scholar] [CrossRef]
- Cheon, S.; Poon, R.; Yu, C.; Khoury, M.; Shenker, R.; Fish, J.; Alman, B.A. Prolonged beta-catenin stabilization and tcf-dependent transcriptional activation in hyperplastic cutaneous wounds. Lab Investig. 2005, 85, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Foote, H.P.; Lechler, T. beta-Catenin protects the epidermis from mechanical stresses. J.Cell Biol. 2013, 202, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Ng, A.; Kim, B.H.; Bianco, A.; Xavier, R.J.; Elledge, S.J. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010, 24, 1507–1518. [Google Scholar] [CrossRef]
- Sato, M. Upregulation of the Wnt/beta-catenin pathway induced by transforming growth factor-beta in hypertrophic scars and keloids. Acta Derm.-Venereol. 2006, 86, 300–307. [Google Scholar] [CrossRef]
- Cheon, S.S.; Wei, Q.; Gurung, A.; Youn, A.; Bright, T.; Poon, R.; Whetstone, H.; Guha, A.; Alman, B.A. Beta-catenin regulates wound size and mediates the effect of TGF-beta in cutaneous healing. FASEB J. 2006, 20, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef]
- Nakajima, H.; Yamamoto, K.; Agarwala, S.; Terai, K.; Fukui, H.; Fukuhara, S.; Ando, K.; Miyazaki, T.; Yokota, Y.; Schmelzer, E.; et al. Flow-Dependent Endothelial YAP Regulation Contributes to Vessel Maintenance. Dev. Cell. 2017, 40, 523–536.e6. [Google Scholar] [CrossRef]
- Lee, M.J.; Byun, M.R.; Furutani-Seiki, M.; Hong, J.H.; Jung, H.S. YAP and TAZ regulate skin wound healing. J. Investig. Dermatol. 2014, 134, 518–525. [Google Scholar] [CrossRef]
- Beverdam, A.; Claxton, C.; Zhang, X.; James, G.; Harvey, K.F.; Key, B. Yap controls stem/progenitor cell proliferation in the mouse postnatal epidermis. J. Investig. Dermatol. 2013, 133, 1497–1505. [Google Scholar] [CrossRef]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef]
- Li, B.S.; Guo, W.J.; Hong, L.; Liu, Y.D.; Liu, C.; Hong, S.S.; Wu, D.B.; Min, J. Role of mechanical strain-activated PI3K/Akt signaling pathway in pelvic organ prolapse. Mol. Med. Rep. 2016, 14, 243–253. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, Y.Y.; Sun, J.E.; Lin, W.H.; Zhou, R.X. ILK-PI3K/AKT pathway participates in cutaneous wound contraction by regulating fibroblast migration and differentiation to myofibroblast. Lab. Investig. 2016, 96, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Calautti, E.; Li, J.; Saoncella, S.; Brissette, J.L.; Goetinck, P.F. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J. Biol. Chem. 2005, 280, 32856–32865. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Komine, M.; Fujimoto, M.; Okochi, H.; Tamaki, K. Activation of Akt by mechanical stretching in human epidermal keratinocytes. Exp. Dermatol. 2006, 15, 356–361. [Google Scholar] [CrossRef]
- Paterno, J.; Vial, I.N.; Wong, V.W.; Rustad, K.C.; Sorkin, M.; Shi, Y.; Bhatt, K.A.; Thangarajah, H.; Glotzbach, J.P.; Gurtner, G.C. Akt-mediated mechanotransduction in murine fibroblasts during hypertrophic scar formation. Wound Repair Regen. 2011, 19, 49–58. [Google Scholar] [CrossRef]
- Gao, Y.L.; Liu, C.S.; Zhao, R.; Wang, L.L.; Li, S.S.; Liu, M.; Zhang, M.; Jiang, S.K.; Tian, Z.L.; Wang, M.; et al. Effects of PI3K/Akt Pathway in Wound Healing Process of Mice Skin. Fa Yi Xue Za Zhi 2016, 32, 7–12. [Google Scholar]
- Rahimi, R.A.; Andrianifahanana, M.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Blenis, J.; Leof, E.B. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res. 2009, 69, 84–93. [Google Scholar] [CrossRef]
- Bujor, A.M.; Pannu, J.; Bu, S.; Smith, E.A.; Muise-Helmericks, R.C.; Trojanowska, M. Akt blockade downregulates collagen and upregulates MMP1 in human dermal fibroblasts. J. Investig. Dermatol. 2008, 128, 1906–1914. [Google Scholar] [CrossRef]
- Lessey, E.C.; Guilluy, C.; Burridge, K. From mechanical force to RhoA activation. Biochemistry 2012, 51, 7420–7432. [Google Scholar] [CrossRef]
- Zhao, X.H.; Laschinger, C.; Arora, P.; Szászi, K.; Kapus, A.; McCulloch, C.A. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J. Cell Sci. 2007, 120, 1801–1809. [Google Scholar] [CrossRef]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Kato, T.; Fujita, A.; Ishizaki, T.; Narumiya, S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1999, 1, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, K.X.; Li, Y.J.; Guo, B.Y.; Wang, M. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses high glucose-induced proliferation and collagen synthesis in rat cardiac fibroblasts. Clin. Exp. Pharm. Physiol. 2011, 38, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.E.; Kokosis, G.; Ren, L.; Selim, M.A.; Bergeron, A.; Levinson, H. Wound contraction is attenuated by fasudil inhibition of Rho-associated kinase. Plast Reconstr. Surg. 2011, 128, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Huveneers, S.; Danen, E.H. Adhesion signaling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef]
- Scheraga, R.G.; Olman, M.A. A Focus on “Eye on” Channels in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 132–133. [Google Scholar] [CrossRef]
- Rahaman, S.; Grove, L.; Paruchuri, S.; Southern, B.; Abraham, S.; Niese, K.; Scheraga, R.; Ghosh, S.; Thodeti, C.; Zhang, D.; et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Investig. 2014, 124, 5225–5238. [Google Scholar] [CrossRef]
- Frank, S.; Stallmeyer, B.; Kämpfer, H.; Kolb, N.; Pfeilschifter, J. Nitric oxide triggers enhanced induction of vascular endothelial growth factor expression in cultured keratinocytes (HaCaT) and during cutaneous wound repair. FASEB J. 1999, 13, 2002–2014. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ayaub, E.A.; Murphy, J.; Lu, C.; Kolb, M.; Ask, K.; Janssen, L.J. Disruption of Calcium Signaling in Fibroblasts and Attenuation of Bleomycin-Induced Fibrosis by Nifedipine. Am. J. Respir. Cell Mol. Biol. 2015, 53, 450–458. [Google Scholar] [CrossRef]
- Bagheri, M.; Jahromi, B.M.; Mirkhani, H.; Solhjou, Z.; Noorafshan, A.; Zamani, A.; Amirghofran, Z. Azelnidipine, a new calcium channel blocker, promotes skin wound healing in diabetic rats. J. Surg. Res. 2011, 169, 101–107. [Google Scholar] [CrossRef]
- Bhaskar, H.N.; Udupa, S.L.; Udupa, A.L. Effect of nifedipine and amlodipine on wound healing in rats. Indian J. Physiol. Pharm. 2004, 48, 111–114. [Google Scholar]
- Ashkani-Esfahani, S.; Hosseinabadi, O.K.; Moezzi, P.; Moafpourian, Y.; Kardeh, S.; Rafiee, S.; Fatheazam, R.; Noorafshan, A.; Nadimi, E.; Mehrvarz, S.; et al. Verapamil, a Calcium-Channel Blocker, Improves the Wound Healing Process in Rats with Excisional Full-Thickness Skin Wounds Based on Stereological Parameters. Adv. Skin Wound Care 2016, 29, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, A.A.; Mojiri Forushani, H.; Mohammad Asgari, H. Wound healing potential of topical amlodipine in full thickness wound of rabbit. Jundishapur J. Nat. Pharm. Prod. 2014, 9, e15638. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Akaishi, S.; Longaker, M.T.; Gurtner, G.C. Pushing back: Wound mechanotransduction in repair and regeneration. J. Investig. Dermatol. 2011, 131, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Haak, A.J.; Ducharme, M.T.; Diaz Espinosa, A.M.; Tschumperlin, D.J. Targeting GPCR Signaling for Idiopathic Pulmonary Fibrosis Therapies. Trends Pharm. Sci. 2020, 41, 172–182. [Google Scholar] [CrossRef]
- D’Souza, K.M.; Malhotra, R.; Philip, J.L.; Staron, M.L.; Theccanat, T.; Jeevanandam, V.; Akhter, S.A. G protein-coupled receptor kinase-2 is a novel regulator of collagen synthesis in adult human cardiac fibroblasts. J. Biol. Chem. 2011, 286, 15507–15516. [Google Scholar] [CrossRef]
- Roberts, M.J.; Broome, R.E.; Kent, T.C.; Charlton, S.J.; Rosethorne, E.M. The inhibition of human lung fibroblast proliferation and differentiation by Gs-coupled receptors is not predicted by the magnitude of cAMP response. Respir. Res. 2018, 19, 56. [Google Scholar] [CrossRef]
- Dooling, L.J.; Discher, D.E. Inhibiting Tumor Fibrosis and Actomyosin through GPCR activation. Trends Cancer 2019, 5, 197–199. [Google Scholar] [CrossRef]
- Bayat, A.; Bock, O.; Mrowietz, U.; Ollier, W.E.; Ferguson, M.W. Genetic susceptibility to keloid disease and hypertrophic scarring: Transforming growth factor beta1 common polymorphisms and plasma levels. Plast Reconstr. Surg. 2003, 111, 535–543. [Google Scholar] [CrossRef]
- Chike-Obi, C.J.; Cole, P.D.; Brissett, A.E. Keloids: Pathogenesis, clinical features, and management. Semin. Plast Surg. 2009, 23, 178–184. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuehlmann, B.; Bonham, C.A.; Zucal, I.; Prantl, L.; Gurtner, G.C. Mechanotransduction in Wound Healing and Fibrosis. J. Clin. Med. 2020, 9, 1423. https://doi.org/10.3390/jcm9051423
Kuehlmann B, Bonham CA, Zucal I, Prantl L, Gurtner GC. Mechanotransduction in Wound Healing and Fibrosis. Journal of Clinical Medicine. 2020; 9(5):1423. https://doi.org/10.3390/jcm9051423
Chicago/Turabian StyleKuehlmann, Britta, Clark A. Bonham, Isabel Zucal, Lukas Prantl, and Geoffrey C. Gurtner. 2020. "Mechanotransduction in Wound Healing and Fibrosis" Journal of Clinical Medicine 9, no. 5: 1423. https://doi.org/10.3390/jcm9051423
APA StyleKuehlmann, B., Bonham, C. A., Zucal, I., Prantl, L., & Gurtner, G. C. (2020). Mechanotransduction in Wound Healing and Fibrosis. Journal of Clinical Medicine, 9(5), 1423. https://doi.org/10.3390/jcm9051423