Clinical Presentations, Pathogenesis, and Therapy of Sarcoidosis: State of the Art


Abstract
:1. Definition and Clinical Presentations
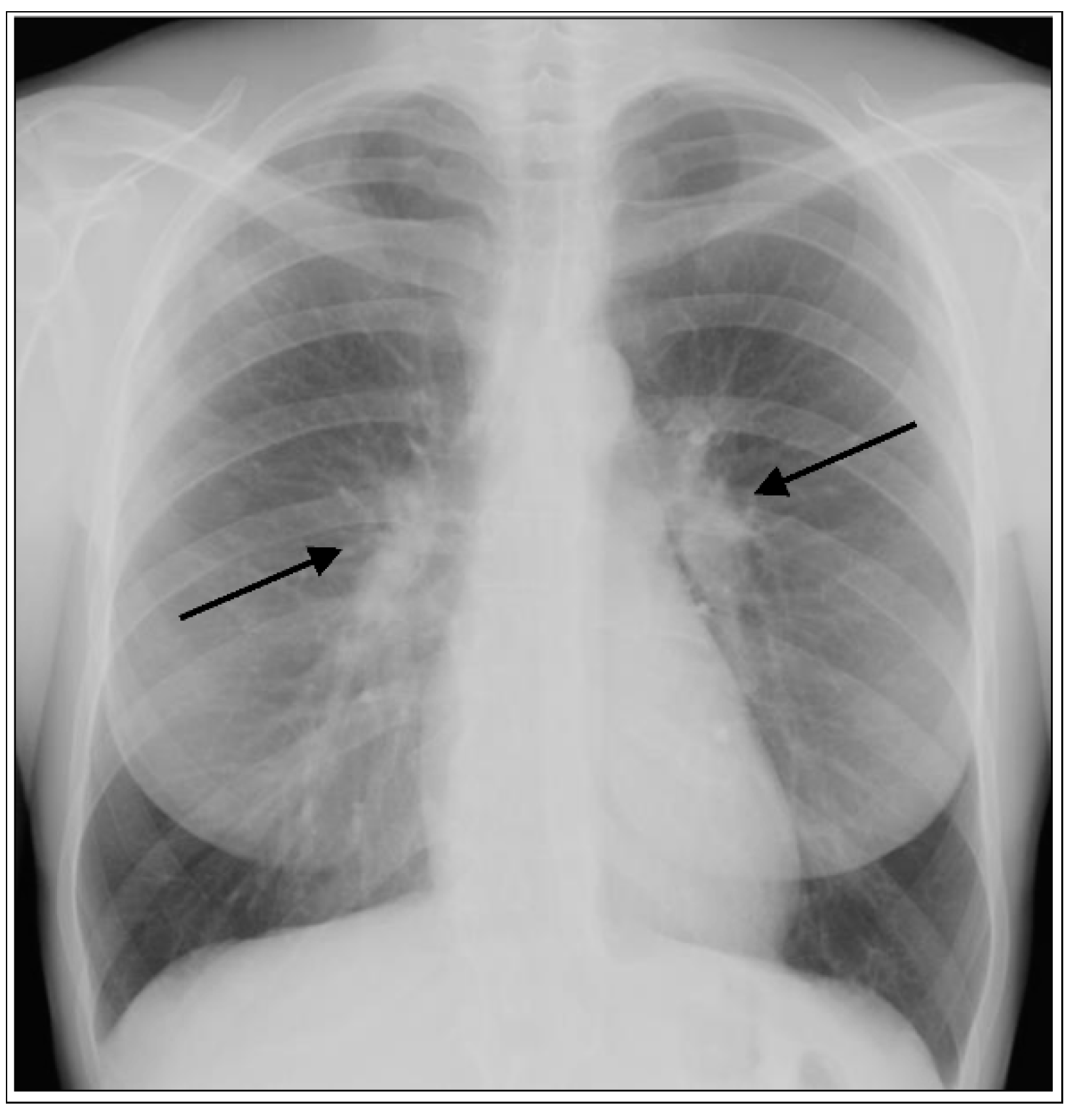
1.1. Lung
1.2. Skin
1.3. Eye
1.4. Heart
1.5. Brain
2. Subtypes of Sarcoidosis and Outcomes
2.1. Lofgren’s Syndrome (LS)
2.2. Subacute/Chronic Sarcoidosis
2.3. Mortality in Sarcoidosis
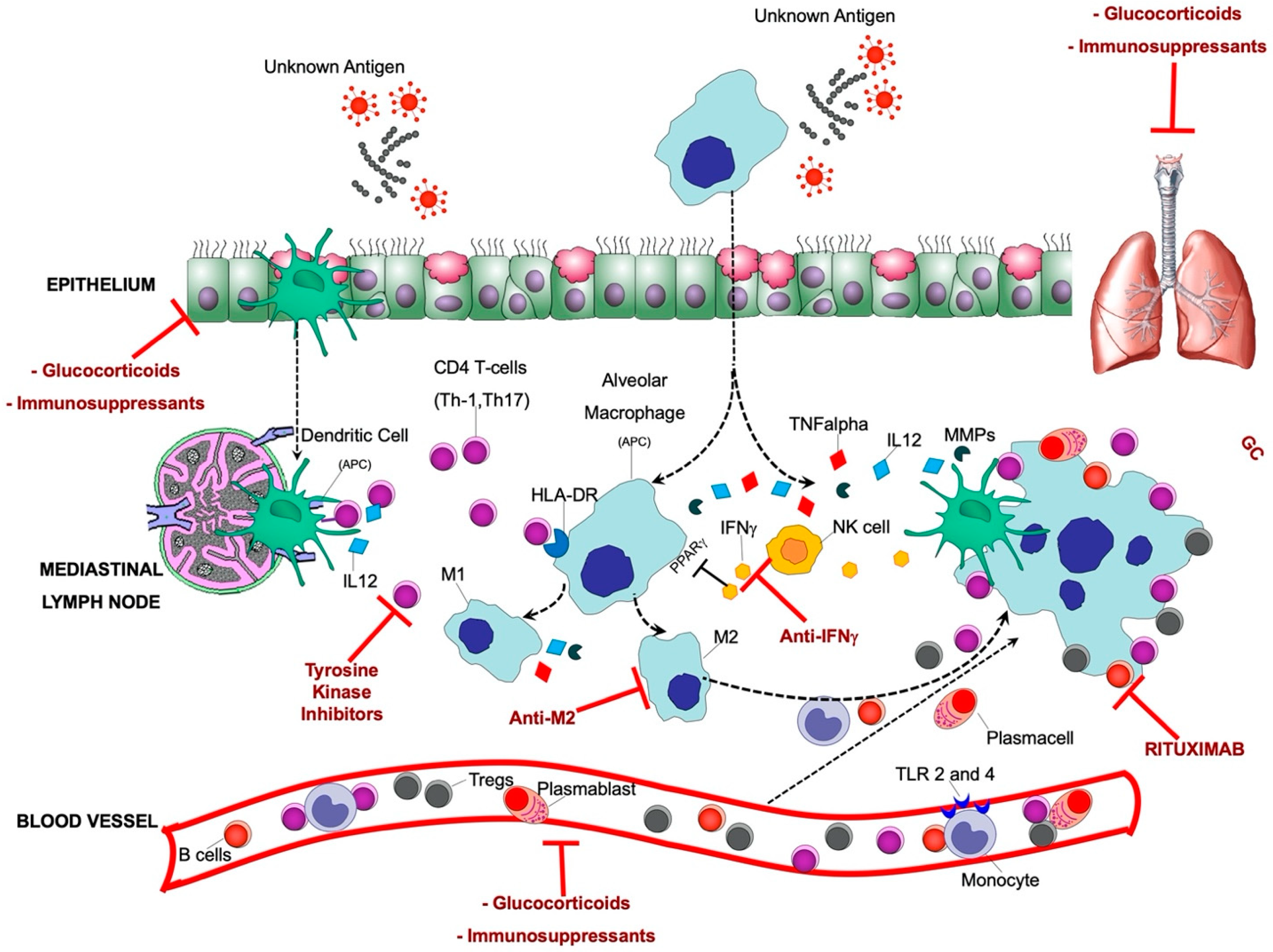
3. Pathogenesis of Sarcoidosis
3.1. Putative Antigens
3.2. Innate Immune Cells
3.3. Adaptive Immune Cells
3.3.1. T Cells
3.3.2. B Cells
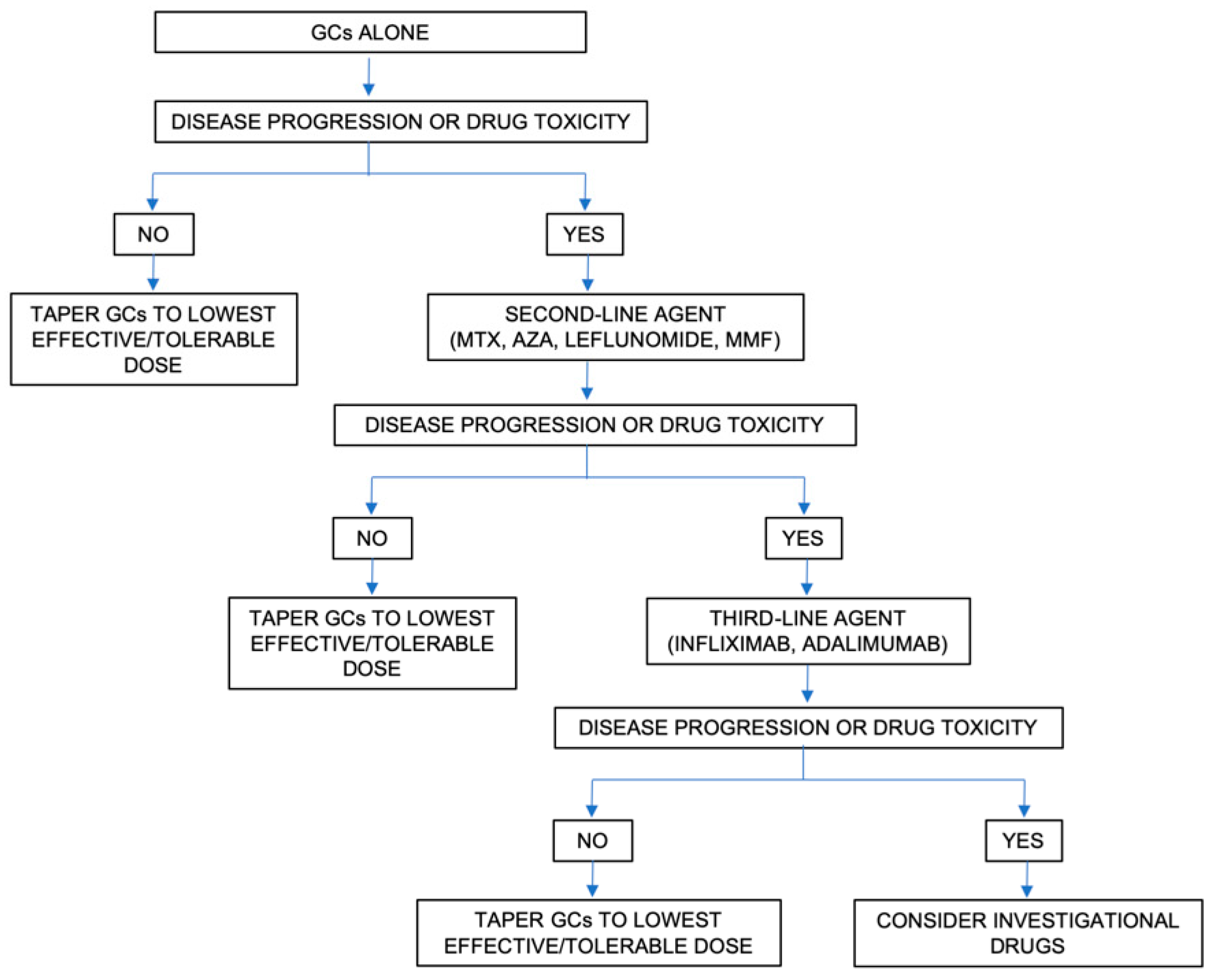
4. Pharmacological Treatment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hunninghake, G.W.; Costabel, U.; Ando, M.; Baughman, R.; Cordier, J.F.; Du Bois, R.; Eklund, A.; Kitaichi, M.; Lynch, J.; Rizzato, G.; et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis. 1999, 16, 149–173. [Google Scholar] [PubMed]
- Arkema, E.V.; Grunewald, J.; Kullberg, S.; Eklund, A.; Askling, J. Sarcoidosis incidence and prevalence: A nationwide register-based assessment in Sweden. Eur. Respir. J. 2016, 48, 1690–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valeyre, D.; Prasse, A.; Nunes, H.; Uzunhan, Y.; Brillet, P.Y.; Muller-Quernheim, J. Sarcoidosis. Lancet 2014, 383, 1155–1167. [Google Scholar] [CrossRef]
- Baughman, R.P.; Teirstein, A.S.; A Hudson, M.; Rossman, M.D.; Yeager, H.; Bresnitz, E.A.; DePalo, L.; Hunninghake, G.; Iannuzzi, M.C.; Johns, C.J.; et al. Clinical Characteristics of Patients in a Case Control Study of Sarcoidosis. Am. J. Respir. Crit. Care Med. 2001, 164, 1885–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mana, J.; Marcoval, J. Skin manifestations of sarcoidosis. Presse Med. 2012, 41, e355–e374. [Google Scholar] [CrossRef]
- Hubail, A.; Belkharoeva, R.; Tepluk, N.; Belerosova, T. Lupus pernio (Besnier-Tenneson syndrome): A rare form of sarcoidosis. Dermatol. Rep. 2018, 10, 7696. [Google Scholar] [CrossRef]
- Pasadhika, S.; Rosenbaum, J.T. Ocular Sarcoidosis. Clin. Chest Med. 2015, 36, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Bonfioli, A.A.; Orefice, F. Sarcoidosis. Semin. Ophthalmol. 2005, 20, 177–182. [Google Scholar] [CrossRef]
- Hamzeh, N.; Steckman, D.A.; Sauer, W.H.; Judson, M.A. Pathophysiology and clinical management of cardiac sarcoidosis. Nat. Rev. Cardiol. 2015, 12, 278–288. [Google Scholar] [CrossRef]
- Kandolin, R.; Lehtonen, J.; Airaksinen, J.; Vihinen, T.; Miettinen, H.; Ylitalo, K.; Kaikkonen, K.; Tuohinen, S.; Haataja, P.; Kerola, T.; et al. Cardiac sarcoidosis: Epidemiology, characteristics, and outcome over 25 years in a nationwide study. Circulation 2015, 131, 624–632. [Google Scholar] [CrossRef]
- Chapelon-Abric, C.; de Zuttere, D.; Duhaut, P.; Veyssier, P.; Wechsler, B.; Huong, D.L.T.; de Gennes, C.; Papo, T.; Blétry, O.; Godeau, P.; et al. Cardiac sarcoidosis: A retrospective study of 41 cases. Medicine (Baltimore) 2004, 83, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Lubitz, S.A.; Frankel, Z.; Wisnivesky, J.P.; Einstein, A.J.; Goldman, M.; Machac, J.; Teirstein, A. Cardiac Involvement in Patients with Sarcoidosis. Chest 2008, 133, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Judson, M.A.; Donnino, R.; Gold, M.; Cooper, L.T., Jr.; Prystowsky, E.N.; Prystowsky, S. Cardiac sarcoidosis. Am. Heart J. 2009, 157, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Handa, T.; Nagai, S.; Miki, S.; Fushimi, Y.; Ohta, K.; Mishima, M.; Izumi, T. Incidence of Pulmonary Hypertension and Its Clinical Relevance in Patients with Sarcoidosis. Chest 2006, 129, 1246–1252. [Google Scholar] [CrossRef] [Green Version]
- Ungprasert, P.; Matteson, E.L. Neurosarcoidosis. Rheum. Dis. Clin. N. Am. 2017, 43, 593–606. [Google Scholar] [CrossRef]
- Kellinghaus, C.; Schilling, M.; Ludemann, P. Neurosarcoidosis: Clinical experience and diagnostic pitfalls. Eur. Neurol. 2004, 51, 84–88. [Google Scholar] [CrossRef]
- Lower, E.E.; Weiss, K.L. Neurosarcoidosis. Clin. Chest Med. 2008, 29, 475–492. [Google Scholar] [CrossRef]
- Ungprasert, P.; Crowson, C.S.; Matteson, E.L. Characteristics and Long-Term Outcome of Neurosarcoidosis: A Population-Based Study from 1976–2013. Neuroepidemiology 2017, 48, 87–94. [Google Scholar] [CrossRef]
- Neville, E.; Walker, A.N.; James, D.G. Prognostic factors predicting the outcome of sarcoidosis: An analysis of 818 patients. Q. J. Med. 1983, 52, 525–533. [Google Scholar]
- Grunewald, J.; Brynedal, B.; Darlington, P.; Nisell, M.; Cederlund, K.; Hillert, J.; Eklund, A. Different HLA-DRB1 allele distributions in distinct clinical subgroups of sarcoidosis patients. Respir. Res. 2010, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Judson, M.A.; Marchell, R.M.; Mascelli, M.; Piantone, A.; Barnathan, E.S.; Petty, K.J.; Chen, D.; Fan, H.; Grund, H.; Ma, K.; et al. Molecular profiling and gene expression analysis in cutaneous sarcoidosis: The role of interleukin-12, interleukin-23, and the T-helper 17 pathway. J. Am. Acad. Dermatol. 2012, 66, 901–910.e2. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Sato, H.; Grunewald, J.; Brynedal, B.; Hillert, J.; Mañá, J.; Wells, A.U.; Eklund, A.; Welsh, K.I.; Du Bois, R.M. A common haplotype of the C-C chemokine receptor 2 gene andHLA-DRB1*0301are independent genetic risk factors for Löfgren’s syndrome. J. Intern. Med. 2008, 264, 433–441. [Google Scholar] [CrossRef]
- Smith, M.J.; Turton, C.W.; Mitchell, D.N.; Turner-Warwick, M.; Morris, L.M.; Lawler, S.D. Association of HLA B8 with spontaneous resolution in sarcoidosis. Thorax 1981, 36, 296–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, J.; Spagnolo, P.; Wahlstrom, J.; Eklund, A. Immunogenetics of Disease-Causing Inflammation in Sarcoidosis. Clin. Rev. Allergy Immunol. 2015, 49, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Rossman, M.; Thompson, B.; Frederick, M.; Iannuzzi, M.C.; A Rybicki, B.; Pander, J.P.; Newman, L.S.; Rose, C.; Magira, E.; Monos, D.S. HLA and environmental interactions in sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2008, 25, 125–132. [Google Scholar]
- Sato, H.; Woodhead, F.; Ahmad, T.; Grutters, J.C.; Spagnolo, P.; Bosch, J.M.V.D.; Maier, L.A.; Newman, L.S.; Nagai, S.; Izumi, T.; et al. Sarcoidosis HLA class II genotyping distinguishes differences of clinical phenotype across ethnic groups. Hum. Mol. Genet. 2010, 19, 4100–4111. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, J.; Eklund, A. Lofgren’s syndrome: Human leukocyte antigen strongly influences the disease course. Am. J. Respir. Crit. Care Med. 2009, 179, 307–312. [Google Scholar] [CrossRef]
- Drent, M.; Lower, E.E.; De Vries, J. Sarcoidosis-associated fatigue. Eur. Respir. J. 2012, 40, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Wahlström, J.; Katchar, K.; Wigzell, H.; Olerup, O.; Eklund, A.; Grunewald, J. Analysis of Intracellular Cytokines in CD4 + and CD8 + Lung and Blood T Cells in Sarcoidosis. Am. J. Respir. Crit. Care Med. 2001, 163, 115–121. [Google Scholar] [CrossRef]
- Conron, M.; Du Bois, R.M. Immunological mechanisms in sarcoidosis. Clin. Exp. Allergy 2001, 31, 543–554. [Google Scholar] [CrossRef]
- Nunes, H.; Humbert, M.; Capron, F.; Brauner, M.; Sitbon, O.; Battesti, J.-P.; Simonneau, G.; Valeyre, D. Pulmonary hypertension associated with sarcoidosis: Mechanisms, haemodynamics and prognosis. Thorax 2005, 61, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Sarcoidosis, race, and short-term outcomes following lung transplantation. Chest 2004, 125, 990–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, H.; Uzunhan, Y.; Gille, T.; Lamberto, C.; Valeyre, M.; Brillet, P.-Y. Imaging of sarcoidosis of the airways and lung parenchyma and correlation with lung function. Eur. Respir. J. 2012, 40, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Nardi, A.; Brillet, P.-Y.; Letoumelin, P.; Girard, F.; Brauner, M.; Uzunhan, Y.; Naccache, J.-M.; Valeyre, D.; Nunes, H. Stage IV sarcoidosis: Comparison of survival with the general population and causes of death. Eur. Respir. J. 2011, 38, 1368–1373. [Google Scholar] [CrossRef]
- Swigris, J.J.; Olson, A.L.; Huie, T.J.; Fernandez-Perez, E.R.; Solomon, J.; Sprunger, D.; Brown, K.K. Sarcoidosis-related mortality in the United States from 1988 to 2007. Am. J. Respir. Crit. Care Med. 2011, 183, 1524–1530. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Maucort-Boulch, D.; Kerever, S.; Gerfaud-Valentin, M.; Broussolle, C.; Eb, M.; Valeyre, M.; Seve, P. Sarcoidosis-related mortality in France: A multiple-cause-of-death analysis. Eur. Respir. J. 2016, 48, 1700–1709. [Google Scholar] [CrossRef] [Green Version]
- Rossides, M.; Kullberg, S.; Askling, J.; Eklund, A.; Grunewald, J.; Arkema, E. Sarcoidosis mortality in Sweden: A population-based cohort study. Eur. Respir. J. 2018, 51, 1701815. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, T.; Azuma, A.; Abe, S.; Usuki, J.; Kudoh, S.; Sugisaki, K.; Oritsu, M.; Nukiwa, T. Epidemiology of sarcoidosis in Japan. Eur. Respir. J. 2008, 31, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Iannuzzi, M.C.; Rybicki, B.A.; Teirstein, A.S. Sarcoidosis. N. Eng. J. Med. 2007, 357, 2153–2165. [Google Scholar] [CrossRef]
- Mathew, S.; Bauer, K.L.; Fischoeder, A.; Bhardwaj, N.; Oliver, S.J.; Saladino, K. The anergic state in sarcoidosis is associated with diminished dendritic cell function1. J. Immunol. 2008, 181, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Eishi, Y.; Suga, M.; Ishige, I.; Kobayashi, D.; Yamada, T.; Takemura, T.; Takizawa, T.; Koike, M.; Kudoh, S.; Costabel, U.; et al. Quantitative Analysis of Mycobacterial and Propionibacterial DNA in Lymph Nodes of Japanese and European Patients with Sarcoidosis. J. Clin. Microbiol. 2002, 40, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, J.; Kaiser, Y.; Ostadkarampour, M.; Rivera, N.V.; Vezzi, F.; Lötstedt, B.; Olsen, R.-A.; Sylwan, L.; Lundin, S.; Käller, M.; et al. T-cell receptor–HLA-DRB1 associations suggest specific antigens in pulmonary sarcoidosis. Eur. Respir. J. 2015, 47, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, C.; Thillai, M.; Parker, R.; Siddiqui, N.; Potiphar, L.; Goldin, R.; Timms, J.F.; Wells, A.U.; Kon, O.M.; Wickremasinghe, M.; et al. Proteomic Analysis of Kveim Reagent Identifies Targets of Cellular Immunity in Sarcoidosis. PLoS ONE 2017, 12, e0170285. [Google Scholar] [CrossRef]
- Ryan, G.B.; Spector, W.G. Natural selection of long-lived macrophages in experimental granulomata. J. Pathol. 1969, 99, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, H.; Zissel, G.; Goldmann, T.; Tschernig, T.; Vollmer, E.; Pabst, R.; Quernheim, J.M. Alveolar macrophages are the main source for tumour necrosis factor-α in patients with sarcoidosis. Eur. Respir. J. 2003, 21, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiken, M.; Grunewald, J.; Eklund, A.; Wahlstrom, J. Higher monocyte expression of TLR2 and TLR4, and enhanced pro-inflammatory synergy of TLR2 with NOD2 stimulation in sarcoidosis. J. Clin. Immunol. 2009, 29, 78–89. [Google Scholar] [CrossRef]
- Linke, M.; Pham, H.T.T.; Katholnig, K.; Schnöller, T.; Miller, A.; Demel, F.; Schütz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat. Immunol. 2017, 18, 293–302. [Google Scholar] [CrossRef]
- Dubaniewicz, A.; Typiak, M.; Wybieralska, M.; Szadurskaa, M.; Nowakowskia, S.; Staniewicz-Panasika, A.; Rogoza, K.; Sternau, A.; Deeg, P.; Trzonkowski, P. Changed phagocytic activity and pattern of Fcgamma and complement receptors on blood monocytes in sarcoidosis. Hum. Immunol. 2012, 73, 788–794. [Google Scholar] [CrossRef]
- Gabrilovich, M.I.; Walrath, J.; Van Lunteren, J.; Nethery, D.; Seifu, M.; Kern, J.A.; Harding, C.V.; Tuscano, L.; Lee, H.; Williams, S.D.; et al. Disordered Toll-like receptor 2 responses in the pathogenesis of pulmonary sarcoidosis. Clin. Exp. Immunol. 2013, 173, 512–522. [Google Scholar] [CrossRef]
- Wojtan, P.; Mierzejewski, M.; Osińska, I.; Domagała-Kulawik, J. Macrophage polarization in interstitial lung diseases. Central Eur. J. Immunol. 2016, 41, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Shamaei, M.; Mortaz, E.; Porabdollah, M.; Garssen, J.; Tabarsie, P.; Velayati, A.; Adcock, I.M. Evidence for M2 macrophages in granulomas from pulmonary sarcoidosis: A new aspect of macrophage heterogeneity. Hum. Immunol. 2018, 79, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Medbury, H.J.; James, V.; Ngo, J.; Hitos, K.; Wang, Y.; Harris, D.C.; Fletcher, J.P. Differing association of macrophage subsets with atherosclerotic plaque stability. Int. Angiol. 2013, 32, 74–84. [Google Scholar] [PubMed]
- Robinson, B.W.; McLemore, T.L.; Crystal, R.G. Gamma interferon is spontaneously released by alveolar macrophages and lung T lymphocytes in patients with pulmonary sarcoidosis. J. Clin. Investig. 1985, 75, 1488–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culver, D.A.; Barna, B.P.; Raychaudhuri, B.; Bonfield, T.L.; Abraham, S.; Malur, A.; Farver, C.F.; Kavuru, M.S.; Thomassen, M.J. Peroxisome Proliferator–Activated Receptor γ Activity Is Deficient in Alveolar Macrophages in Pulmonary Sarcoidosis. Am. J. Respir. Cell Mol. Boil. 2004, 30, 1–5. [Google Scholar] [CrossRef]
- Zaba, L.C.; Smith, G.P.; Sanchez, M.; Prystowsky, S.D. Dendritic Cells in the Pathogenesis of Sarcoidosis. Am. J. Respir. Cell Mol. Boil. 2010, 42, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Ringkowski, S.; Thomas, P.S.; Herbert, C. Interleukin-12 family cytokines and sarcoidosis. Front. Pharmacol. 2014, 5, 233. [Google Scholar] [CrossRef] [Green Version]
- Forman, J.D.; Klein, J.T.; Silver, R.F.; Liu, M.C.; Greenlee, B.M.; Moller, D.R. Selective activation and accumulation of oligoclonal V beta-specific T cells in active pulmonary sarcoidosis. J. Clin. Investig. 1994, 94, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Pang, C.; Wu, Y.; Li, D.; Wan, C.; Liao, Z.; Yang, T.; Chen, L.; Wen, F. Diagnostic Performance of Bronchoalveolar Lavage Fluid CD4/CD8 Ratio for Sarcoidosis: A Meta-analysis. EBioMedicine 2016, 8, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Oswald-Richter, K.A.; Richmond, B.W.; Braun, N.A.; Isom, J.; Abraham, S.; Taylor, T.R.; Drake, J.M.; Culver, D.A.; Wilkes, D.S.; Drake, W.P. Reversal of global CD4+ subset dysfunction is associated with spontaneous clinical resolution of pulmonary sarcoidosis. J. Immunol. 2013, 190, 5446–5453. [Google Scholar] [CrossRef] [Green Version]
- Miyara, M.; Amoura, Z.; Parizot, C.; Badoual, C.; Dorgham, K.; Trad, S.; Kambouchner, M.; Valeyre, D.; Chapelon-Abric, C.; Debré, P.; et al. The immune paradox of sarcoidosis and regulatory T cells. J. Exp. Med. 2006, 203, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, C.; Shaginurova, G.; Shelton, D.A.; Herazo-Maya, J.D.; Oswald-Richter, K.A.; Rotsinger, J.; Young, A.; Celada, L.J.; Kaminski, N.; Sevin, C.; et al. Local and Systemic CD4+ T Cell Exhaustion Reverses with Clinical Resolution of Pulmonary Sarcoidosis. J. Immunol. Res. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, G.; Liu, X.; Ngo, K.; De Leon-Tabaldo, A.; Zhao, S.; Luna-Roman, R.; Yu, J.; Cao, T.; Kuhn, R.; Wilkinson, P.; et al. RORgammat and RORalpha signature genes in human Th17 cells. PLoS ONE 2017, 12, e0181868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Dai, J.; Cai, H.; Gao, Q.; Wen, Y. Extensively disturbance of regulatory T cells - Th17 cells balance in stage II pulmonary sarcoidosis. Int. J. Med. Sci. 2017, 14, 1136–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taflin, C.; Miyara, M.; Nochy, D.; Valeyre, M.; Naccache, J.-M.; Altare, F.; Salek-Peyron, P.; Badoual, C.; Bruneval, P.; Haroche, J.; et al. FoxP3+ Regulatory T Cells Suppress Early Stages of Granuloma Formation but Have Little Impact on Sarcoidosis Lesions. Am. J. Pathol. 2009, 174, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappl, G.; Pabst, S.; Riemann, D.; Schmidt, A.; Wickenhauser, C.; Schütte, W.; Hombach, A.; Seliger, B.; Grohe, C.; Abken, H. Regulatory T cells with reduced repressor capacities are extensively amplified in pulmonary sarcoid lesions and sustain granuloma formation. Clin. Immunol. 2011, 140, 71–83. [Google Scholar] [CrossRef]
- Greaves, S.A.; Atif, S.M.; Fontenot, A.P. Adaptive Immunity in Pulmonary Sarcoidosis and Chronic Beryllium Disease. Front. Immunol. 2020, 11, 474. [Google Scholar] [CrossRef] [Green Version]
- Atif, S.M.; Mack, D.G.; McKee, A.S.; Rangel-Moreno, J.; Martin, A.K.; Getahun, A.; Maier, L.; Cambier, J.C.; Tuder, R.M.; Fontenot, A.P. Protective role of B cells in sterile particulate–induced lung injury. JCI Insight 2019, 4, 5. [Google Scholar] [CrossRef]
- Hunninghake, G.W.; Crystal, R.G. Mechanisms of hypergammaglobulinemia in pulmonary sarcoidosis. Site of increased antibody production and role of T lymphocytes. J. Clin. Investig. 1981, 67, 86–92. [Google Scholar] [CrossRef]
- Kamphuis, L.S.; Van Zelm, M.C.; Lam, K.H.; Rimmelzwaan, G.F.; Baarsma, G.S.; Dik, W.A.; Thio, H.B.; Van Daele, P.L.; Van Velthoven, M.E.; Batstra, M.R.; et al. Perigranuloma Localization and Abnormal Maturation of B Cells. Am. J. Respir. Crit. Care Med. 2013, 187, 406–416. [Google Scholar] [CrossRef]
- Starshinova, A.A.; Malkova, A.M.; Basantsova, N.Y.; Zinchenko, Y.S.; Kudryavtsev, I.V.; Ershov, G.A.; Soprun, L.A.; Mayevskaya, V.A.; Churilov, L.P.; Yablonskiy, P.K. Sarcoidosis as an Autoimmune Disease. Front. Immunol. 2020, 10, 2933. [Google Scholar] [CrossRef]
- El Jammal, T.; Jamilloux, Y.; Gerfaud-Valentin, M.; Valeyre, D.; Sève, P. Refractory Sarcoidosis: A Review. Ther. Clin. Risk Manag. 2020, 16, 323–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnell, J.; Kumar, V.; Wang, J.; Wang, S.; Voynova, E.; Ettinger, R. Role of CD11c + T-bet + B cells in human health and disease. Cell. Immunol. 2017, 321, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, M.; Alter, S.; Frasca, D.; Blomberg, B.B.; Riley, R.L. In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell 2013, 12, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Phalke, S.; Aviszus, K.; Rubtsova, K.; Rubtsov, A.; Barkes, B.; Powers, L.; Warner, B.; Crooks, J.L.; Kappler, J.W.; Fernández-Pérez, E.R.; et al. Age Associated B Cells Appear in Patients with Granulomatous Lung Diseases. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; A Hudson, M.; Teirstein, A.; Yeager, H.; Rossman, M.; Knatterud, G.; Thompson, B. Presenting characteristics as predictors of duration of treatment in sarcoidosis. QJM 2006, 99, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Rossi, G.; Trisolini, R.; Sverzellati, N.; Baughman, R.P.; Wells, A.U. Pulmonary sarcoidosis. Lancet Respir. Med. 2018, 6, 389–402. [Google Scholar] [CrossRef]
- Baughman, R.P.; Lower, E.E. Treatment of Sarcoidosis. Clin. Rev. Allergy Immunol. 2015, 49, 79–92. [Google Scholar] [CrossRef]
- Eklund, A.; Du Bois, R.M. Approaches to the treatment of some of the troublesome manifestations of sarcoidosis. J. Intern. Med. 2014, 275, 335–349. [Google Scholar] [CrossRef]
- Judson, M.A. The treatment of pulmonary sarcoidosis. Respir. Med. 2012, 106, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Judson, M.A. An Approach to the Treatment of Pulmonary Sarcoidosis with Corticosteroids. Chest 1999, 115, 1158–1165. [Google Scholar] [CrossRef]
- Rizzato, G.; Montemurro, L.; Colombo, P. The late follow-up of chronic sarcoid patients previously treated with corticosteroids. Sarcoidosis Vasc. Diffus. Lung Dis. 1998, 15, 52–58. [Google Scholar]
- Paramothayan, S.; Lasserson, T. Treatments for pulmonary sarcoidosis. Respir. Med. 2008, 102, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zic, J.A.; Horowitz, D.H.; Arzubiaga, C.; King, L.E. Treatment of Cutaneous Sarcoidosis With Chloroquine. Arch. Dermatol. 1991, 127, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Korsten, P.; Mirsaeidi, M.; Sweiss, N.J. Nonsteroidal therapy of sarcoidosis. Curr. Opin. Pulm. Med. 2013, 19, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Schutt, A.C.; Bullington, W.M.; A Hudson, M. Pharmacotherapy for pulmonary sarcoidosis: A delphi consensus study. Respir. Med. 2010, 104, 717–723. [Google Scholar] [CrossRef] [Green Version]
- Baughman, R.P.; Grutters, J.C. New treatment strategies for pulmonary sarcoidosis: Antimetabolites, biological drugs, and other treatment approaches. Lancet Respir. Med. 2015, 3, 813–822. [Google Scholar] [CrossRef]
- Vorselaars, A.D.M.; Wuyts, W.A.; Vorselaars, V.M.M.; Zanen, P.; Deneer, V.H.M.; Veltkamp, M.; Thomeer, M.; Van Moorsel, C.H.M.; Grutters, J.C. Methotrexate vs Azathioprine in Second-line Therapy of Sarcoidosis. Chest 2013, 144, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Lower, E.; Baughman, R.P. Prolonged use of methotrexate for sarcoidosis. Arch. Intern. Med. 1995, 155, 846–851. [Google Scholar] [CrossRef]
- Hamzeh, N.; Voelker, A.; Forssen, A.; Gottschall, E.B.; Rose, C.; Mroz, P.; Maier, L.A. Efficacy of mycophenolate mofetil in sarcoidosis. Respir. Med. 2014, 108, 1663–1669. [Google Scholar] [CrossRef] [Green Version]
- Baughman, R.P.; Drent, M.; Kavuru, M.; Judson, M.A.; Costabel, U.; du Bois, R.; Albera, C.; Brutsche, M.; Davis, G.; Donohue, J.F.; et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am. J. Respir. Crit. Care Med. 2006, 174, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Vorselaars, A.D.; Crommelin, H.A.; Deneer, V.H.; Meek, B.; Claessen, A.M.; Keijsers, R.G.; Van Moorsel, C.H.; Grutters, J.C. Effectiveness of infliximab in refractory FDG PET-positive sarcoidosis. Eur. Respir. J. 2015, 46, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweiss, N.J.; Noth, I.; Mirsaeidi, M.; Zhang, W.; Naureckas, E.T.; Hogarth, D.K.; Strek, M.; Caligiuri, P.; Machado, R.F.; Niewold, T.B.; et al. Efficacy Results of a 52-week Trial of Adalimumab in the Treatment of Refractory Sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2014, 31, 46–54. [Google Scholar]
- Crommelin, H.A.; Van Der Burg, L.M.; Vorselaars, A.D.; Drent, M.; Van Moorsel, C.H.; Rijkers, G.T.; Deneer, V.H.; Grutters, J.C. Efficacy of adalimumab in sarcoidosis patients who developed intolerance to infliximab. Diffus. Parenchymal. Lung Dis. 2016, 48, PA820. [Google Scholar] [CrossRef]
- Utz, J.P.; Limper, A.H.; Kalra, S.; Specks, U.; Scott, J.P.; Vuk-Pavlovic, Z.; Schroeder, D.R. Etanercept for the treatment of stage II and III progressive pulmonary sarcoidosis. Chest 2003, 124, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moravan, M.; Segal, B.M. Treatment of CNS sarcoidosis with infliximab and mycophenolate mofetil. Neurology 2009, 72, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Bradshaw, M.J.; Stern, B.J.; Clifford, D.B.; Wang, Y.; Cho, T.; Koth, L.L.; Hauser, S.L.; Dierkhising, J.; Vu, N.; et al. Infliximab for the treatment of CNS sarcoidosis. Neurology 2017, 89, 2092–2100. [Google Scholar] [CrossRef]
- Harper, L.J.; McCarthy, M.; Neto, M.L.R.; Hachamovitch, R.; Pearson, K.; Bonanno, B.; Shaia, J.; Brunken, R.; Joyce, E.; Culver, D.A.; et al. Infliximab for Refractory Cardiac Sarcoidosis. Am. J. Cardiol. 2019, 124, 1630–1635. [Google Scholar] [CrossRef]
- Sweiss, N.J.; Lower, E.E.; Mirsaeidi, M.; Dudek, S.; Garcia, J.G.N.; Perkins, D.; Finn, P.W.; Baughman, R.P. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur. Respir. J. 2014, 43, 1525–1528. [Google Scholar] [CrossRef]
- Reinisch, W.; de Villiers, W.; Bene, L.; Simon, L.; Rácz, I.; Katz, S.; Altorjay, I.; Feagan, B.; Riff, D.; Bernstein, C.N.; et al. Fontolizumab in moderate to severe Crohn’s disease: A phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm. Bowel Dis. 2010, 16, 233–242. [Google Scholar] [CrossRef]
- Sahoo, D.H.; Bandyopadhyay, D.; Xu, M.; Pearson, K.; Parambil, J.G.; Lazar, C.A.; Chapman, J.T.; Culver, D.A. Effectiveness and safety of leflunomide for pulmonary and extrapulmonary sarcoidosis. Eur. Respir. J. 2011, 38, 1145–1150. [Google Scholar] [CrossRef]
- Le, V.; Crouser, E.D. Potential immunotherapies for sarcoidosis. Expert Opin. Biol. Ther. 2018, 18, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C. Lung transplantation for pulmonary sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2019, 36, 92–107. [Google Scholar]
- Taimeh, Z.; I Hertz, M.; Shumway, S.; Pritzker, M. Lung transplantation for pulmonary sarcoidosis. Twenty-five years of experience in the USA. Thorax 2016, 71, 378–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingaraju, R.; Pochettino, A.; Blumenthal, N.P.; Mendez, J.; Lee, J.; Christie, J.D.; Kotloff, R.M.; Ahya, V.N.; Hadjiliadis, D. Lung Transplant Outcomes in White and African American Recipients: Special Focus on Acute and Chronic Rejection. J. Hear. Lung Transplant. 2009, 28, 8–13. [Google Scholar] [CrossRef]
- Perkel, D.; Czer, L.; Morrissey, R.; Ruzza, A.; Rafiei, M.; Awad, M.; Patel, J.; Kobashigawa, J. Heart Transplantation for End-Stage Heart Failure Due to Cardiac Sarcoidosis. Transplant. Proc. 2013, 45, 2384–2386. [Google Scholar] [CrossRef]
- Theofilogiannakos, E.K.; Pettit, S.J.; Ghazi, A.; Rassl, R.; Lewis, C.; Parameshwar, J. Heart transplantation for advanced heart failure due to cardiac sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2015, 32, 208–214. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ Involvement | Frequency, % |
|---|---|
| Lung | 90 |
| Skin (excluding EN) | 16 |
| Erythema nodosum (EN) | 8 |
| Eye | 12 |
| Extrathoracic lymph node | 15.2 |
| Liver | 12 |
| Spleen | 7 |
| Neurologic | 5 |
| Cardiac | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polverino, F.; Balestro, E.; Spagnolo, P. Clinical Presentations, Pathogenesis, and Therapy of Sarcoidosis: State of the Art. J. Clin. Med. 2020, 9, 2363. https://doi.org/10.3390/jcm9082363
Polverino F, Balestro E, Spagnolo P. Clinical Presentations, Pathogenesis, and Therapy of Sarcoidosis: State of the Art. Journal of Clinical Medicine. 2020; 9(8):2363. https://doi.org/10.3390/jcm9082363
Chicago/Turabian StylePolverino, Francesca, Elisabetta Balestro, and Paolo Spagnolo. 2020. "Clinical Presentations, Pathogenesis, and Therapy of Sarcoidosis: State of the Art" Journal of Clinical Medicine 9, no. 8: 2363. https://doi.org/10.3390/jcm9082363
APA StylePolverino, F., Balestro, E., & Spagnolo, P. (2020). Clinical Presentations, Pathogenesis, and Therapy of Sarcoidosis: State of the Art. Journal of Clinical Medicine, 9(8), 2363. https://doi.org/10.3390/jcm9082363