Impact of Short-Term Hypoxia on Sirtuins as Regulatory Elements in HUVECs

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Cell Culture
2.2. Hypoxia and Normoxia Conditions
2.3. RNA Isolation
2.4. cDNA Synthesis
2.5. Quantitative Real-Time PCR for mRNA Expression Analysis
2.6. Western Blot Analysis
2.7. SIRT1 and SIRT3 Activity Assay
2.8. Determination of NAD+
2.9. Statistical Methods
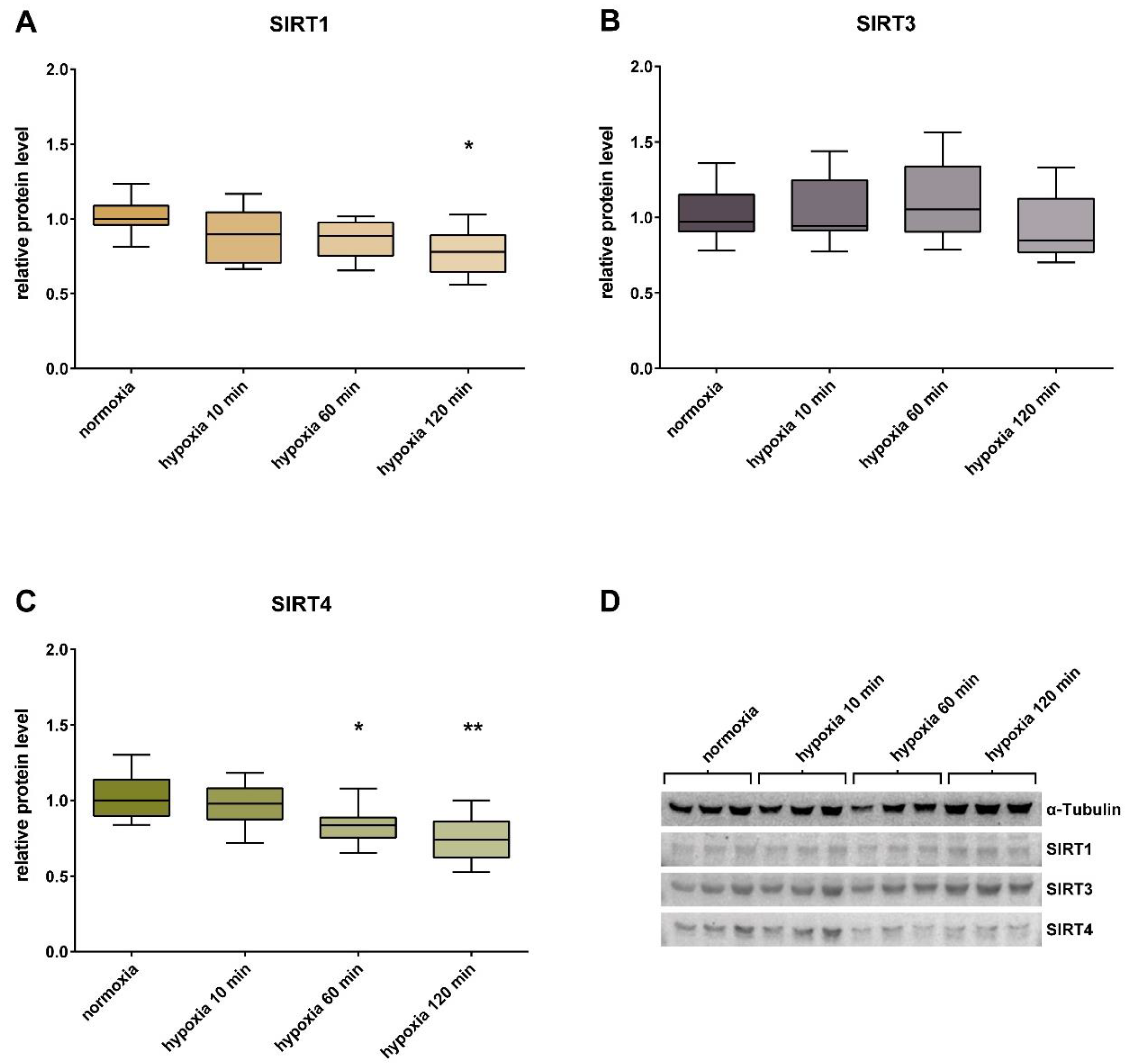
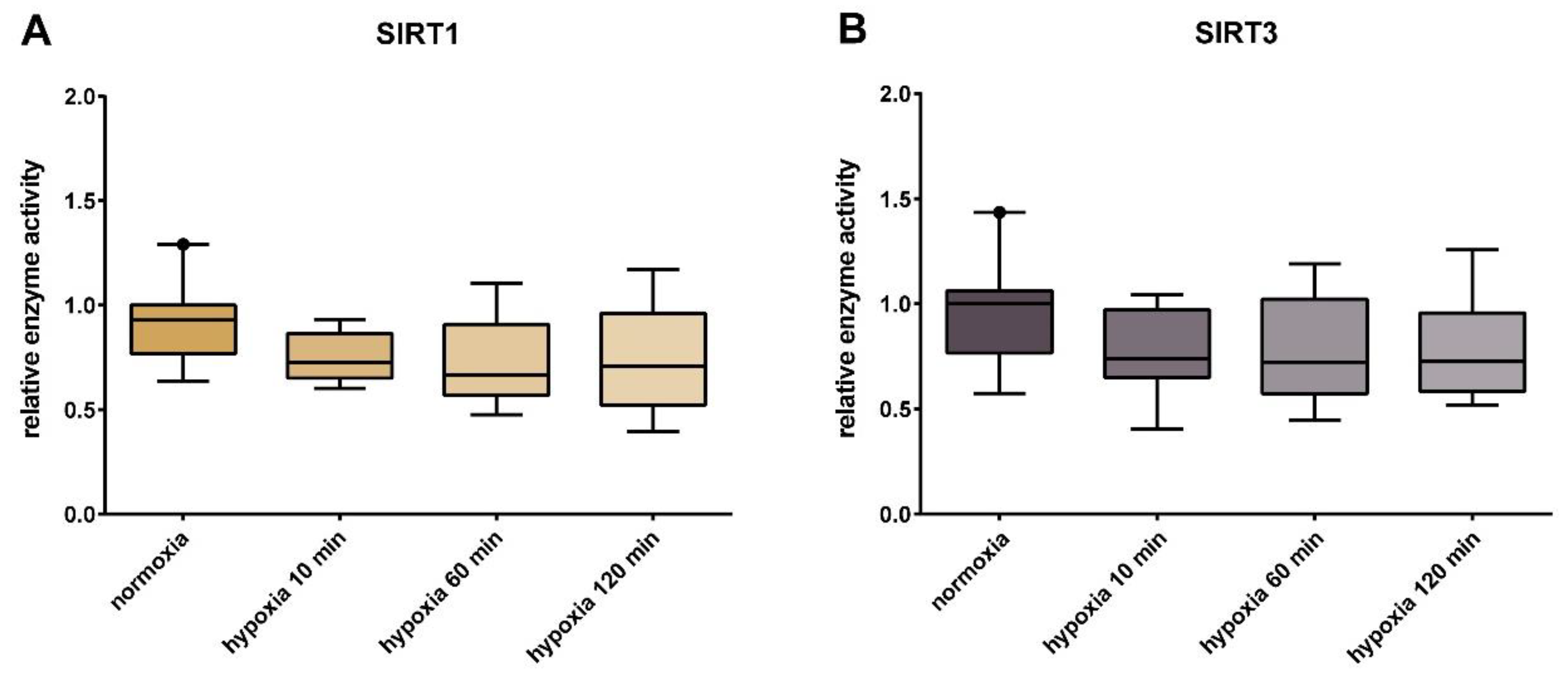
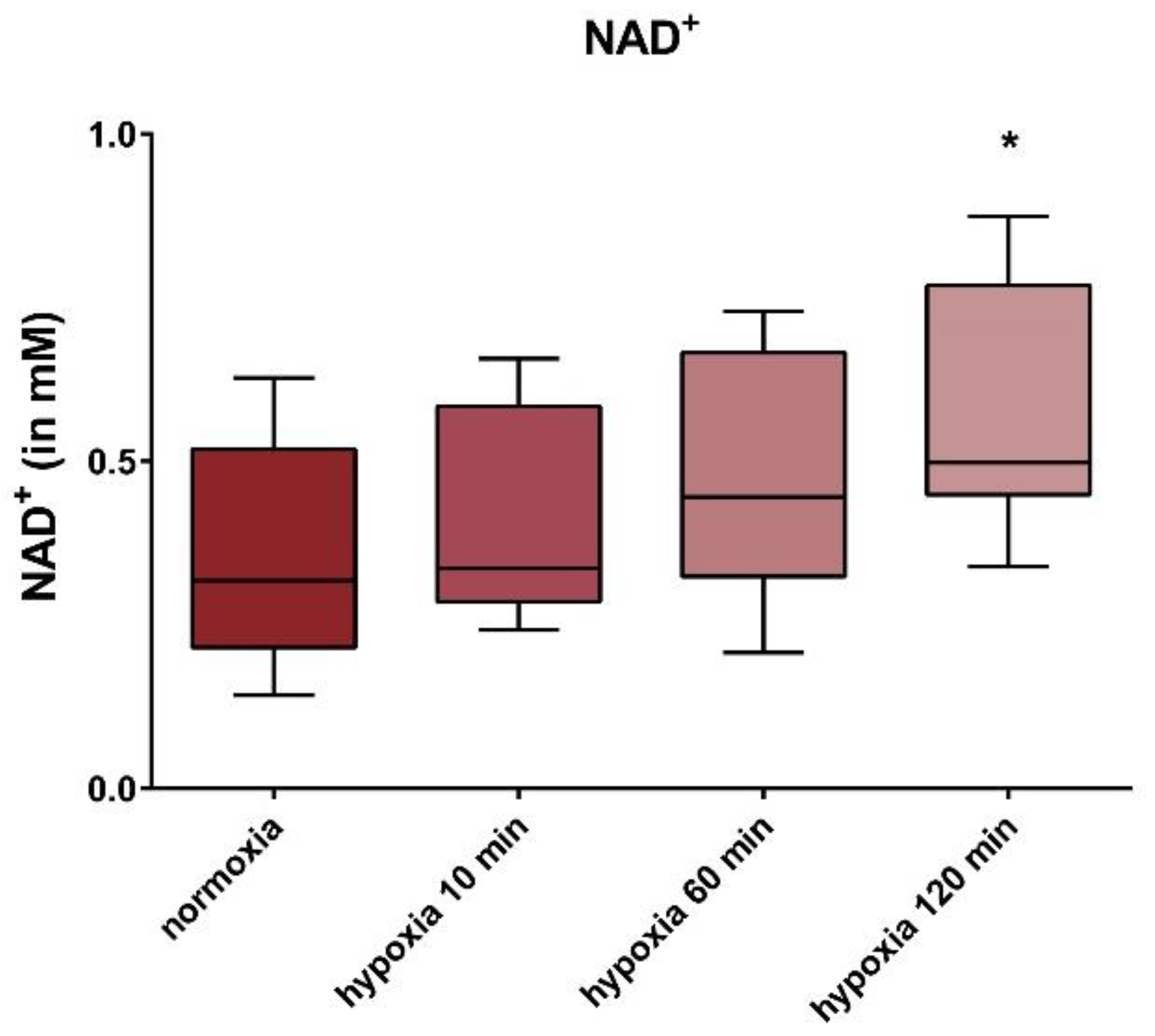
3. Results
4. Discussion
5. A Clinical Medical Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Kida, Y.; Goligorsky, M.S. Sirtuins, cell senescence, and vascular aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, A.W.C.; Li, H.; Xia, N. The role of sirtuin1 in regulating endothelial function, arterial remodeling and vascular aging. Front. Physiol. 2019, 10, 1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Zeng, H.; Chen, J.X. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J. Cell. Physiol. 2019, 234, 2252–2265. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xia, N.; Hasselwander, S.; Daiber, A. Resveratrol and vascular function. Int. J. Mol. Sci. 2019, 20, 2155. [Google Scholar] [CrossRef] [Green Version]
- Braunstein, M.; Rose, A.B.; Holmes, S.G.; Allis, C.D.; Broach, J.R. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 1993, 7, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Shore, D.; Squire, M.; Nasmyth, K.A. Characterization of two genes required for the position-effect control of yeast mating-type genes. EMBO J. 1984, 3, 2817–2823. [Google Scholar] [CrossRef]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin catalysis and regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, G.A.; Golino, M.; Villano, A.; Lanza, O.; Lamendola, P.; Fusc, A.; Leggio, M. Cardiac rehabilitation and endothelial function. J. Clin. Med. 2020, 9, 2487. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, J.G. NAD(+), sirtuins, and cardiovascular disease. Curr. Pharm. Des. 2009, 15, 110–117. [Google Scholar] [CrossRef]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef]
- Ham, P.B., 3rd; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef] [Green Version]
- Winnik, S.; Auwerx, J.; Sinclair, D.A.; Matter, C.M. Protective effects of sirtuins in cardiovascular diseases: From bench to bedside. Eur. Heart J. 2015, 36, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Sosnowska, B.; Mazidi, M.; Penson, P.; Gluba-Brzozka, A.; Rysz, J.; Banach, M. The sirtuin family members SIRT1, SIRT3 and SIRT6: Their role in vascular biology and atherogenesis. Atherosclerosis 2017, 265, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, X.F.; Chen, H.Z.; Liu, D.P. Mitochondrial sirtuins in cardiometabolic diseases. Clin. Sci. (Lond.) 2017, 131, 2063–2078. [Google Scholar] [CrossRef]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 2011, 589, 4545–4554. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging (Albany N. Y.) 2016, 8, 2290–2307. [Google Scholar] [CrossRef] [Green Version]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Laina, A.; Stellos, K.; Stamatelopoulos, K. Vascular ageing: Underlying mechanisms and clinical implications. Exp. Gerontol. 2018, 109, 16–30. [Google Scholar] [CrossRef]
- Verdin, E. NAD⁺ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Dabke, P. Sirtuins, Mitochondria and Exercise in Health and Diseases; Kenneth, M., Ed.; Sirtuin Biology in Cancer and Metabolic Diseases; Elsevier: London, UK, 2020; in press. [Google Scholar]
- Stein, S.; Matter, C.M. Protective roles of SIRT1 in atherosclerosis. Cell. Cycle 2011, 10, 640–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Sandvoss, M.; Potthast, A.B.; von Versen-Hoynck, F.; Das, A.M. HELLP syndrome. Reprod. Sci. 2017, 24, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, B. Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol. 2012, 64, 309–320. [Google Scholar]
- Griesmacher, A.; Windischbauer, A.; Muller, M.M. Endothelial cells’ responses to hypoxia and reperfusion. Adv. Exp. Med. Biol. 1994, 370, 299–302. [Google Scholar]
- Berna, N.; Arnould, T.; Remacle, J.; Michiels, C. Hypoxia-induced increase in intracellular calcium concentration in endothelial cells: Role of the Na(+)-glucose cotransporter. J. Cell. Biochem. 2001, 84, 115–131. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Li, X. SIRT1 performs a balancing act on the tight-rope toward longevity. Aging (Albany N. Y.) 2009, 1, 669–673. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, S.; Arimoto, A.; Kuramoto, Y.; Kozako, T.; Honda, S.; Shimeno, H.; Soeda, S. Fasting promotes the expression of SIRT1, an NAD+-dependent protein deacetylase, via activation of PPARalpha in mice. Mol. Cell. Biochem. 2010, 339, 285–292. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef] [Green Version]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M., Jr.; Sindler, A.L.; Seals, D.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.A.; Hirschey, M.D. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012, 52, 23–25. [Google Scholar] [CrossRef]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [Green Version]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [Green Version]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potthast, A.B.; Nebl, J.; Wasserfurth, P.; Haufe, S.; Eigendorf, J.; Hahn, A.; Das, A. Impact of nutrition on short-term exercise-induced sirtuin regulation: Vegans differ from omnivores and lacto-ovo vegetarians. Nutrients 2020, 12, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimen, H.; Han, M.J.; Yang, Y.; Tong, Q.; Koc, H.; Koc, E.C. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 2010, 49, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.T.; Lee, H.C.; Liao, C.C.; Wei, Y.H. Regulation of mitochondrial F(o)F(1)ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977bp deletion of mitochondrial DNA. Biochim. Biophys. Acta 2013, 1832, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [Green Version]
- Tseng, A.H.; Wu, L.H.; Shieh, S.S.; Wang, D.L. SIRT3 interactions with FOXO3 acetylation, phosphorylation and ubiquitinylation mediate endothelial cell responses to hypoxia. Biochem. J. 2014, 464, 157–168. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany N. Y.) 2010, 2, 914–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.; Titus, A.S.; Banerjee, K.K.; George, S.; Lin, W.; Deota, S.; Saha, A.K.; Nakamura, K.; Gut, P.; Verdin, E.; et al. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging (Albany N. Y.) 2013, 5, 835–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich-Merzenich, G.; Metzner, C.; Bhonde, R.R.; Malsch, G.; Schiermeyer, B.; Vetter, H. Simultaneous isolation of endothelial and smooth muscle cells from human umbilical artery or vein and their growth response to low-density lipoproteins. In Vitro Cell. Dev. Biol. Anim. 2002, 38, 265–272. [Google Scholar] [CrossRef]
- Illsinger, S.; Janzen, N.; Sander, S.; Schmidt, K.H.; Bednarczyk, J.; Mallunat, L.; Bode, J.; Hagebolling, F.; Hoy, L.; Lucke, T.; et al. Preeclampsia and HELLP syndrome: Impaired mitochondrial function in umbilical endothelial cells. Reprod. Sci. 2010, 17, 219–226. [Google Scholar] [CrossRef]
- Brodowski, L.; Schröder-Heurich, B.; Hubel, C.A.; Vu, T.H.; von Kaisenberg, C.S.; von Versen-Höynck, F. Role of vitamin D in cell-cell interaction of fetal endothelial progenitor cells and umbilical cord endothelial cells in a preeclampsia-like model. Am. J. Physiol. Cell. Physiol. 2019, 317, C348–C357. [Google Scholar] [CrossRef]
- Brunssen, C.; Korten, S.; Brux, M.; Seifert, S.; Roesler, J.; Bornstein, S.R.; Morawietz, H.; Goettsch, W. COUP-TFII is regulated by high glucose in endothelial cells. Horm. Metab. Res. 2010, 42, 81–87. [Google Scholar] [CrossRef]
- Wein, Y.; Shira, E.B.; Friedman, A. Increased serum levels of advanced glycation end products due to induced molting in hen layers trigger a proinflammatory response by peripheral blood leukocytes. Poult. Sci. 2020, 99, 3452–3462. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Münzel, T. Endothelial dysfunction: Pathophysiology, diagnosis and prognosis. Dtsch. Med. Wochenschr. 2008, 133, 2465–2470. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Ortiz, K.; Perez-Vazquez, V.; Macias-Cervantes, M.H. Exercise and Sirtuins: A Way to Mitochondrial Health in Skeletal Muscle. Int. J. Mol. Sci. 2019, 20, 2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merksamer, P.I.; Liu, Y.; He, W.; Hirschey, M.D.; Chen, D.; Verdin, E. The sirtuins, oxidative stress and aging: An emerging link. Aging (Albany N. Y.) 2013, 5, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Gui, J.; Potthast, A.; Rohrbach, A.; Borns, K.; Das, A.M.; von Versen-Hoynck, F. Gestational diabetes induces alterations of sirtuins in fetal endothelial cells. Pediatr. Res. 2016, 79, 788–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J. Biol. Chem. 2011, 286, 13869–13878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Xu, M.; Hogg, R.T.; Li, J.; Little, B.; Gerard, R.D.; Garcia, J.A. The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. J. Biol. Chem. 2012, 287, 30800–30811. [Google Scholar] [CrossRef] [Green Version]
- Thirupathi, A.; de Souza, C.T. Multi-regulatory network of ROS: The interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 2017, 73, 487–494. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Hattori, Y.; Okamoto, Y.; Nagatsuka, K.; Takahashi, R.; Kalaria, R.N.; Kinoshita, M.; Ihara, M. SIRT1 attenuates severe ischemic damage by preserving cerebral blood flow. Neuroreport 2015, 26, 113–117. [Google Scholar] [CrossRef]
- Hattori, Y.; Okamoto, Y.; Maki, T.; Yamamoto, Y.; Oishi, N.; Yamahara, K.; Nagatsuka, K.; Takahashi, R.; Kalaria, R.N.; Fukuyama, H.; et al. Silent information regulator 2 homolog 1 counters cerebral hypoperfusion injury by deacetylating endothelial nitric oxide synthase. Stroke 2014, 45, 3403–3411. [Google Scholar] [CrossRef] [Green Version]
- Potthast, A.B.; Heuer, T.; Warneke, S.J.; Das, A.M. Alterations of sirtuins in mitochondrial cytochrome c-oxidase deficiency. PLoS ONE 2017, 12, e0186517. [Google Scholar] [CrossRef] [Green Version]
- Xiong, S.; Salazar, G.; Patrushev, N.; Alexander, R.W. FoxO1 mediates an autofeedback loop regulating SIRT1 expression. J. Biol. Chem. 2011, 286, 5289–5299. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar] [CrossRef]
- Lombard, D.B.; Tishkoff, D.X.; Bao, J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb. Exp. Pharmacol. 2011, 206, 163–188. [Google Scholar] [CrossRef] [Green Version]
- Cross, R.L.; Muller, V. The evolution of A-, F-, and V-type ATP synthases and ATPases: Reversals in function and changes in the H+/ATP coupling ratio. FEBS Lett. 2004, 576, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Di Lisa, F.; Fogolari, F.; Lippe, G. From ATP to PTP and back. A dual function for the mitochondrial ATP synthase. Circ. Res. 2015, 116, 1850–1862. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, D.; Goldstein, A.C.; El-Khoury, R.; Rak, M.; Edmunds, L.; Rustin, P.; Vockley, J.; Schiff, M. ANT2-defective fibroblasts exhibit normal mitochondrial bioenergetics. Mol. Genet. Metab. Rep. 2015, 3, 43–46. [Google Scholar] [CrossRef]
- Tabatabaie, T.; Potts, J.D.; Floyd, R.A. Reactive oxygen species-mediated inactivation of pyruvate dehydrogenase. Arch. Biochem. Biophys. 1996, 336, 290–296. [Google Scholar] [CrossRef]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.X.; Tang, X.; An, X.Z.; Xie, X.M.; Chen, X.F.; Zhao, X.; Hao, D.L.; Chen, H.Z.; Liu, D.P. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur. Heart J. 2017, 38, 1389–1398. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Huang, C.; Huang, Y.; Hong, L.; Wang, H.; Zhou, Z.; Qiu, Y. SIRT4 suppresses inflammatory responses in human umbilical vein endothelial cells. Cardiovasc. Toxicol. 2015, 15, 217–223. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecher, S.J.; Potthast, A.B.; von Versen-Höynck, F.; Das, A.M. Impact of Short-Term Hypoxia on Sirtuins as Regulatory Elements in HUVECs. J. Clin. Med. 2020, 9, 2604. https://doi.org/10.3390/jcm9082604
Pecher SJ, Potthast AB, von Versen-Höynck F, Das AM. Impact of Short-Term Hypoxia on Sirtuins as Regulatory Elements in HUVECs. Journal of Clinical Medicine. 2020; 9(8):2604. https://doi.org/10.3390/jcm9082604
Chicago/Turabian StylePecher, Simone Johanna, Arne Björn Potthast, Frauke von Versen-Höynck, and Anibh Martin Das. 2020. "Impact of Short-Term Hypoxia on Sirtuins as Regulatory Elements in HUVECs" Journal of Clinical Medicine 9, no. 8: 2604. https://doi.org/10.3390/jcm9082604