
Transcriptomic Analysis Reveals the Mechanism of MtLOX24 in Response to Methyl Jasmonate Stress in Medicago truncatula

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Cloning of MtLOX24 and Its Genetic Transformation
2.3. MeJA Treatment of M. truncatula
2.4. Measurement of Physiological Parameters
2.5. Sampling and RNA Extraction
2.6. Transcriptome Sequencing and De Novo Assembly
2.7. qRT-PCR Analysis
2.8. Differential Expression Gene Screening and Annotation Analysis
2.9. Statistical Analysis
3. Results
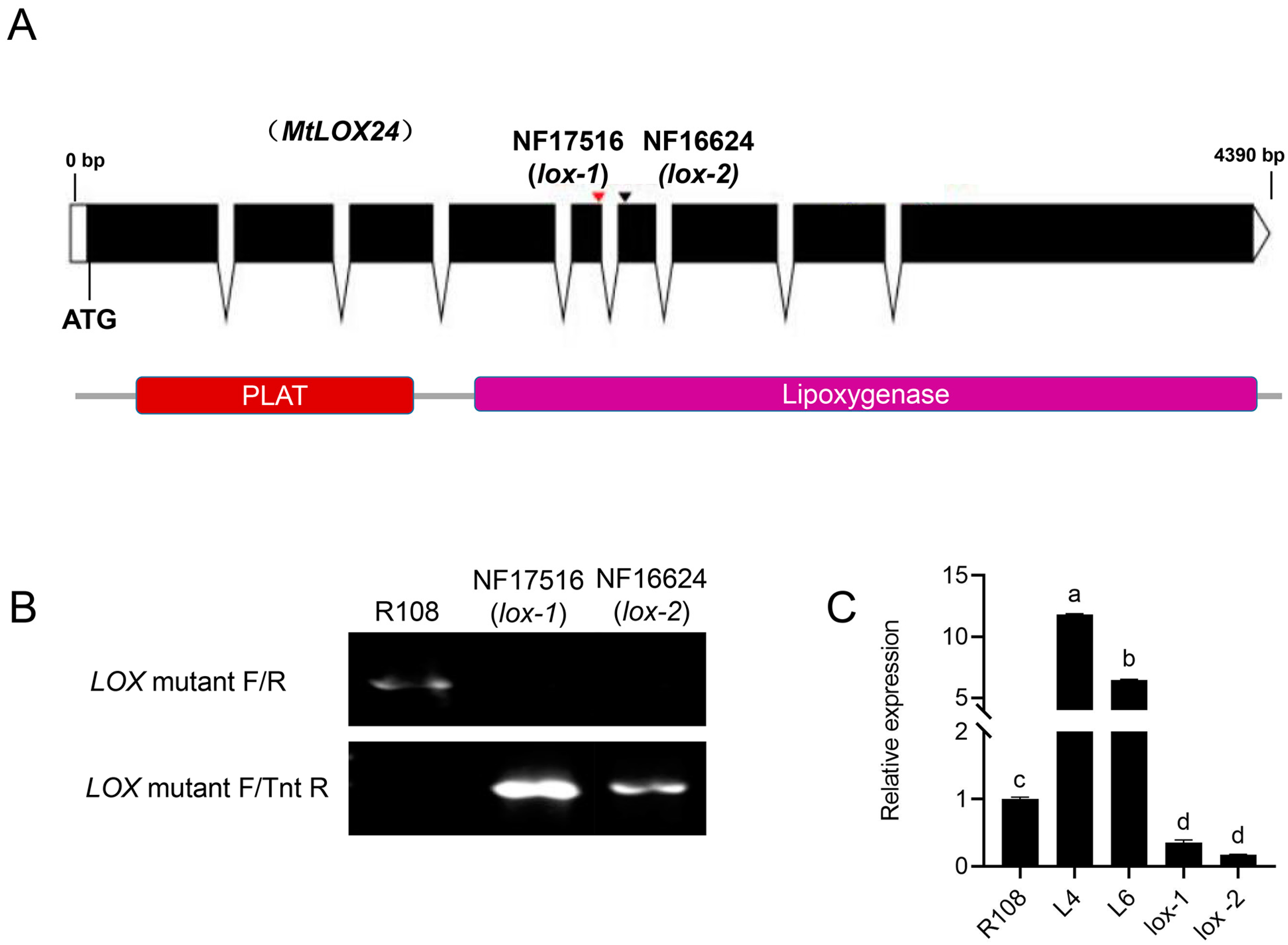
3.1. Identification of Lox Mutants and Generation of MtLOX24 Overexpression Lines
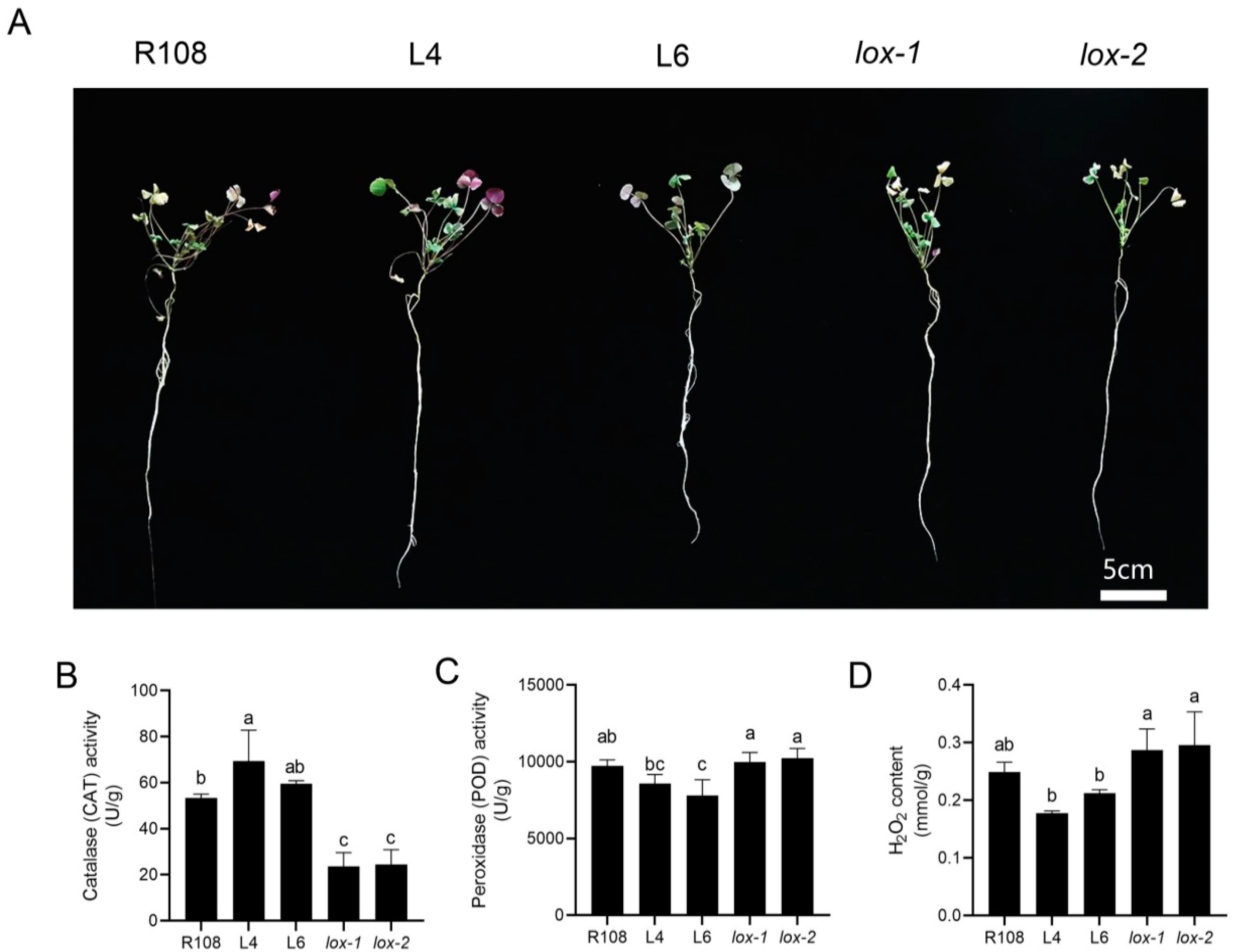
3.2. Physiological Characteristics of Medicago truncatula Treated with Exogenous MeJA
3.3. Sequencing Data Quality Analysis
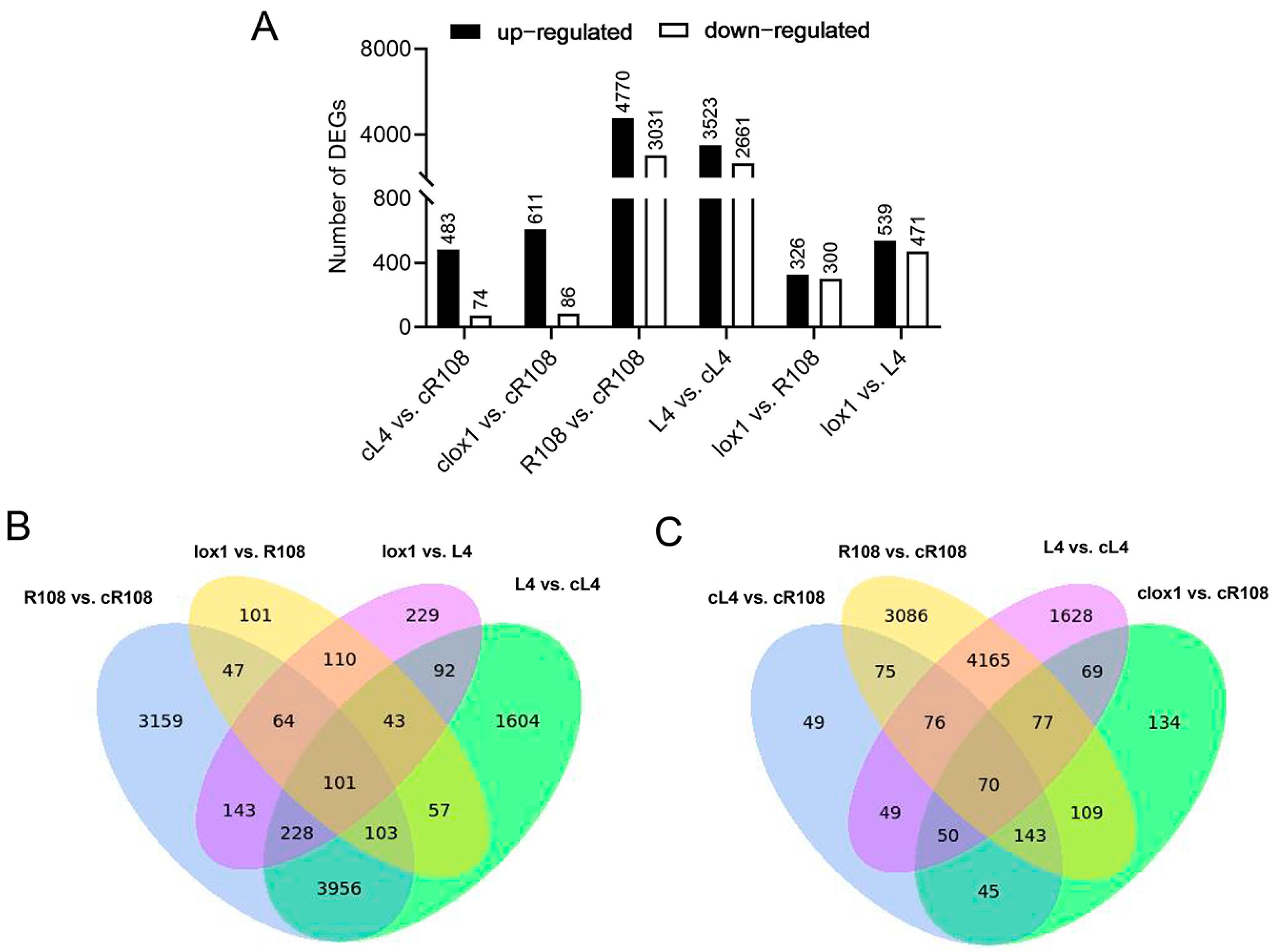
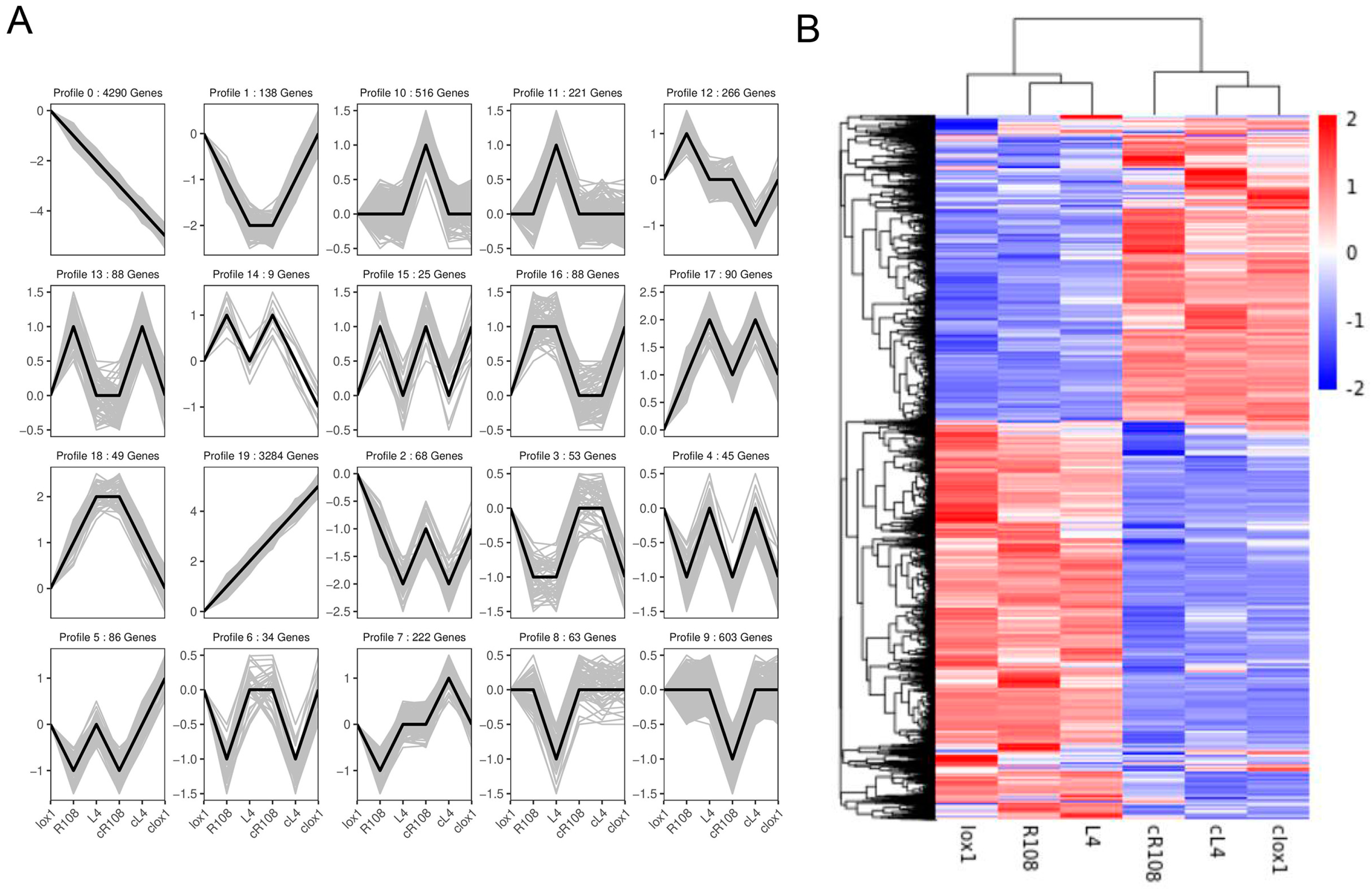
3.4. Expression Patterns and Cluster Analysis of Differentially Expressed Genes (DEGs)
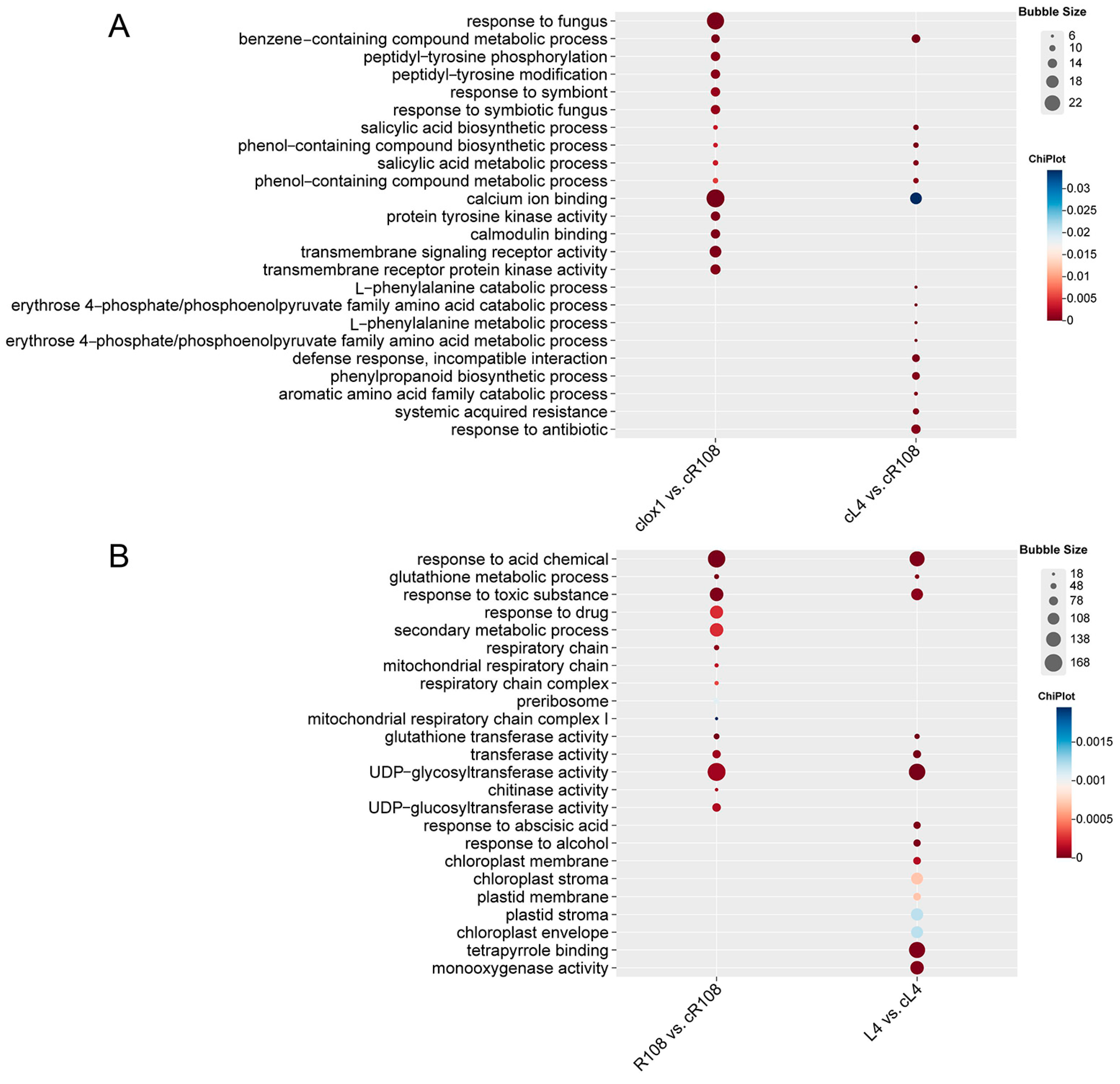
3.5. GO Enrichment Analysis of DEGs
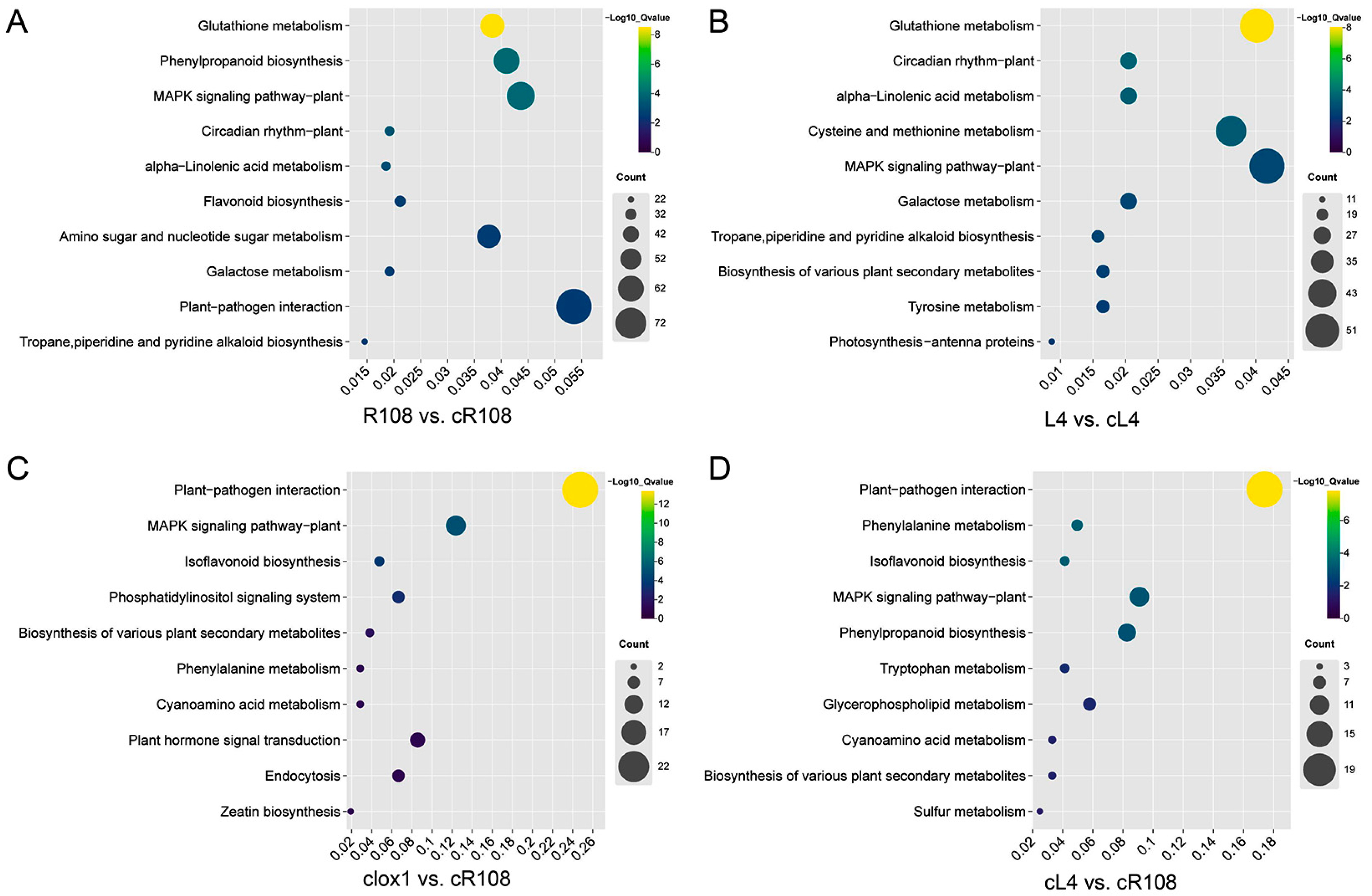
3.6. KEGG Enrichment Analysis of DEGs
3.7. Analysis of DEGs and Related Pathways in Response to MeJA Stress in M. truncatula
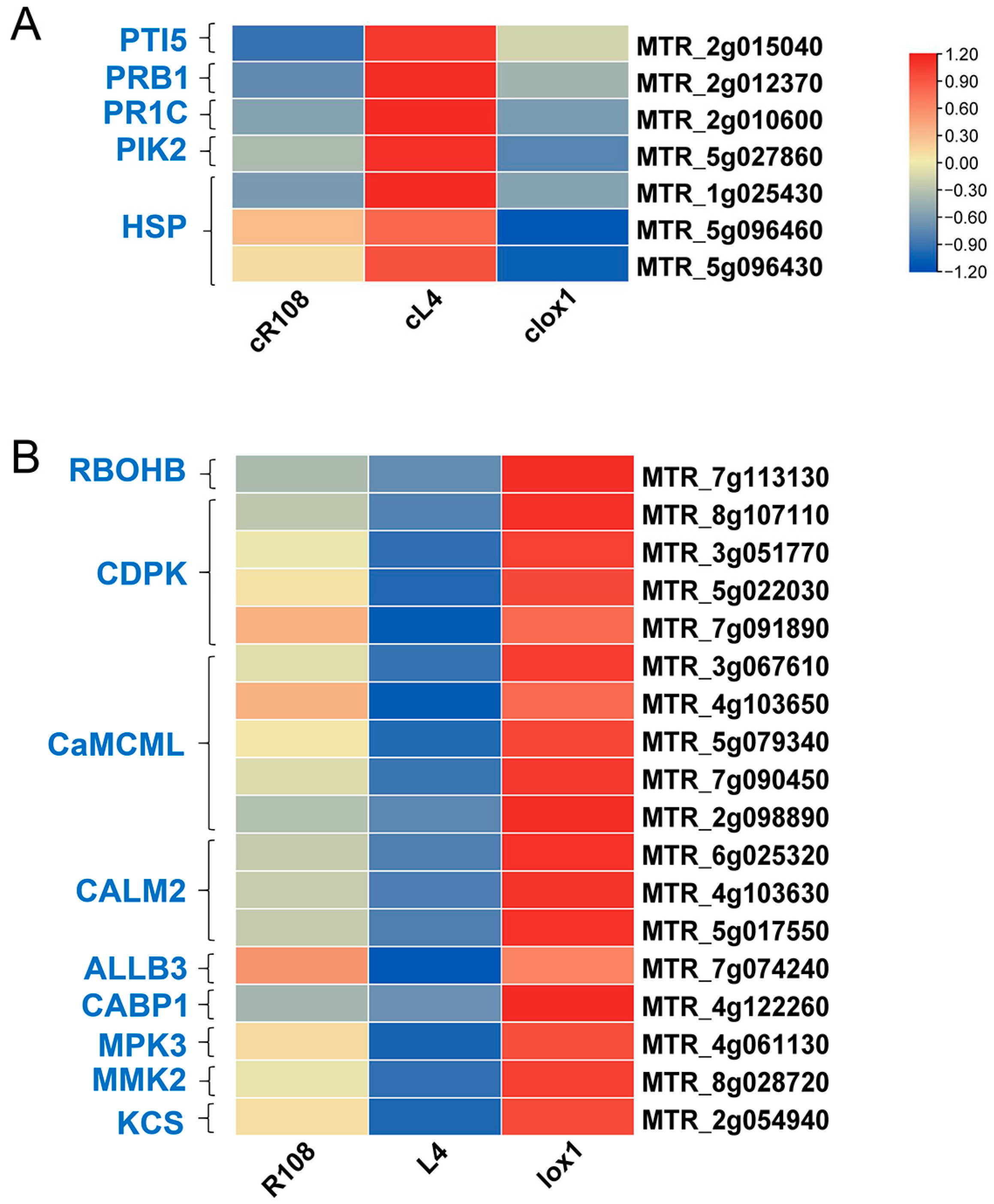
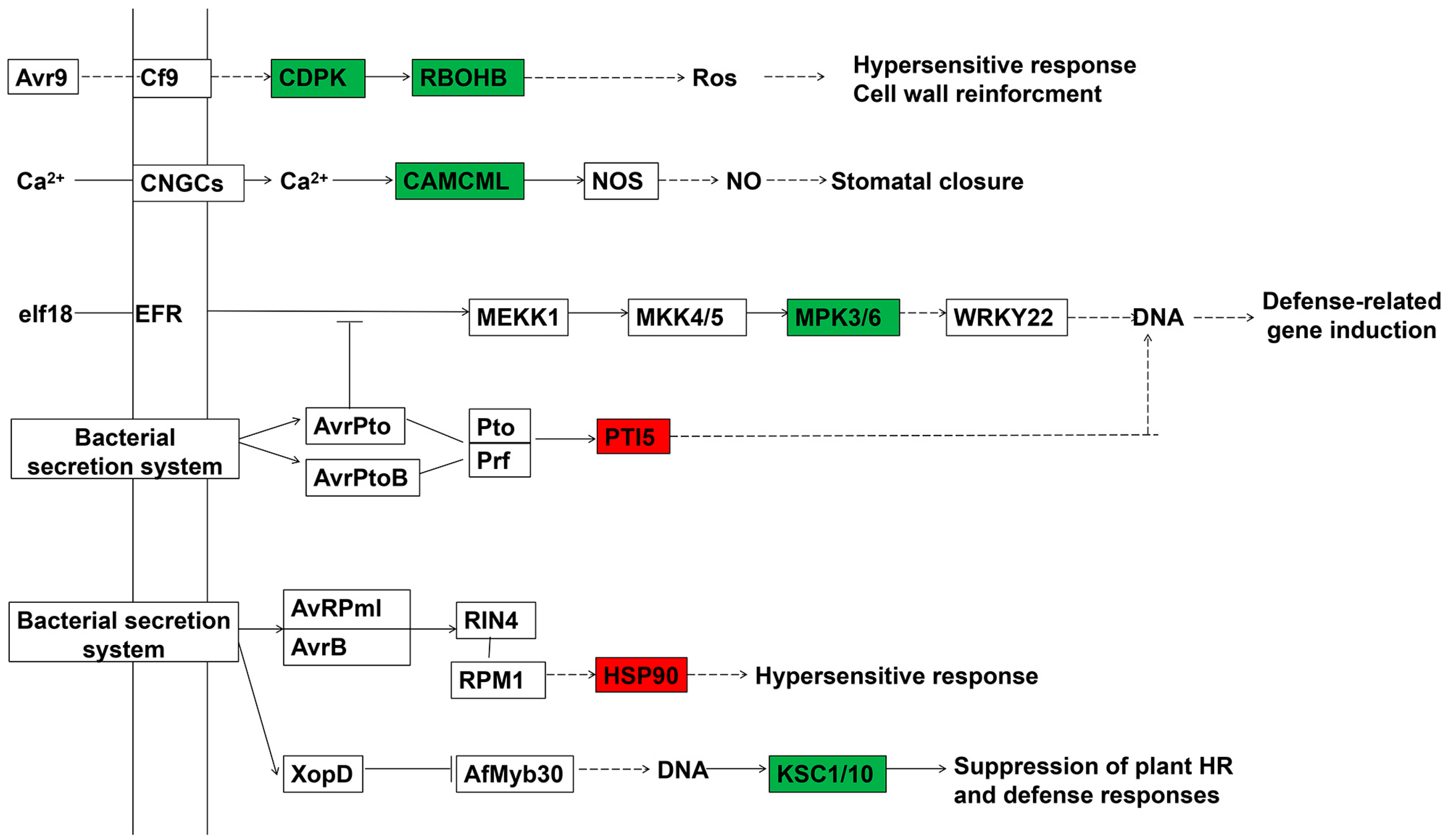
3.7.1. Analysis of DEGs Related to Plant–Pathogen Interactions
3.7.2. Analysis of DEGs Related to Phenylpropanoid Biosynthesis
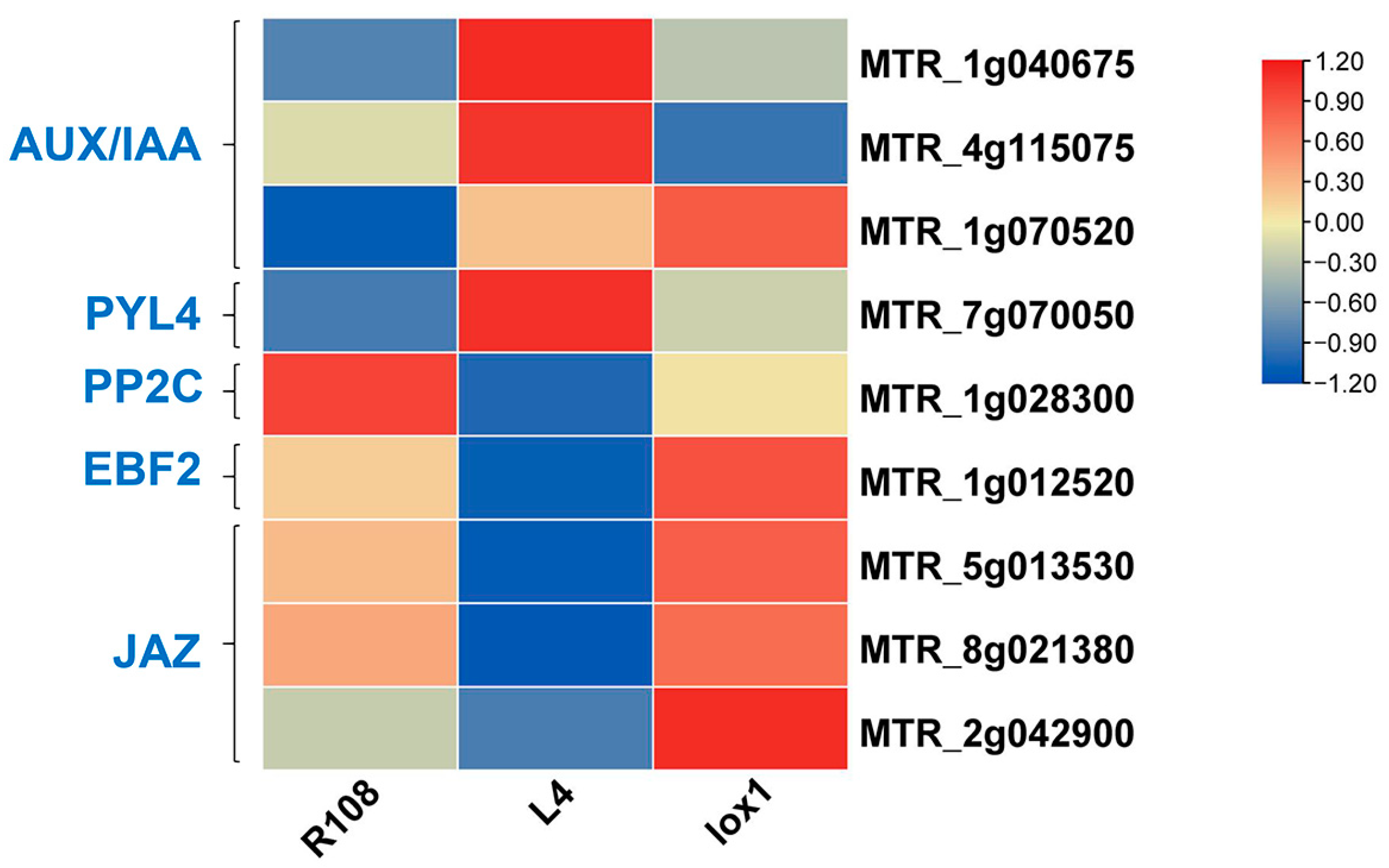
3.7.3. Analysis of DEGs Related to Plant Hormones
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, B.; Zhao, Y.; Guo, Z.F. Research Progress and Prospect of Alfalfa Resistance to Pathogens and Pests. Plants 2022, 11, 2008. [Google Scholar] [CrossRef]
- Li, J.; Shang, Q.; Liu, Y.; Dai, W.; Li, X.; Wei, S.; Hu, G.; McNeill, M.R.; Ban, L. Occurrence, Distribution, and Transmission of Alfalfa Viruses in China. Viruses 2022, 14, 1519. [Google Scholar] [CrossRef]
- Howe, G.A.; Major, I.T.; Koo, A.J. Modularity in Jasmonate Signaling for Multistress Resilience. Annu. Rev. Plant Biol. 2018, 69, 387–415. [Google Scholar] [CrossRef]
- Staswick, P.E.; Su, W.; Howell, S.H. Methyl Jasmonate Inhibition of Root Growth and Induction of a Leaf Protein are Decreased in an Arabidopsis thaliana mutant. Proc. Natl. Acad. Sci. USA 1992, 89, 6837–6840. [Google Scholar] [CrossRef]
- Hung, K.T.; Kao, C.H. Involvement of Lipid Peroxidation in Methyl Jasmonate-promoted Senescence in Detached Rice Leaves. Plant Growth Regul. 1998, 24, 17–21. [Google Scholar] [CrossRef]
- Wang, F.B.; Wan, C.Z.; Wu, W.Y.; Zhang, Y.N.; Pan, Y.X.; Chen, X.M.; Li, C.; Pi, J.L.; Wang, Z.X.; Ye, Y.X. Methyl Jasmonate (MeJA) Enhances Salt Tolerance of Okra (Abelmoschus esculentus L.) Plants by Regulating ABA Signaling, Osmotic Adjustment Substances, Photosynthesis and ROS Metabolism. Sci. Hortic. 2023, 319, 112145. [Google Scholar] [CrossRef]
- Wasternack, C.; Parthier, B. Jasmonate-signalled Plant Gene Expression. Trends Plant Sci. 1997, 2, 302–307. [Google Scholar] [CrossRef]
- Chang, L.J.; Wu, S.; Tian, L. Methyl Jasmonate Elicits Distinctive Hydrolyzable Tannin, Flavonoid, and Phyto-oxylipin Responses in Pomegranate (Punica granatum L.) Leaves. Planta 2021, 254, 89. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.B.; Guan, W.L.; Yang, Y.J.; Shao, Y.L.; Mao, L.C. Methyl Jasmonate Promotes Wound Healing by Activation of Phenylpropanoid Metabolism in Harvested Kiwifruit. Postharvest Biol. Technol. 2021, 175, 111472. [Google Scholar] [CrossRef]
- Thaler, J.S.; Stout, M.J.; Karban, R.; Duffey, S.S. Exogenous Jasmonates Simulate Insect Wounding in Tomato Plants (Lycopersicon esculentum) in the Laboratory and Field. J. Chem. Ecol. 1996, 22, 1767. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, X.; Yan, H.W.; Li, W.W.; Li, Y.; Cai, R.H.; Xiang, Y. The Lipoxygenase Gene Family in Poplar: Identification, Classification, and Expression in Response to MeJA Treatment. PLoS ONE 2015, 10, e0125526. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.H.; Zhang, W.; Shen, Y.B.; Fu, H.J.; Su, X.H.; Zhang, Z.Y. Activities of Lipoxygenase and Phenylalanine Ammonia Lyase in Poplar Leaves Induced by Insect Herbivory and Volatiles. J. For. Res. 2009, 20, 372. [Google Scholar] [CrossRef]
- Ogunola, O.F.; Hawkins, L.K.; Mylroie, E.; Kolomiets, M.V.; Borrego, E.; Tang, J.D.; Williams, W.P.; Warburton, M.L. Characterization of the Maize Lipoxygenase Gene Family in Relation to Aflatoxin Accumulation Resistance. PLoS ONE 2017, 12, e0181265. [Google Scholar] [CrossRef] [PubMed]
- Grimes, H.D.; Koetje, D.S.; Franceschi, V.R. Expression, Activity, and Cellular Accumulation of Methyl Jasmonate-responsive Lipoxygenase in Soybean Seedlings. Plant Physiol. 1992, 100, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zeng, H.; Hu, Z.L.; Qv, X.X.; Chen, G.P. Overexpression of the Tomato 13-lipoxygenase Gene TomloxD Increases Generation of Endogenous Jasmonic Acid and Resistance to Cladosporium fulvum and High Temperature. Plant Mol. Biol. Rep. 2013, 31, 1141–1149. [Google Scholar] [CrossRef]
- Yuan, P.G.; Borrego, E.; Park, Y.S.; Gorman, Z.; Huang, P.C.; Tolley, J.; Christensen, S.A.; Blanford, J.; Kilaru, A.; Meeley, R. 9,10-KODA, An α-ketol Produced by the Tonoplast-localized 9-lipoxygenase ZmLOX5, Plays a Signaling Role in Maize Defense Against Insect Herbivory. Mol. Plant 2023, 16, 1283–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.D.; Borrego, E.J.; Kenerley, C.M.; Kolomiets, M.V. Oxylipins Other Than Jasmonic Acid Are Xylem-resident Signals Regulating Systemic Resistance Induced by Trichoderma virens In Maize. Plant Cell 2020, 32, 166–185. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, S.; Hashad, N.; Ali, S.; Ibrahim, R.; Said, A.M.; Moharram, F.A.; Mady, M. Polyphenolic Profile of Callistemon viminalis Aerial Parts: Antioxidant, Anticancer and In Silico 5-LOX Inhibitory Evaluations. Molecules 2021, 6, 2481. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Du, M.M.; Deng, L.; Shen, J.F.; Fang, M.M.; Chen, Q.; Liu, Y.H.; Wang, Q.M.; Lu, Y.H.; Wang, Q.M.; et al. MYC2 Regulates the Termination of Jasmonate Signaling via an Autoregulatory Negative Feedback Loop. Plant Cell 2019, 31, 106–127. [Google Scholar] [CrossRef]
- Schaller, F. Enzymes of the Biosynthesis of Octadecanoid-derived Signalling Molecules. J. Exp. Bot. 2001, 52, 11–23. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, X.X.; Yi, F.Y.; Liu, Y.J.; Sod, B.; Li, M.N.; Chen, L.; Kang, J.M.; Yang, Q.C.; Long, R.C. A Genome-wide Study of the Lipoxygenase Gene Families in Medicago truncatula and Medicago sativa Reveals that MtLOX24 Participates in the Methyl Jasmonate Response. BMC Genom. 2024, 25, 195. [Google Scholar] [CrossRef]
- Chabaud, M.; Ratet, P.; Duque, S.; Araújo, S. Agrobacterium tumefaciens-mediated Transformation and in Vitro Plant Regeneration of M. truncatula. In Medicago truncatula Handbook; The Samuel Roberts Noble Foundation, Inc.: Ardmore, OK, USA, 2007. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data using Real-time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods Companion Methods Enzymol. 2001, 4, 25. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Páll, M.; Pachter, L. Near-optimal RNA-Seq quantification. Computer. Sci. 2015, 1505, 02710. [Google Scholar]
- Dangl, J.L.; Jones, D.G. Plant Pathogens and Integrated Defence Responses to Infection. Nature 2001, 411, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Farmer, E.E.; Ryan, C.A. Interplant Communication: Airborne Methyl Jasmonate Induces Synthesis of Proteinase Inhibitors in Plant leaves. Proc. Natl. Acad. Sci. USA 1990, 87, 7713–7716. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Pamanick, B.; Venugopalan, V.K.; Ibrahimova, U.; Rahman, M.A.; Siyal, A.L.; Maitra, S.; Chatterjee, S.; Aftab, T. Emerging Roles of Plant Growth Regulators for Plants Adaptation to Abiotic Stress-induced Oxidative Stress. In Emerging Plant Growth Regulators in Agriculture; Academic Press: Cambridge, MA, USA, 2022; pp. 1–72. [Google Scholar]
- Lang, D.Y.; Yu, X.X.; Jia, X.X.; Li, Z.X.; Zhang, X.H. Methyl Jasmonate Improves Metabolism and Growth of NaCl-stressed Glycyrrhiza uralensis Seedlings. Sci. Hortic. 2020, 266, 109287. [Google Scholar] [CrossRef]
- Pan, L.Y.; Zhao, X.Y.; Chen, M.; Fu, Y.Q.; Xiang, M.L.; Chen, J.Y. Effect of Exogenous Methyl Jasmonate Treatment on Disease Resistance of Postharvest Kiwifruit. Food Chem. 2020, 305, 125483. [Google Scholar] [CrossRef]
- Zhu, L.J.; Yu, H.T.; Dai, X.M.; Yu, M.L.; Yu, Z.F. Effect of Methyl Jasmonate on the Quality and Antioxidant Capacity by Modulating Ascorbate-glutathione Cycle in Peach Fruit. Sci. Hortic. 2022, 303, 111216. [Google Scholar] [CrossRef]
- Liao, Z.; Liu, X.; Zheng, J.; Zhao, C.; Wang, D.; Xu, Y.; Sun, C. A Multifunctional True Caffeoyl Coenzyme A O-methyltransferase Enzyme Participates in the Biosynthesis of Polymethoxylated Flavones in Citrus. Plant Physiol. 2023, 192, 2049–2066. [Google Scholar] [CrossRef]
- Barakat, A.; Bagniewska-Zadworna, A.; Choi, A.; Plakkat, U.; DiLoreto, D.S.; Yellanki, P.; Carlson, J.E. The Cinnamyl Alcohol Dehydrogenase Gene Family in Populus: Phylogeny, Organization, and Expression. BMC Plant Biol. 2009, 9, 26. [Google Scholar] [CrossRef]
- Herrero, J.; Carrasco, A.E.; Zapata, J.M. Arabidopsis thaliana Peroxidases Involved in Lignin Biosynthesis: In Silico Promoter Analysis and Hormonal Regulation. Plant Physiol. Biochem. 2014, 80, 192–202. [Google Scholar] [CrossRef]
- Naoumkina, M.; Farag, M.A.; Sumner, L.W.; Tang, Y.H.; Liu, C.J.; Dixon, R.A. Different Mechanisms for Phytoalexin Induction by Pathogen and Wound Signals in Medicago truncatula. Proc. Natl. Acad. Sci. USA 2007, 104, 17909–17915. [Google Scholar] [CrossRef]
- Yin, Y.Q.; Xue, J.Y.; Hu, J.J.; Yang, Z.F.; Fang, W.M. Exogenous Methyl Jasmonate Combined with Ca2+ Promote Resveratrol Biosynthesis and Stabilize Sprout Growth for the Production of Resveratrol-rich Peanut Sprouts. Plant Physiol. Biochem. 2023, 203, 107988. [Google Scholar] [CrossRef]
- Yuan, P.G.; Tanaka, K.; Poovaiah, B.W. Calcium/Calmodulin-mediated Defense Signaling: What Is Looming on the Horizon for AtSR1/CAMTA3-mediated Signaling in Plant Immunity. Front. Plant Sci. 2022, 12, 795353. [Google Scholar] [CrossRef]
- Taga, Y.; Takai, R.; Kaneda, T.; Matsui, H.; Isogai, A.; Che, F.S. Role of OsHSP90 and IREN, Ca2+ Dependent Nuclease, in Plant Hypersensitive Cell Death Induced by Transcription Factor OsNAC4. Plant Signal. Behav. 2009, 4, 740–742. [Google Scholar] [CrossRef]
- Chen, J.Y.; Mumtaz, A.; Gonzales-Vigil, E. Evolution and Molecular Basis of Substrate Specificity in A 3-ketoacyl-CoA Synthase Gene Cluster from Populus trichocarpa. J. Biol. Chem. 2022, 298, 102496. [Google Scholar] [CrossRef]
- Zhang, H.H.; Wang, F.M.; Song, W.Q.; Yang, Z.H.; Li, L.L.; Ma, Q.; Tan, X.X.; Wei, Z.Y.; Li, Y.J.; Li, J.M. Different Viral Effectors Suppress Hormone-mediated Antiviral Immunity of Rice Coordinated by OsNPR1. Nat. Commun. 2023, 14, 3011. [Google Scholar] [CrossRef]
- He, X.; Jiang, J.S.; Wang, C.Q.; Dehesh, K. ORA59 and EIN3 Interaction Couples Jasmonate-ethylene Synergistic Action to Antagonistic Salicylic Acid Regulation of PDF Expression. J. Integr. Plant Biol. 2017, 59, 275–287. [Google Scholar] [CrossRef]
- Guo, H.; Ecker, J.R. Plant Responses to Ethylene Gas are Mediated by SCF(EBF1/EBF2)-dependent Proteolysis of EIN3 Transcription Factor. Cell 2003, 115, 667–677. [Google Scholar] [CrossRef]
- Wang, G.M.; Guo, L.; Guo, Z.H. The Involvement of Ein3-binding F-box protein PbrEBF3 in Regulating Ethylene Signaling during Cuiguan Pear Fruit Ripening. Plant Sci. 2023, 329, 111600. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, Y.; Wang, Y.B.; Liu, X.L.; Ma, L.; Zhang, Z.J.; Mu, C.; Zhang, Y.; Peng, L.; Xie, S.J. Initiation and Amplification of SnRK2 Activation in Abscisic Acid Signaling. Nat. Commun. 2021, 12, 2456. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Xu, Y.; Lv, H.; Xu, Y.; Wen, J.; Li, M.; Kang, J.; Liu, Z.; Yang, Q.; Long, R. Transcriptomic Analysis Reveals the Mechanism of MtLOX24 in Response to Methyl Jasmonate Stress in Medicago truncatula. Agriculture 2024, 14, 1076. https://doi.org/10.3390/agriculture14071076
Xu L, Xu Y, Lv H, Xu Y, Wen J, Li M, Kang J, Liu Z, Yang Q, Long R. Transcriptomic Analysis Reveals the Mechanism of MtLOX24 in Response to Methyl Jasmonate Stress in Medicago truncatula. Agriculture. 2024; 14(7):1076. https://doi.org/10.3390/agriculture14071076
Chicago/Turabian StyleXu, Lei, Yanchao Xu, Huanhuan Lv, Yanran Xu, Jiangqi Wen, Mingna Li, Junmei Kang, Zhipeng Liu, Qingchuan Yang, and Ruicai Long. 2024. "Transcriptomic Analysis Reveals the Mechanism of MtLOX24 in Response to Methyl Jasmonate Stress in Medicago truncatula" Agriculture 14, no. 7: 1076. https://doi.org/10.3390/agriculture14071076