
Combined Physiology and Transcriptome Analyses Provide Insights into Malformed Fruit of Cocos nucifera L.


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Observing the Microstructure of Coconut Pulp
2.3. Measurement of Nutrient Substance
2.4. Measurement of Cell Wall Related to Enzyme Activity
2.5. Measurement of Phytohormones
2.6. Transcriptomics Analysis
2.7. Validation of DEGs in Coconut Fruit via qRT-PCR
2.8. Statistical Analysis of Data
3. Results
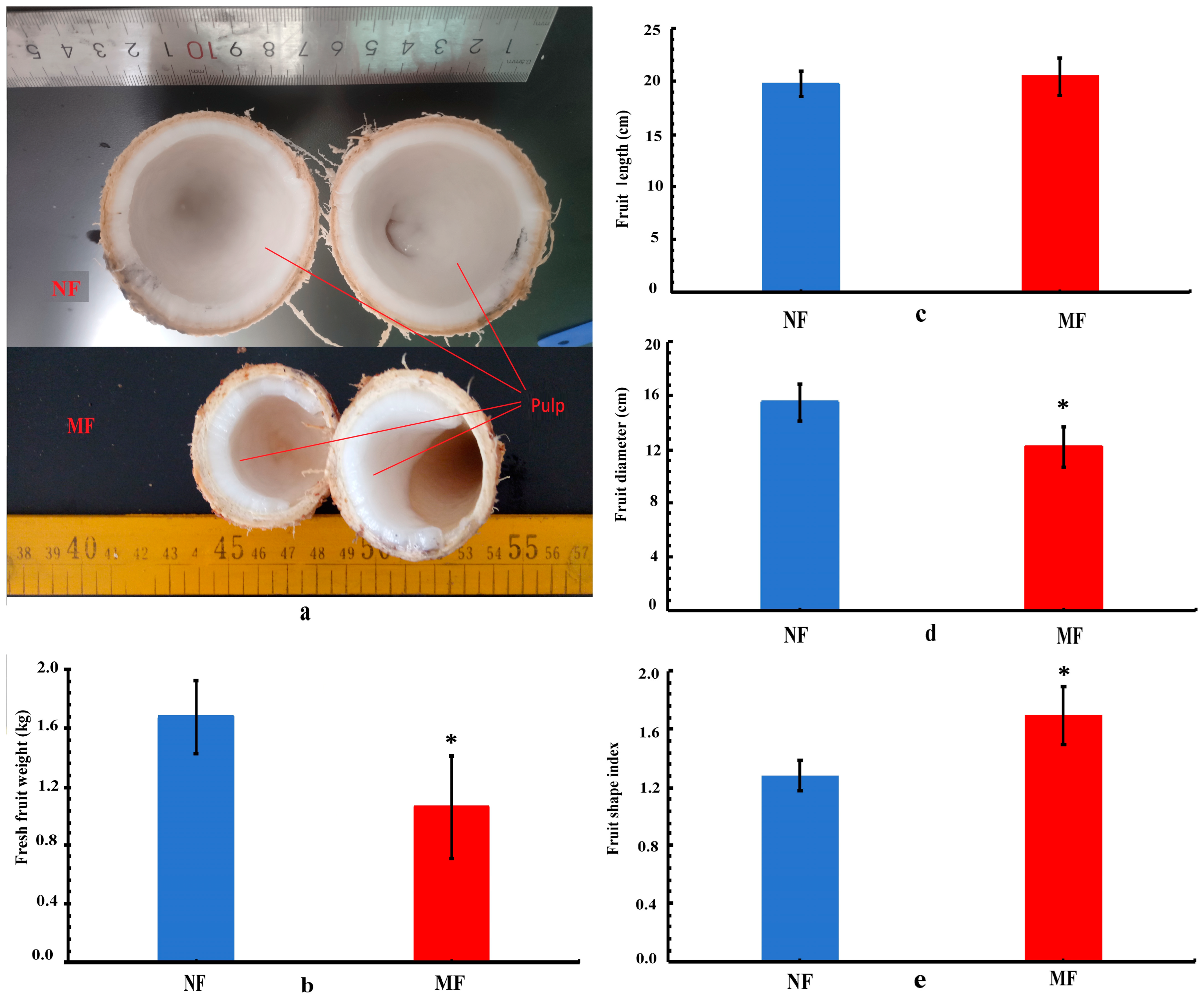
3.1. Morphological Comparison of Fruit
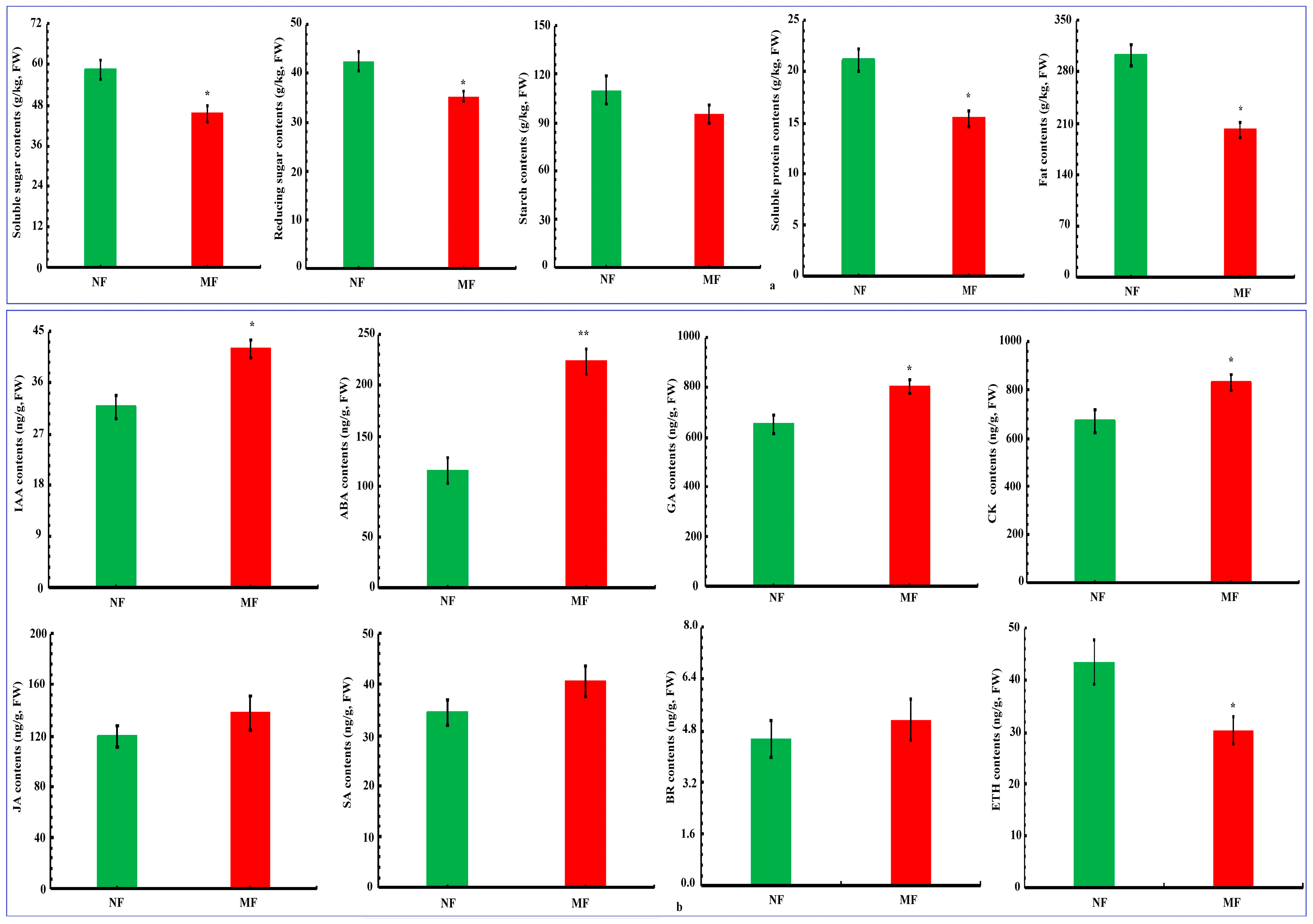
3.2. Nutrient Substance, Phytohormone Content in Fruit
3.3. Transcriptome
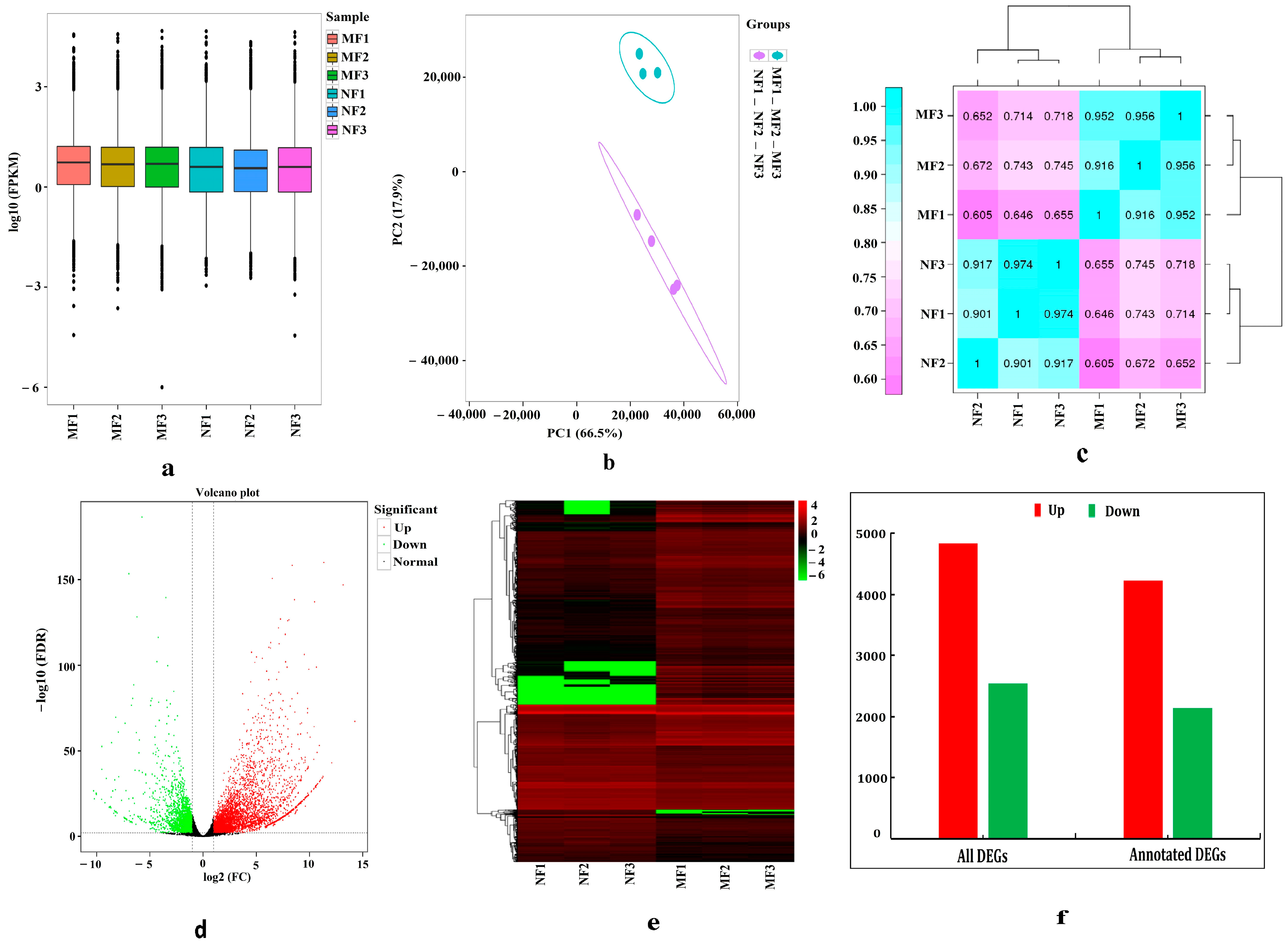
3.3.1. Evaluation of Transcriptome Sequencing Data
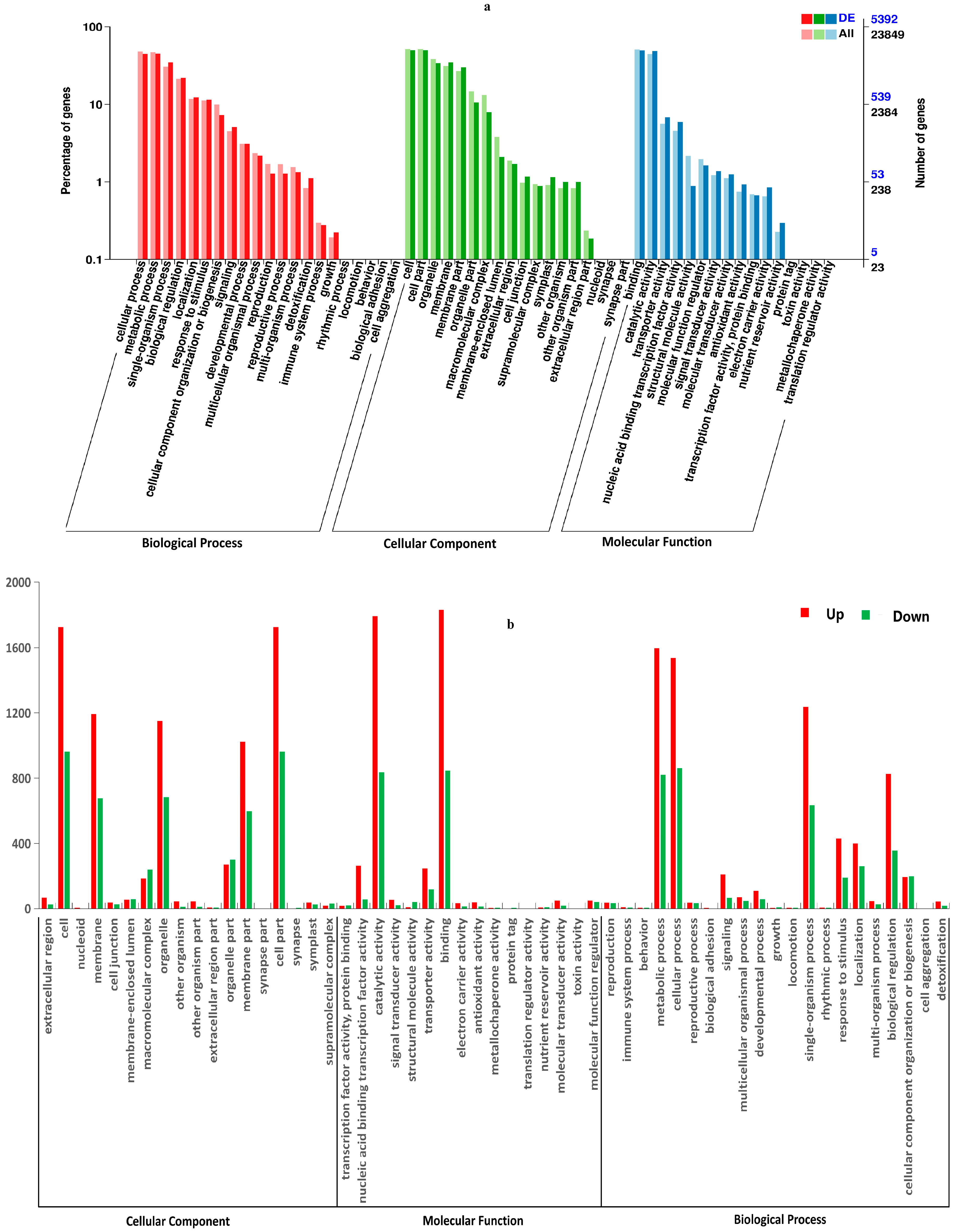
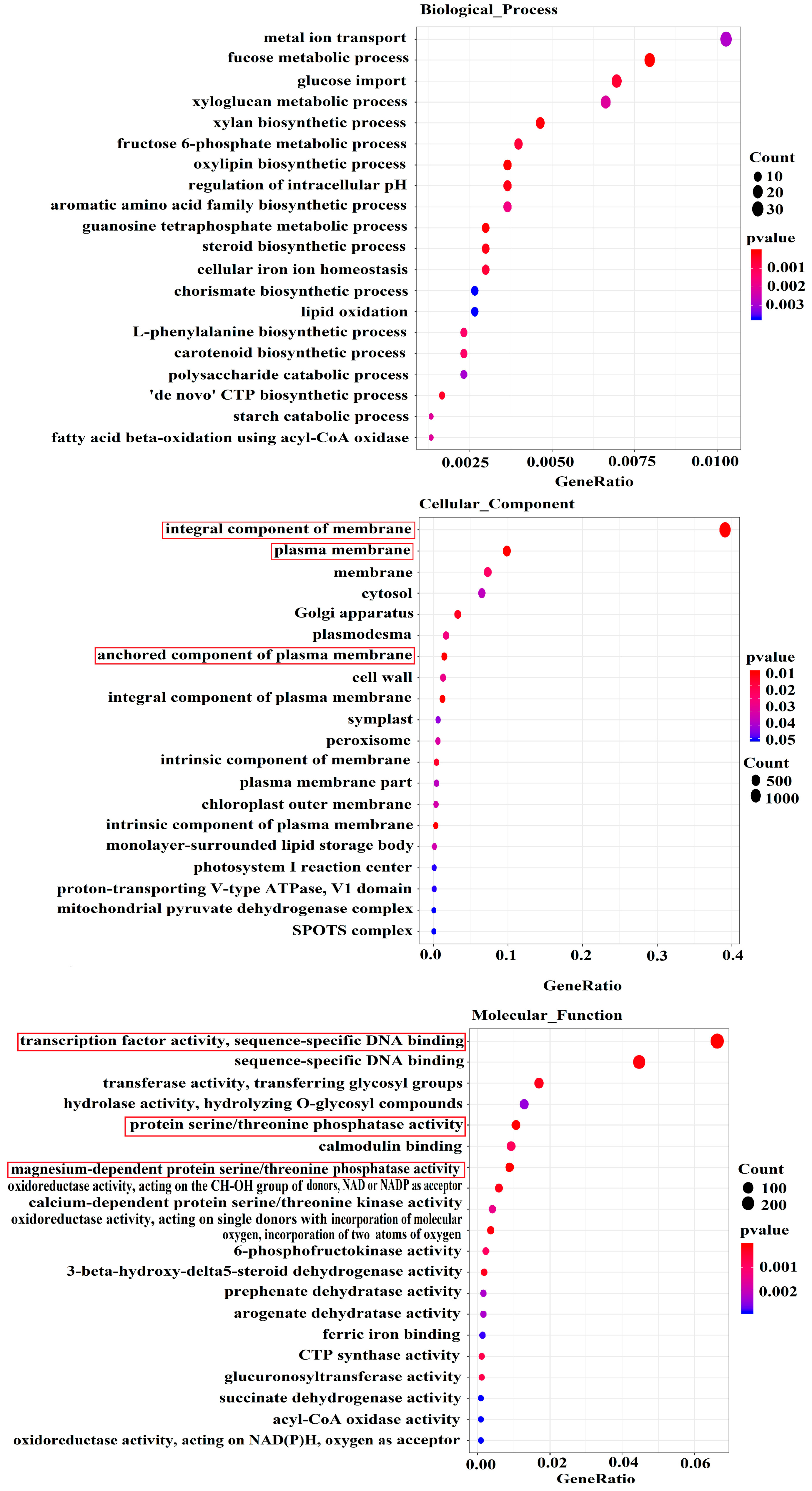
3.3.2. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)
Analysis of DEGs
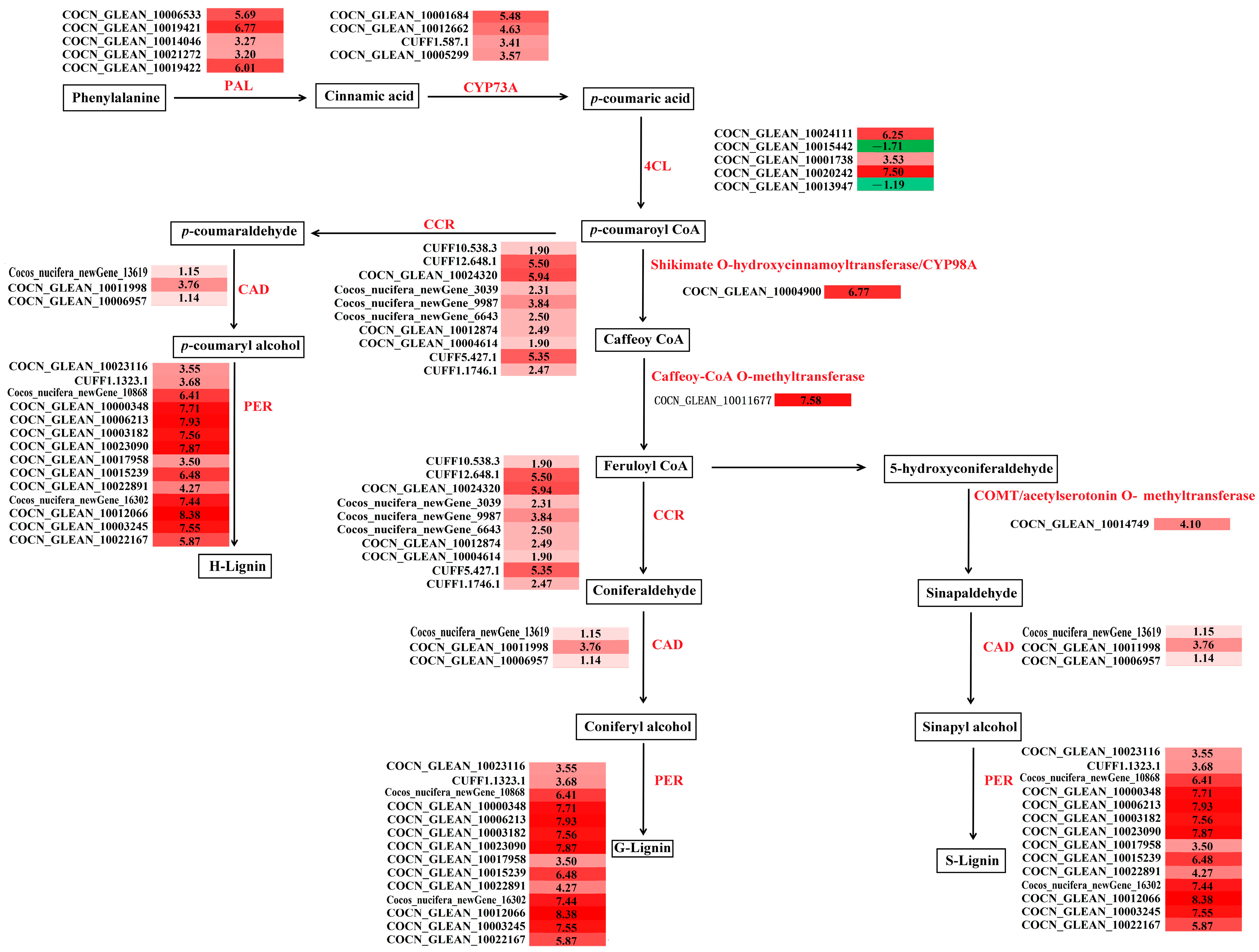
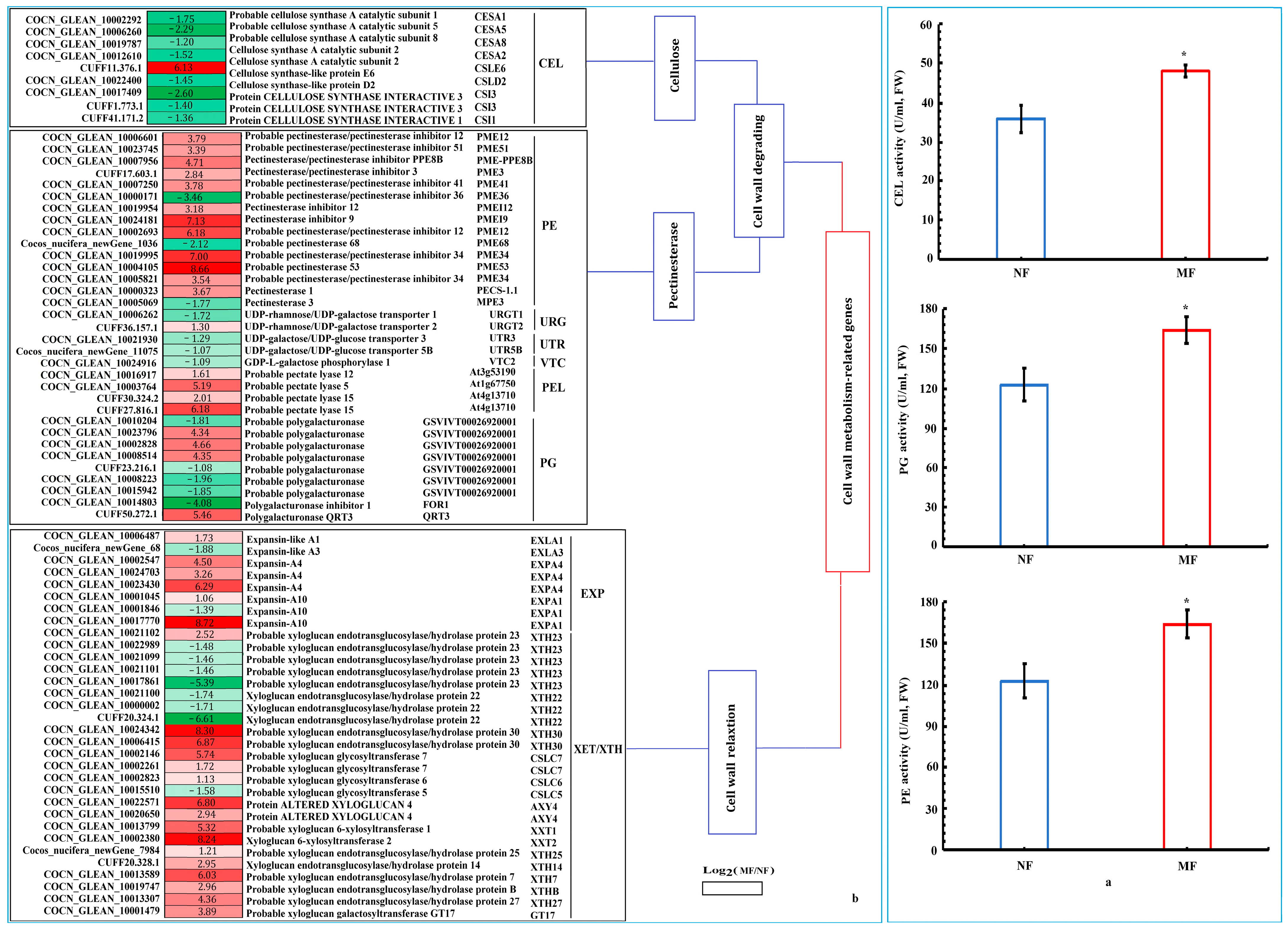
3.3.3. Expression of Cell Wall-Related Genes During the Occurrence of Malformed Fruits
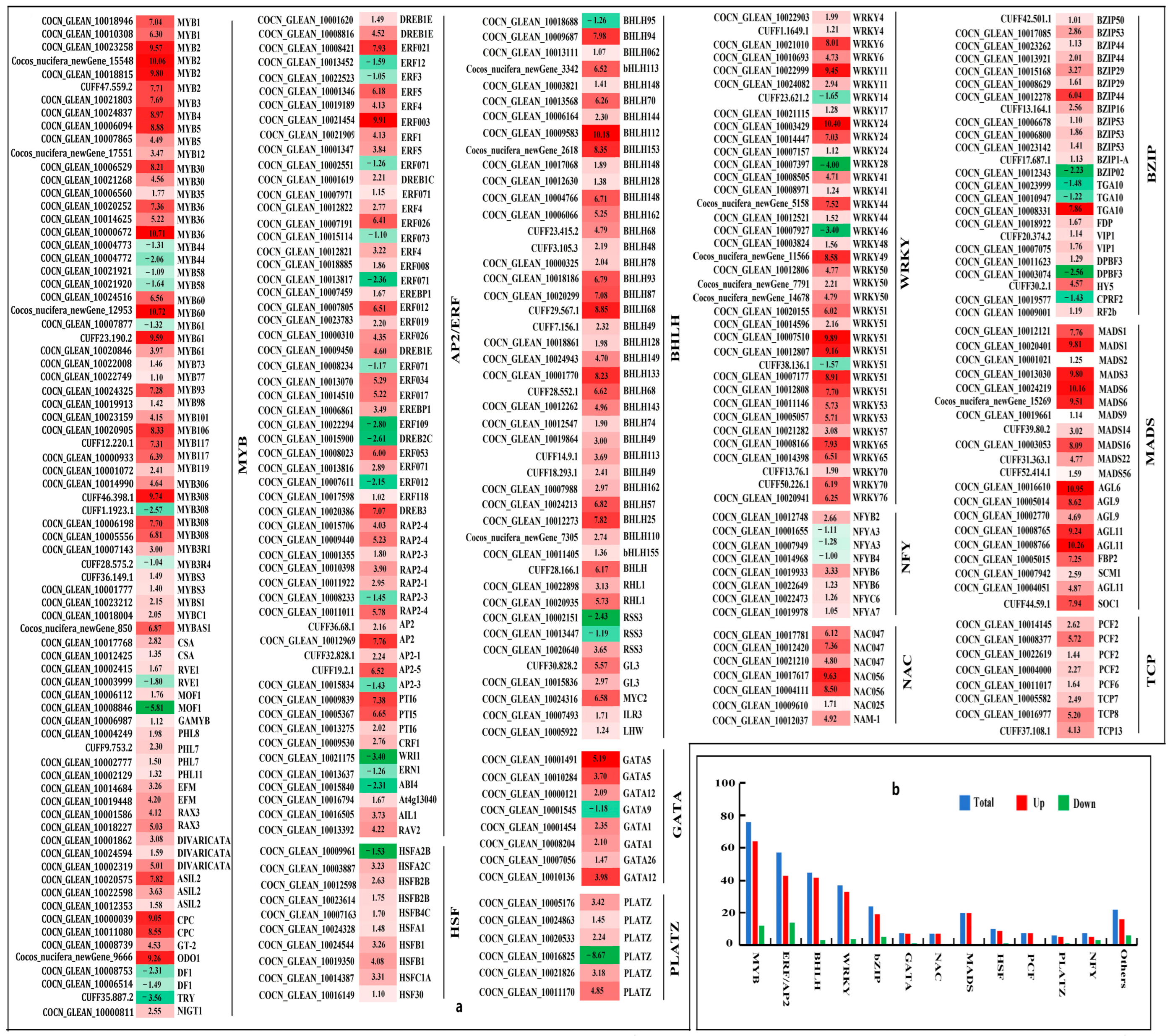
3.3.4. Transcription Factors
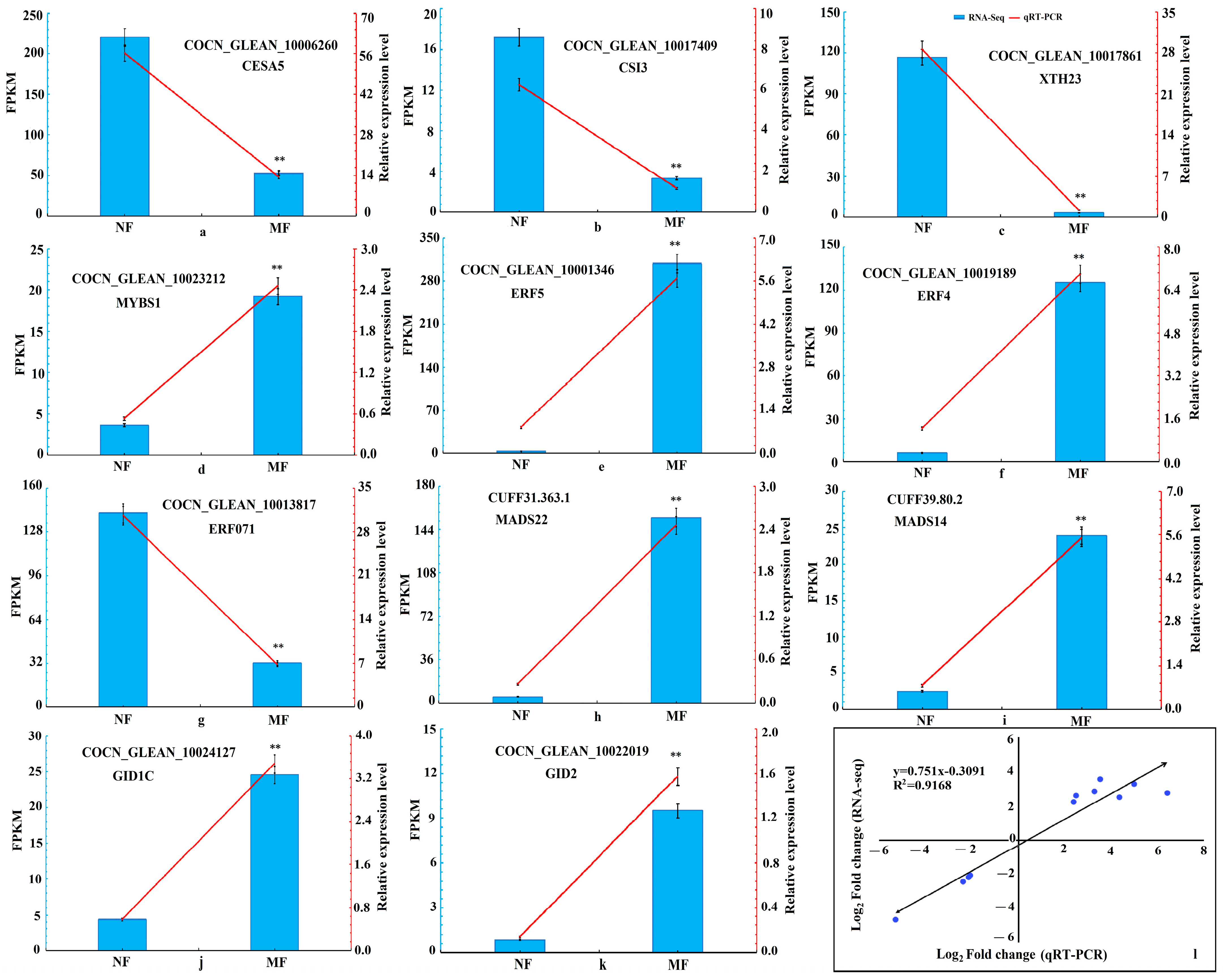
3.4. DEGs Validation by qRT-PCR
4. Discussion
4.1. The Effects of Crucial Enrichment Pathways on Malformed Fruits
4.2. The Effect of Crucial Phytohormones on Malformed Fruits
4.3. The Effects of Crucial Cell Wall Remodeling-Related Genes on Malformed Fruits
4.4. The Effect of Crucial Transcription Factors on Malformed Fruits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ashok, Y.; Jayaswa, P.K.; Raman, K.V.; Singh, B.; Singh, N.K.; Usha, K. Correction to: Transcriptome analysis of flowering genes in mango (Mangifera indica L.) in relation to floral malformation. J. Plant Biochem. Biotechnol. 2020, 29, 571–572. [Google Scholar] [CrossRef]
- Liu, K.; Li, H.; Li, W.; Zhong, J.; Chen, Y.; Shen, C.; Yuan, C. Comparative transcriptomic analyses of normal and malformed flowers in sugar apple (Annona squamosa L.) to identify the differential expressed genes between normal and malformed flowers. BMC Plant Biol. 2017, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- Jong, M.; Wolters-Arts, M.; Feron, R.; Mariani, C.; Vriezen, W.H. The Solanum lycopersicum auxin response factor 7 (SlARF7) regulates auxin signaling during tomato fruit set and development. Plant J. 2009, 57, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, Y.; Jing, L.; Jiang, B.; Wang, Q.; Wang, Y. Preliminary Study on the Formation Mechanism of Malformed Sweet Cherry (Prunus avium L.) Fruits in Southern China Using Transcriptome and Metabolome Data. Int. J. Mol. Sci. 2024, 25, 153. [Google Scholar] [CrossRef]
- Davis, S.J. Integrating hormones into the floral-transition pathway of Arabidopsis thaliana. Plant Cell Environ. 2009, 32, 1201–1210. [Google Scholar] [CrossRef]
- Mesejo, C.; Yuste, R.; Reig, C.; Martínez-Fuentes, A.; Iglesias, D.J.; Muñoz-Fambuena, N.; Bermejo, A.; Germanà, M.A.; Primo-Millo, E.; Agustí, M. Gibberellin reactivates and maintains ovary-wall cell division causing fruit set in parthenocarpic Citrus species. Plant Sci. 2016, 247, 13–24. [Google Scholar] [CrossRef]
- Lu, L.; Liang, J.J.; Chang, X.; Yang, H.T.; Li, T.Z.; Hu, J.F. Enhanced vacuolar invertase activity and capability for carbohydrate import in GA-treated inflorescence correlate with increased fruit set in grapevine. Tree Genet. Geno. 2017, 13, 21–33. [Google Scholar] [CrossRef]
- Jiang, S.; Luo, J.; Xu, F.J.; Zhang, X.Y. Transcriptome Analysis Reveals Candidate Genes Involved in Gibberellin-Induced Fruit Setting in Triploid Loquat (Eriobotrya japonica). Front. Plant Sci. 2016, 7, 1924. [Google Scholar] [CrossRef]
- Wen, B.B.; Song, W.L.; Sun, M.Y.; Chen, M.; Mu, Q.; Zhang, X.H.; Wu, Q.J.; Chen, X.D.; Gao, D.S.; Wu, H.Y. Identification and characterization of cherry (Cerasus pseudocerasus G. Don) genes responding to parthenocarpy induced by GA3 through transcriptome analysis. BMC Genet. 2019, 20, 65. [Google Scholar] [CrossRef]
- Srivastava, A.; Handa, A.K. Hormonal regulation of tomato fruit development: A molecular perspective. J. Plant Growth Regul. 2005, 24, 67–82. [Google Scholar] [CrossRef]
- Bassel, G.W.; Mullen, R.T.; Bewley, J.D. Procera is a putative DELLA mutant in tomato (Solanum lycopersicum): Effects on the seed and vegetative plant. J. Exp. Bot. 2008, 59, 585–593. [Google Scholar] [CrossRef]
- Audran-Delalande, C.; Bassa, C.; Mila, I.; Regad, F.; Zouine, M.; Bouzayen, M. Genome-wide identification, functional analysis and expression profiling of the Aux/IAA gene family in Tomato. Plant Cell Physiol. 2012, 53, 659–672. [Google Scholar] [CrossRef]
- Wang, H.; Jones, B.; Li, Z.; Frasse, P.; Delalande, C.; Regad, F.; Chaabouni, S.; Latché, A.; Pech, J.C.; Bouzayen, M. The tomato Aux/IAA transcription factor IAA9 is involved in fruit development and leaf morphogenesis. Plant Cell 2005, 17, 2676–2692. [Google Scholar] [CrossRef] [PubMed]
- Jong, M.; Mariani, C.; Vriezen, W.H. The role of auxin and gibberellin in tomato fruit set. J. Exp. Bot. 2009, 60, 1523–1532. [Google Scholar] [CrossRef]
- Nitsch, L.M.; Oplaat, C.; Feron, R.; Ma, Q.; Wolters-Arts, M.; Hedden, P.; Mariani, C.; Vriezen, W.H. Abscisic acid levels in tomato ovaries are regulated by LeNCED1 and SlCYP707A1. Planta 2009, 229, 1335–1346. [Google Scholar] [CrossRef] [PubMed]
- Vriezen, W.H.; Feron, R.; Maretto, F.; Keijman, J.; Mariani, C. Changes in tomato ovary transcriptome demonstrate complex hormonal regulation of fruit set. New Phytol. 2008, 177, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, N.; Serrano, A.; Abello, C.; Arce, A.; Espinoza, C.; Gouthu, S.; Deluc, L.; Arce-Johnson, P. Regulation of polar auxin transport in grapevine fruitlets (Vitis vinifera L.) and the proposed role of auxin homeostasis during fruit abscission. BMC Plant Biol. 2016, 16, 234. [Google Scholar] [CrossRef]
- Eccher, G.; Begheldo, M.; Boschetti, A.; Ruperti, B.; Botton, A. Roles of ethylene production and ethylene receptor expression in regulating apple fruitlet abscission. Plant Physiol. 2015, 169, 125–137. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Zhang, F.; Liu, Y.; Wang, G. Comparative Analysis of the Transcriptomes of Persisting and Abscised Fruitlets: Insights into Plant Hormone and Carbohydrate Metabolism Regulated Self-Thinning of Pecan Fruitlets during the Early Stage. Curr. Issues Mol. Biol. 2022, 44, 176–193. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, W.J.; Li, J.G.; Jiang, Y.M. Differential expression of two expansin genes in developing fruit of cracking-susceptible and resistant Litchi Cultivars. J. Amer. Soc. Hort. Sci. 2006, 131, 118–121. [Google Scholar] [CrossRef]
- Kasai, S.; Hayama, H.; Kashimura, Y.; Kudo, S.; Osanai, Y. Relationship between fruit cracking and expression of the expansin gene MdEXPA3 in ‘Fuji’ apples (Malus domestica Borkh.). Sci. Hortic. 2008, 116, 194–198. [Google Scholar] [CrossRef]
- Jiang, F.; Lopez, A.; Jeon, S.; Freitas, S.T.; Yu, Q.; Wu, Z.; Labavitch, J.M.; Tian, S.; Powell, A.L.T.; Mitcham, E. Disassembly of the fruit cell wall by the ripening-associated polygalacturonase and expansin influences tomato cracking. Hortic. Res. 2019, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Moctezuma, E.; Smith, D.L.; Gross, K.C. Antisense suppression of a beta-galactosidase gene (TBG6) in tomato increases fruit cracking. J. Exp. Bot. 2003, 54, 2025–2033. [Google Scholar] [CrossRef]
- Moctezuma, E.; Smith, D.L.; Gross, K.C. Effect of ethylene on mRNA abundance of three β-galactosidase genes in wild type and mutant tomato fruit. Postharvest Biol. Technol. 2003, 28, 207–217. [Google Scholar] [CrossRef]
- Lu, W.; Wang, Y.; Jiang, Y.; Li, J.; Liu, H.; Duan, X.; Song, L. Differential expression of litchi XET genes in relation to fruit growth. Plant Physiol. Biochem. 2006, 44, 707–713. [Google Scholar] [CrossRef]
- Mishra, A.; Khare, S.; Trivedi, P.K.; Nath, P. Effect of ethylene, 1-MCP, ABA and IAA on break strength, cellulase and polygalacturonase activities during cotton leaf abscission. S. Afr. J. Bot. 2008, 74, 282–287. [Google Scholar] [CrossRef]
- Roberts, J.A.; Gonzalez-Carranza, Z.H. Pectinase functions in abscission. Stewart Postharvest Rev. 2009, 5, 1–4. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Satoh, S.; Iwai, H. Distribution of XTH, expansin, and secondary-wall-related CesA in floral and fruit abscission zones during fruit development in tomato (Solanum lycopersicum). Front. Plant Sci. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Patterson, S.E.; Bleecker, A.B. Ethylene-Dependent and -Independent Processes Associated with Floral Organ Abscission in Arabidopsis. Plant Physiol. 2004, 134, 194–203. [Google Scholar] [CrossRef]
- Honma, T.; Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 2001, 409, 525–529. [Google Scholar] [CrossRef]
- Vrebalov, J.; Ruezinsky, D.; Padmanabhan, V.; White, R.; Medrano, D.; Drake, R.; Schuch, W.; Giovannoni, J. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (rin) locus. Science 2002, 296, 343–346. [Google Scholar] [CrossRef]
- Molesini, B.; Dusi, V.; Pennisi, F.; Pandolfini, T. How hormones and mads-box transcription factors are involved in controlling fruit set and parthenocarpy in tomato. Genes 2020, 11, 1441. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Fujisawa, M.; Shima, Y.; Ito, Y. The AP2/ERF transcription factor SlERF52 functions in flower pedicel abscission in tomato. J. Exp. Bot. 2014, 65, 3111–3119. [Google Scholar] [CrossRef]
- Ma, C.; Meir, S.; Xiao, L.; Tong, J.; Liu, Q.; Reid, M.S.; Jiang, C.Z. A KNOTTED1-LIKE HOMEOBOX protein, KD1, regulates abscission in tomato by modulating the auxin pathway. Plant Physiol. 2015, 167, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Jinn, T.L.; Stone, J.M.; Walker, J.C. HAESA, an Arabidopsis leucine-rich repeat receptor kinase, controls floral organ abscission. Genes Dev. 2000, 14, 108–117. [Google Scholar] [CrossRef]
- Cheng, C.; Zhang, L.; Yang, X.; Zhong, G. Profiling gene expression in citrus fruit calyx abscission zone (AZ-C) treated with ethylene. Mol. Genet. Genom. 2015, 290, 1991–2006. [Google Scholar] [CrossRef] [PubMed]
- Merelo, P.; Agustí, J.; Arbona, V.; Costa, M.L.; Estornell, L.H.; Gómez-Cadenas, A.; Coimbra, S.; Gómez, M.D.; Pérez-Amador, M.A.; Domingo, C. Cell wall remodeling in abscission zone cells during ethylene promoted fruit abscission in citrus. Front. Plant Sci. 2017, 8, 1–20. [Google Scholar] [CrossRef]
- Ran, E.; Yermiyahu, U.; Yasuor, H.; Dan, C.C.; Schwartz, A.; Ben-Gal, A.; Dag, A. Phosphorous Nutritional Level, Carbohydrate Reserves and Flower Quality in Olives. PLoS ONE 2016, 11, e0167591. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, X.; Fan, Y.; Wang, H.; Li, J.; Huang, X. Burst of reactive oxygen species in pedicel-mediated fruit abscission after carbohydrate supply was cut off in longan (Dimocarpus longan). Front. Plant Sci. 2015, 6, 360. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Huang, X.; Li, J.; Wang, H.; Li, J. An improved fruit transcriptome and the identification of the candidate genes involved in fruit abscission induced by carbohydrate stress in litchi. Front. Plant Sci. 2015, 6, 439. [Google Scholar] [CrossRef]
- Kuang, J.F.; Wu, J.Y.; Zhong, H.Y.; Li, C.Q.; Chen, J.Y.; Lu, W.J.; Li, J.G. Carbohydrate stress affecting fruitlet abscission and expression of genes related to auxin signal transduction pathway in litchi. Int. J. Mol. Sci. 2012, 13, 16084–16103. [Google Scholar] [CrossRef] [PubMed]
- Celton, J.M.; Dheilly, E.; Guillou, M.C.; Simonneau, F.; Juchaux, M.; Costes, E.; Laurens, F.; Renou, J.P. Additional amphivasal bundles in pedicel pith exacerbate central fruit dominance and induce self-thinning of lateral fruitlets in apple. Plant Physiol. 2014, 164, 1930–1951. [Google Scholar] [CrossRef] [PubMed]
- Kusano, M.; Worarad, K.; Fukushima, A.; Kamiya, K.; Mitani, Y.; Okazaki, Y.; Higashi, Y.; Nakabayashi, R.; Kobayashi, M.; Mori, T.; et al. Transcriptomic, Hormonomic and Metabolomic Analyses Highlighted the Common Modules Related to Photosynthesis, Sugar Metabolism and Cell Division in Parthenocarpic Tomato Fruits during Early Fruit Set. Cells 2022, 11, 1420. [Google Scholar] [CrossRef]
- Rook, F.; Corke, F.; Baier, M.; Holman, R.; May, A.G.; Bevan, M.W. Impaired sucrose induction1 encodes a conserved plant-specific protein that couples carbohydrate availability to gene expression and plant growth. Plant J. 2006, 46, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Rorat, T. Plant dehydrins–tissue location, structure and function. Cell. Mol. Biol. Lett. 2006, 11, 536–556. [Google Scholar] [CrossRef]
- Herrera-Rodriguez, M.B.; Maldonado, J.M.; Perez-Vicente, R. Light and metabolic regulation of HAS1, HAS1.1 and HAS2, three asparagine synthetase genes in Helianthus annuus. Plant Physiol. Biochem. 2004, 42, 511–518. [Google Scholar] [CrossRef]
- Wu, X. Study on the Regulatory Effect of Cytokinin on Tomato Multi Ventricular Malformation Fruit. Ph.D. Thesis, Shenyang Agricultural University, Shenyang, China, 2020. [Google Scholar]
- Wu, J.; Sun, W.; Sun, C.; Xu, C.; Li, S.; Li, P.; Xu, H.; Zhu, D.; Li, M.; Yang, L.; et al. Cold stress induces malformed tomato fruits by breaking the feedback loops of stem cell regulation in floral meristem. New Phytol. 2023, 237, 2268–2283. [Google Scholar] [CrossRef]
- Gao, J.F. Experimental Guidance for Plant Physiology; Higher Education Press: Beijing, China, 2006; pp. 144–200. [Google Scholar]
- Balcke, G.U.; Handrick, V.; Bergau, N.; Fichtner, M.; Henning, A.; Stellmach, H.; Tissier, A.; Hause, B.; Frolov, A. An UPLC-MS/MS method for highly sensitive high-throughput analysis of phytohormones in plant tissues. Plant Method. 2012, 8, 47. [Google Scholar] [CrossRef]
- Owen, S.J.; Abrams, S.R. Measurement of plant hormones by liquid chromatography—Mass spectrometry. In Plant Hormones; Humana Press: Totowa, NJ, USA, 2009; pp. 39–51. [Google Scholar]
- Lu, L.; Dong, Z.; Yin, X.; Chen, S.; Mehvish, A. Integration of Phenotypes, Phytohormones, and Transcriptomes to Elucidate the Mechanism Governing Early Physiological Abscission in Coconut Fruits (Cocos nucifera L.). Forests 2024, 15, 1475. [Google Scholar] [CrossRef]
- Xiao, Y.; Xu, P.X.; Fan, H.K.; Luc, B.; Xia, W.; Stephanie, B.; Xu, J.Y.; Li, Q.; Guo, A.P.; Zhou, L.X.; et al. The genome draft of coconut (Cocos nucifera). GigaScience 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Alex, B.; Jody, C.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment analysis for gene ontology. In R Package Version 2.8; The Pennsylvania State University: University Park, TX, USA, 2010. [Google Scholar]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2-DDCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rong, Y.; Li, J.; Zhang, N.; Yu, Q.; Xu, W. Phenotypically abnormal cotyledonary Vitis vinifera embryos differ in anatomy, endogenous hormone levels and transcriptome proffles. Tree Physiol. 2023, 43, 467–485. [Google Scholar] [CrossRef]
- Li, H.; Suo, Y.; Li, H.; Sun, P.; Han, W.; Fu, J. Cytological, Phytohormone, and Transcriptome Analyses Provide Insights into Persimmon Fruit Shape Formation (Diospyros kaki Thunb.). Int. J. Mol. Sci. 2024, 25, 4812. [Google Scholar] [CrossRef]
- Ye, J.; Hu, T.; Yang, C.; Li, H.; Yang, M.; Ijaz, R.; Ye, Z.; Zhang, Y. Transcriptome Profiling of Tomato Fruit Development Reveals Transcription Factors Associated with Ascorbic Acid, Carotenoid and Flavonoid Biosynthesis. PLoS ONE 2015, 10, e0130885. [Google Scholar] [CrossRef]
- Gou, N.; Chen, C.; Huang, M.; Zhang, Y.; Bai, H.; Li, H.; Wang, L.; Wuyun, T. Transcriptome and Metabolome Analyses Reveal Sugar and Acid Accumulation during Apricot Fruit Development. Int. J. Mol. Sci. 2023, 24, 16992. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tian, C.; Ji, S.; Ni, F.; Fan, X.; Yang, Y.; Sun, C.; Gong, H.; Zhang, A. Integrative analyses of metabolome and transcriptome reveals metabolomic variations and candidate genes involved in sweet cherry (Prunus avium L.) fruit quality during development and ripening. PLoS ONE 2021, 16, e0260004. [Google Scholar] [CrossRef]
- Xie, W.; Xu, D.; Chen, F.; Wang, Z.; Luo, J.; He, Y.; Zheng, Q.; Liu, C. Integrated Cytological, Physiological, and Transcriptome Analyses Provide Insight into the Albino Phenotype of Chinese Plum (Prunus salicina). Int. J. Mol. Sci. 2023, 24, 14457. [Google Scholar] [CrossRef] [PubMed]
- Balbontín, C.; Ayala, H.; Rubilar, J.; Cote, J.; Figueroa, C.R. Transcriptional analysis of cell wall and cuticle related genes during fruit development of two sweet cherry cultivars with contrasting levels of cracking tolerance. Chil. J. Agric. Res. 2014, 74, 162–169. [Google Scholar] [CrossRef]
- Wang, J.; Gao, X.; Ma, Z.; Chen, J.; Liu, Y. Analysis of the molecular basis of fruit cracking susceptibility in Litchi chinensis cv. Baitangying by transcriptome and quantitative proteome profiling. J. Plant Physiol. 2019, 234–235, 106–116. [Google Scholar] [CrossRef]
- Liu, Y.L.; Chen, S.Y.; Liu, G.T.; Jia, X.Y.; Haq, S.; Deng, Z.J.; Luo, D.X.; Li, R.; Gong, Z.H. Morphological, physiochemical, and transcriptome analysis and CaEXP4 identiffcation during pepper (Capsicum annuum L.) fruit cracking. Sci. Hortic. 2022, 297, 110982. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Li, M.; Wang, N.; Qu, X.; Fan, S. Transcriptome Analysis of Elm (Ulmus pumila)Fruit to Identify Phytonutrients Associated Genes and Pathways. Forests 2019, 10, 738. [Google Scholar] [CrossRef]
- Cheng, H.; Kong, W.; Tang, T.; Ren, K.; Zhang, K.; Wei, H.; Lin, T. Identification of Key Gene Networks Controlling Soluble Sugar and Organic Acid Metabolism During Oriental Melon Fruit Development by Integrated Analysis of Metabolic and Transcriptomic Analyses. Front. Plant Sci. 2022, 13, 830517. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GeneID | Description | Symbol | log2 (MF/NF) | p-Value |
|---|---|---|---|---|
| COCN_GLEAN_10001934 | Flavanone 3-dioxygenase F3H1 | F3H-1 | 5.955 | 5.59 × 10−14 |
| COCN_GLEAN_10013702 | Flavanone 3-dioxygenase F3H1 | F3H-1 | 6.504 | 2.03 × 10−151 |
| COCN_GLEAN_10017999 | p-coumarate 3-hydroxylase | C3H | 3.863 | 6.96 × 10−39 |
| COCN_GLEAN_10010266 | p-coumarate 3-hydroxylase | C3H | 3.508 | 2.71 × 10−8 |
| COCN_GLEAN_10023544 | Chalcone synthase 1 | CHS1 | 5.033 | 6.36 × 10−10 |
| COCN_GLEAN_10000658 | Chalcone synthase 3 | CHS3 | 4.989 | 9.10 × 10−6 |
| COCN_GLEAN_10008211 | Probable chalcone–flavonone isomerase 3 | CHI3 | 2.059 | 3.60 × 10−4 |
| COCN_GLEAN_10003476 | Chalcone–flavonone isomerase | CHI | 5.962 | 9.33 × 10−63 |
| COCN_GLEAN_10004414 | Probable chalcone–flavonone isomerase 3 | CHI3 | 4.521 | 4.26 × 10−11 |
| COCN_GLEAN_10003477 | Chalcone–flavonone isomerase | CHI | 5.557 | 7.83 × 10−105 |
| CUFF8.287.1 | Anthocyanidin reductase | ANR | 3.543 | 1.83 × 10−15 |
| COCN_GLEAN_10000601 | Anthocyanidin reductase | ANR | 3.701 | 2.53 × 10−16 |
| COCN_GLEAN_10006296 | Anthocyanidin reductase | ANR | 3.012 | 6.67 × 10−18 |
| COCN_GLEAN_10022326 | Leucoanthocyanidin dioxygenase | ANS | 5.989 | 3.88 × 10−12 |
| COCN_GLEAN_10024906 | Leucoanthocyanidin reductase | LAR | 1.168 | 1.10 × 10−3 |
| COCN_GLEAN_10012891 | Leucoanthocyanidin reductase | LAR | 4.799 | 3.11 × 10−4 |
| GeneID | Description | Symbol | log2 (MF/NF) | p-Value |
|---|---|---|---|---|
| COCN_GLEAN_10020022 | Beta-amylase 1 | BAM1 | 1.301 | 1.37 × 10−5 |
| COCN_GLEAN_10013727 | Beta-amylase 3 | BAM3 | 2.484 | 1.02 × 10−10 |
| COCN_GLEAN_10014734 | Beta-amylase 2 | BAM2 | 1.010 | 2.05 × 10−3 |
| CUFF24.74.1 | Beta-amylase 8 | BAM8 | 1.563 | 1.03 × 10−9 |
| COCN_GLEAN_10022348 | Beta-amylase 1 | BAM1 | 5.555 | 1.83 × 10−33 |
| COCN_GLEAN_10012930 | Inactive beta-amylase 9 | BAM9 | −2.614 | 1.43 × 10−20 |
| COCN_GLEAN_10017584 | 4-alpha-glucanotransferase DPE2 | DPE2 | 1.015 | 4.20 × 10−4 |
| Cocos_nucifera_newGene_9328 | Beta-glucosidase 26 | BGLU26 | 8.342 | 3.58 × 10−16 |
| COCN_GLEAN_10008735 | Beta-glucosidase 1 | BGLU1 | 8.226 | 1.92 × 10−15 |
| CUFF30.742.1 | Beta-glucosidase 4 | BGLU4 | 4.876 | 1.03 × 10−17 |
| COCN_GLEAN_10009626 | Beta-glucosidase 1 | BGLU1 | 2.641 | 1.74 × 10−10 |
| CUFF9.551.3 | Beta-glucosidase 20 | BGLU20 | 7.658 | 9.59 × 10−13 |
| CUFF28.239.1 | Beta-glucosidase 18 | BGLU18 | 2.872 | 1.51 × 10−9 |
| COCN_GLEAN_10021067 | Beta-glucosidase 18 | BGLU18 | 10.582 | 2.59 × 10−30 |
| COCN_GLEAN_10002326 | Fructokinase-1 | FRK1 | −1.721 | 8.30 × 10−14 |
| CUFF47.25.2 | Probable fructokinase-6 | At1g66430 | −1.747 | 3.50 × 10−28 |
| COCN_GLEAN_10003025 | Fructokinase-2 | FRK2 | 1.413 | 7.48 × 10−5 |
| COCN_GLEAN_10020868 | Sucrose synthase 2 | SUS2 | −1.404 | 3.89 × 10−8 |
| COCN_GLEAN_10006864 | Sucrose synthase 2 | SUS2 | −1.752 | 2.14 × 10−17 |
| COCN_GLEAN_10007458 | Sucrose synthase 1 | SUS1 | −1.721 | 2.68 × 10−33 |
| Cocos_nucifera_newGene_8455 | Sucrose synthase 2 | SUS2 | 2.331 | 9.76 × 10−15 |
| CUFF36.91.2 | Probable sucrose–phosphate synthase 1 | SPS1 | −1.342 | 8.43 × 10−10 |
| COCN_GLEAN_10006080 | Granule-bound starch synthase 1 | GBSS | 2.040 | 5.34 × 10−15 |
| COCN_GLEAN_10009906 | Hexokinase-2 | HXK2 | 2.803 | 1.62 × 10−13 |
| GeneID | Description | Symbol | log2 (MF/NF) | p-Value |
|---|---|---|---|---|
| COCN_GLEAN_10009294 | Phosphoenolpyruvate carboxykinase (ATP) 1 | PCK1 | −1.002 | 2.78 × 10−3 |
| COCN_GLEAN_10017173 | Phosphoenolpyruvate carboxykinase (ATP) 1 | PCK1 | 3.710 | 3.86 × 10−84 |
| CUFF40.301.1 | Alcohol dehydrogenase-like 7 | At5g42250 | −7.930 | 1.24 × 10−35 |
| COCN_GLEAN_10014124 | Alcohol dehydrogenase-like 7 | At5g42250 | 9.574 | 3.83 × 10−23 |
| CUFF40.298.1 | Alcohol dehydrogenase-like 7 | At5g42250 | 6.939 | 1.29 × 10−13 |
| COCN_GLEAN_10019006 | Catalase isozyme 2 | CAT2 | 4.311 | 1.48 × 10−16 |
| COCN_GLEAN_10001533 | Catalase isozyme 1 | CAT1 | −1.806 | 2.20 × 10−22 |
| CUFF10.712.1 | Glucose-6-phosphate 1-dehydrogenase | G6PDH | −1.285 | 7.57 × 10−9 |
| COCN_GLEAN_10004035 | Glucose-6-phosphate isomerase | PGIC1 | −1.084 | 1.25 × 10−9 |
| COCN_GLEAN_10022495 | ATP-dependent 6-phosphofructokinase 2 | PFK2 | −1.784 | 2.13 × 10−12 |
| COCN_GLEAN_10010537 | ATP-dependent 6-phosphofructokinase 3 | PFK3 | 1.404 | 5.06 × 10−10 |
| COCN_GLEAN_10011659 | ATP-dependent 6-phosphofructokinase 2 | PFK2 | 1.899 | 4.70 × 10−10 |
| COCN_GLEAN_10014631 | ATP-dependent 6-phosphofructokinase 3 | PFK3 | 1.995 | 3.12 × 10−16 |
| COCN_GLEAN_10009250 | ATP-dependent 6-phosphofructokinase 3 | PFK3 | −1.410 | 1.40 × 10−17 |
| COCN_GLEAN_10013045 | Cysteine synthase | RCS1 | −1.694 | 7.94 × 10−16 |
| COCN_GLEAN_10021515 | Cysteine synthase | RCS1 | −1.331 | 1.15 × 10−5 |
| COCN_GLEAN_10016963 | Fructose bisphosphate aldolase | ALDP | −1.451 | 2.11 × 10−3 |
| COCN_GLEAN_10017469 | Fructose bisphosphate aldolase 1 | FBA1 | −1.595 | 3.42 × 10−13 |
| COCN_GLEAN_10006409 | Fructose bisphosphate aldolase 1 | FBA1 | −2.179 | 2.46 × 10−19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, L.; Dong, Z.; Zhang, Y.; Chen, S.; Wu, Q. Combined Physiology and Transcriptome Analyses Provide Insights into Malformed Fruit of Cocos nucifera L. Agriculture 2025, 15, 723. https://doi.org/10.3390/agriculture15070723
Lu L, Dong Z, Zhang Y, Chen S, Wu Q. Combined Physiology and Transcriptome Analyses Provide Insights into Malformed Fruit of Cocos nucifera L. Agriculture. 2025; 15(7):723. https://doi.org/10.3390/agriculture15070723
Chicago/Turabian StyleLu, Lilan, Zhiguo Dong, Yuan Zhang, Siting Chen, and Qingxin Wu. 2025. "Combined Physiology and Transcriptome Analyses Provide Insights into Malformed Fruit of Cocos nucifera L." Agriculture 15, no. 7: 723. https://doi.org/10.3390/agriculture15070723
APA StyleLu, L., Dong, Z., Zhang, Y., Chen, S., & Wu, Q. (2025). Combined Physiology and Transcriptome Analyses Provide Insights into Malformed Fruit of Cocos nucifera L. Agriculture, 15(7), 723. https://doi.org/10.3390/agriculture15070723