Abstract
Microbes in coral reef sediments are thought to play an important role in organic matter remineralization and nutrient recycling. Microbial communities also reflect the environmental conditions, such as nutrient status, of an ecosystem. This study investigates the relationship between microbial community diversity in the reef sediments and environmental conditions at Liuqiu Island. We sampled sediments seasonally from four sites around the island, Beauty Cave, Geban Bay, Houshi Fringing Reef, and Lobster Cave, from 2015–2020. The V5–V6 hypervariable region of 16S rRNA was amplified and sequenced using the Illumina MiSeq platform to identify the microbial communities. The results showed that the high abundance of Pseudomonadota, Planctomycetota, and Bacteroidota might reflect the eutrophic environments of the sediments on Liuqiu Island. Second, the identification of putative pathogens and human-related genera suggests that human activities have affected the marine environment of Liuqiu Island. Third, the insignificant spatial differences and the significant temporal differences in the microbial communities of Liuqiu Island indicate that annual or periodical events, such as the Kuroshio Branch Current and South China Sea Surface Current, could shape the microbial communities of Liuqiu Island. Furthermore, the abundance of human-related genera—Cutibacterium, Herbaspirillum, Corynebacterium 1, Escherichia-Shigella, and Kocuria—increased dramatically in the Lobster Cave site in September 2015 and may have been induced by a strong climate event, such as a typhoon or heavy rainfall. Our results revealed that the microbial communities of Liuqiu Island are dynamic and sensitive to adjacent environmental conditions. The sedimented microbial communities could monitor the bacteria and pathogens related to human activities and even reveal the putative events that could affect the ecological environments.
1. Introduction
Tropical coral reefs are highly diverse ecosystems in which complex environments provide diverse habitats and niches over time and space [1,2]. Microbial communities, including bacteria, microbial eukaryotes (fungi), and small eukaryotes, play an important role in nutrient cycling in coral reef ecosystems, encompassing many pelagic and benthic processes [3,4,5,6,7,8]. Vertical community stratification caused by redox gradients enables highly diverse microbial communities in coral reef sediments [9]. The diverse microbial metabolisms remineralize the dissolved and particulate organic matter through the heterotrophic metabolism pathway in the oxygenated upper sediment layer [10,11,12,13]. Inorganic nitrogen is also fixed by bacteria and archaea present in reef sediments and can contribute significantly to the nitrogen budget of the coral reef [14]. The accompanying nutrient cycling is particularly important for coral reefs to maintain high levels of primary production and biomass in extremely nutrient-poor seawater [15,16]. Further, a nutritional link between seasonal dynamics and sediment-associated bacterial communities was demonstrated in a study conducted at the Red Sea [17]. Sediment-associated microbes may provide the seeds for coral to restore their mucus microbes [18,19]. Finally, in the Great Barrier Reef, the bacterial communities in surface sediments were different between inshore and offshore and could be used as a biological indicator of environmental perturbations in coral reef ecosystems [20,21].
The microbial community structures of sediment samples from different environments exhibit various characteristics. Microbial community examination of sediments from different regions—including the Sea of Japan, the South China Sea, the Sea of Okhotsk, the Marginal Sea of Peru, the Red Sea, the Bohai Sea, and Okinawa—has shown that the structure and diversity of their microbial communities are divergent [17,22,23,24,25,26,27]. For example, in some polluted areas, the petroleum-eating bacteria—which belong to the phyla Pseudomonadota, Bacillota, Actinomycetota, and Bacteroidota—are abundant in coastal sediments [28,29,30,31,32]. Several studies on coastal marine sediments further show that the abundance and diversity of pathogenic micro-organisms, such as the genera Vibrio, Pseudomonas, and Bacillus, correlate with the degree of pollution [33,34]. In many areas contaminated and disturbed by humans, the genera Firmicutes and Bacilli dominate the microbial community in coastal sediments [30,35]. In addition to urban pollution, mariculture also alters the microbial communities in surrounding sediments by accumulating organic matter and increasing the abundance of fish-gut-related bacteria [36]. The degree of community shift and diversity decline in sediment bacteria was inversely related to the distance and time of the mariculture sites [37]. Many bacterial biomarkers on the genus level, such as Sulfurovum, Desulfobacter, and Fusobacterium, are documented as being associated with mariculture.
Liuqiu Island, also known as Little Liuqiu, is located to the southwest of Taiwan Island and faces the mouth of the Gaoping River about eight nautical miles away. It is the only coral reef island in Taiwan. The whole island covers an area of 6.8 km2 with a 12 km coastline and has a tropical monsoon climate. The waters around Liuqiu Island remain above 25 °C all year round. The average water temperature is 25.0 °C in January and is 29.4 °C in July. The sea area around Liuqiu Island is affected by the Kuroshio tributary, the southwest blowing current and the plume of the Gaoping River [38,39]. The environments of the coral reefs around the island are different. The reef check survey [40] showed that the coral coverage on the east side of the island is about 40–69%, while that on the west side is lower, about 6–30%. In addition to coral, the fish flocks were also different between west and east sides of the island. Furthermore, the alga was abundant in the benthic communities, and the scene of algae covering the coral can be observed around the island. The indicators of invertebrates, such as cherry blossom shrimp, devil sea urchin, sea cucumber, giant clam, etc., can be recorded in low numbers. On the other hand, the convenient location for travelers and pleasant climate make Liuqiu Island a popular tourist destination in Taiwan, much like Taiwan’s well-known Turtle Island, where numerous sea turtles reside. In the last decade, the number of tourists has risen, with about 3 million tourists visiting Liuqiu Island in 2018. Large numbers of tourists are known to bring with them environmental stress, ecological disturbance, and sewage and garbage pollution. The environment around Liuqiu Island is experiencing these effects as a result of the increased tourism.
In this study, the microbial communities of the coral reef sediments from four selected sites around Liuqiu Island from 2015 to 2020 were investigated to elucidate the microbial response to human activities and the role of the microbial communities as indicators of pollution. The goals of this study were: (1) to identify the spatial and temporal distribution of the microbial community structure in sediments among different coral reefs; (2) to understand the role of sediment micro-organisms on Liuqiu Island as indicators of environmental pollution associated with human activities; (3) to monitor the microbial community for signs of events affecting the ecological environments. The results of this study could provide valuable microbial indicators for monitoring the environmental changes of Liuqiu Island.
2. Materials and Methods
2.1. Samples Collection and DNA Extraction
The coral reef sediments were collected seasonally from 4 fixed sites around Liuqiu Island (Figure 1), from 2015–2020. They were Beauty Cave (BC, northwest), Geban Bay (GB, west), Houshi Fringing Reef (HFR, southeast), and Lobster Cave (LC, northeast), and the GPS information was listed as the following: BC, 22°21′15.8″ N, 120°22′22.3″ E; GB, 22°20′34.8″ N 120°21′38.2″ E; HFR, 22°19′37.6″ N 120°22′10.6″ E; LC, 22°20′52.4″ N 120°23′27.9″ E. The benthic communities of the four sites were different based on the reef check [40]. In two west sites, BC and GB, the benthic communities were dominated by algal, followed by coral. On the other hand, on the east side, the soft coral dominated in LC, and the reef coral dominated in HFR. We collected the surface sediments by SCUBA diving at a depth of about 10 m, pushed a 15 mL sterile plastic tube into the sediments (~3 cm), preserved the sediments with 95% ETOH immediately [41], and stored them in a −20 °C freezer until the analysis began. We collected 87 sediment samples in total during 2015–2020. We used a DNeasy Power Soil DNA kit (Qiagen, Germantown, MD, USA) to extract the total genomic DNA from 500 mg of sediments from each sample following the manufacturer’s instructions.
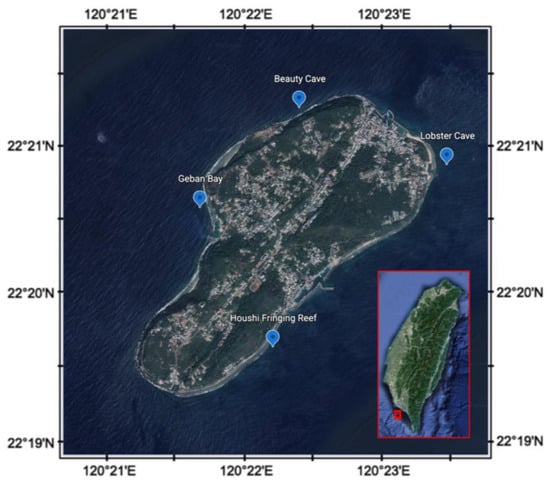
Figure 1.
Sampling sites of coral reef areas around Liuqiu Island, southwestern Taiwan.
2.2. PCR Amplification and Sequencing
The 16S rRNA V5-V6 hypervariable region was chosen as the marker of microbial community analysis. We used the primers U789F (5′-TAG ATA CCC SSG TAG TCC-3′) and U1071R (5′-GAR CTG RCG RCR RCC ATG CA-3′) [42] to construct amplicon libraries using the following PCR program: 94 °C for 3 min and then 35 cycles of 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 30 s. Each PCR reaction had a total volume of 20 μL and contained 1 μL (10 ng/μL) of total genomic DNA, 0.5 μL (5 mM) of each forward and reverse primer, 10 μL of 2× HotStarTaq Plus Master Mix Kit (Qiagen, Germantown, MD, USA), and 8 μL of PCR-grade water. We pooled five separate PCR reactions per sample to eliminate the potential for early cycle PCR induced error. All PCR reactions for sequencing produced bands of the correct size (~280 bp), whereas no bands or smears were observed for the negative controls. We sequenced the pooled amplicons using an Illumina MiSeq platform (Illumina, San Diego, CA, USA).
2.3. Data Analysis
The software package of CLC Genomic Workbench and Microbial Genomics Module (QIAGEN, Aarhus, Denmark, http://www.clcbio.com, accessed on 11 October 2017) was used to analyze the sequence data. High-throughput raw reads were imported into CLC Genomics Workbench version 11 according to Illumina pipelines 1.8. We used a sequence trim setting of 0.05 for the limit for quality scores and 2 for the maximum number of ambiguities. The trimmed reads were merged with the default settings (mismatch cost = 1, minimum score = 40, gap cost = 4, and maximum unaligned end mismatch = 1). CLC Microbial Genomics Module version 3.0 was used to analyze operational taxonomic unit (OTU) clustering and microbial community structure. The sequence similarity of OTU clustering and chimeric sequence detecting was set at 97%. The SILVA 132 rRNA database provided reference OTU data for this study [43]. The diversity indices (Shannon entropy and Simpson’s index) and heat map of abundance were also calculated using CLC Microbial Genomics Module. The formulas of the two indices were listed as the following:
Shannon entropy
Simpson’s index
where n is the number of features, and pi is the fraction of reads that belong to feature i [44,45]. A permutational multivariate analysis of variance (PERMANOVA) test based on Bray–Curtis [46] distance was used to test the differences of microbial community structures among the sampling sites, years, or seasons. The test of differential abundance at the genus level among years or sampling locations was conducted using the CLC Microbial Genomics Module, and the p-value was checked using false discovery rate (FDR). Non-metric multidimensional scaling (NMDS) analysis and an analysis of similarities (ANOSIM) test based on Bray–Curtis [46] distance were deployed to examine the similarities among samples and to calculate the significant differences among the microbial communities of the year or location groups, respectively. The tests were conducted using Paleontological Statistics (PAST4.0) software [47].
3. Results and Discussion
3.1. Diverse and Dynamic Microbial Community Structure on Liuqiu Island
The 16S rRNA amplicon of 87 sediment samples was seasonally collected from 10 m deep at the four sites (BC, GB, HRF, and LC) from 2015 to 2020. A total of 11,438 OTUs were identified from these samples. Table S1 summarizes the predicted OTUs, reads in OTUs, filtered reads, and Shannon entropy and Simpson’s index values of each sample. The two diversity indices, the Shannon entropy and Simpson’s indices, exhibited high values of 9.182 ± 0.525 and 0.992 ± 0.006, respectively (Figure 2). The boxplot of Shannon and Simpson indices of different years are shown in Figure 2b,c. The ANOVA test showed that the two diversity indices among years were different, while that among the locations were not different (Table S2). The Shannon indices showed that species richness was relatively low in 2015, 2017, and 2020, while the Simpson indices exhibited low species evenness in 2015 and 2017.
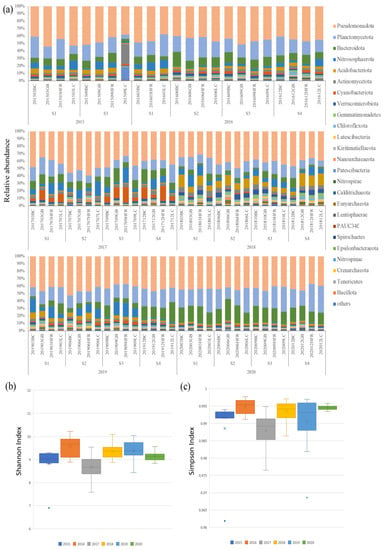
Figure 2.
(a) The relative abundances of top 25 microbial phyla; (b) boxplot of Shannon indices calculated using the OTUs data of different years; (c) boxplot of Simpson indices calculated using the OTUs data of different years on Liuqiu Island, Pingtung County, Taiwan. The BC, GB, HFR, and LC in (a) indicate the sites Beauty Cave, Geban Bay, Houshi Fringing Reef, and Lobster Cave, accordingly. The x in (b,c) indicates the average value of biodiversity index of each group, while the dot was the outsider of each group.
The OTU annotation based on the SILVA 132 database classified 11,438 OTUs into different levels of taxonomic precision, including 65 phyla, 204 classes, 558 orders, 1096 families, and 2122 genera. At the phylum level, the top ten ubiquitous phyla were Pseudomonadota, Planctomycetota, Bacteroidota, Nitrososphaerota, Acidobacteriota, Actinomycetota, Cyanobacteriota, Verrucomicrobiota, Gemmatimonadetes, and Chloroflexota (Figure 2 and Table S3). Pseudomonadota (43.5%) and Planctomycetota (19.2%) had the highest relative abundances on Liuqiu Island and dominated the microbial community structures of Liuqiu Island’s sediments across the period we sampled. This phenomenon may have resulted from the eutrophic environment. Previous studies in the Bohai, Yellow, South, and East China Seas [27,48] have shown that the high abundance of Pseudomonadota and Plancytomycetes could be due to the environmental eutrophication. Pseudomonadota is critical for the degradation of the organic nitrogen in sediments [49,50], and Planctomycetota play important biogeochemical roles in anaerobic ammonium oxidation [38], methane oxidation [51], and carbon recycling [42]. Both phyla are chemotrophic bacteria, and their high abundance might reflect the high concentration of relative chemicals in the environment [52]. Furthermore, the third and sixth abundant phyla, Bacteroidota and Actinomycetota, were suggested as degraders of complex organic matters. Bacteroidota are considered to be specialized in the degradation of complex organic matter, especially in polysaccharides and protein [53,54]. The mineralization of high molecular weight organic matter proceeded by Bacteroidota was reported in ubiquitous aquatic environments, such as the coastal sea and deep sea [55,56,57]. The Actinomycetota could breakdown and recycle organic compounds, such as cellulose and chitin [58], and were reported to play a significant role in organic matter mineralization, mineral nutrient immobilization, and nitrogen fixation [59]. The abundance of the Bacteroidota and Actinomycetota might reflect the concentration of Liuqiu Island’s organic compounds. The above results suggest that the sediments in Liuqiu Island might be eutrophic environments for the micro-organisms.
The major microbial communities of each sample were dynamic, and the differential abundance in phyla level was observed over different years. For example, the relative abundance of Actinomycetota and Patescibacteria in 2015, Cyanobacteriota in 2017, and Bacteriodiota and Planctomycetota in 2020 increased significantly. In 2015, the increase of Actinomycetota and Patescibacteria was observed in sample 201509LC, in which the relative abundance of Actinomycetota and Patescibacteria was 26.8% and 16.9%, respectively (Figure 2 and Table S3). Actinomycetota is thought to be a degrader of organic compounds [58], while Patescibacteria consists of ultramicrobacteria with limited metabolic capacities and anaerobic lifestyles [60,61]. The dramatic increase of the two phyla suggests that the environment of Lobster Cave in September 2015 could have been more suitable for Actinomycetota and Patescibacteria. This increase indicates an ecological event that may have changed the habitat environment in 2015 and might have induced the decrease of the diversity of the microbial communities in Liuqiu Island. Furthermore, the increase of Cyanobacteriota in 2017 and Bacteroidota in 2020 might have correlated with the low diversity of the microbial communities in 2017 and 2020, respectively.
At the genus level, the 25 genera with the highest coefficients of variation were Marinomonas, Aureibacter, Endozoicomonas, Zeaxanthinibacter, Woeseia, Blastopirellula, Rhodopirellula, Cutibacterium, Corynebacterium 1, Kocuria, Herbaspirillum, Escherichia-Shigella, and some no-rank bacteria (Figure 3). Figure 3 shows that samples from the same year seemed to share similar abundant microbial genera and had a tendency to cluster. For example, Marinomonas, Aureibacter, and Endozoicomonas were three highly abundant genera in the 2019 samples. The genus Marinomonas, a gammaproteobacterium that inhabits various marine environments [62,63,64,65], is involved in the metabolism of various chemicals, including dimethylsulfoniopropionate, phthalate, and oil compounds [66,67,68]. The genus Endozoicomonas, belonging to gammaproteobacteria, is a symbiont of marine animals in coral reefs, including coral, sponges, tunicates, sea slugs, and some mollusks [69,70,71,72,73,74,75]. The presence of Endozoicomonas is correlated with the health of coral reef systems and contributes to the nitrogen and sulfur cycles of organic molecules [72,76]. The genus Aureibacter, isolated from a coral reef sea squirt [77] and belonging to Bacteroidota, is abundant in the anaerobic digestion process [78]. Next, Zeaxanthinibacter and Woeseia were two highly abundant genera in 2017. The Zeaxanthinibacter, inhabiting seawater and sediments, can produce carotenoid pigments [79,80] and is dominant in oiled sediments [81]. The Woeseia could be widely involved in the cycling of detrital proteins in both coastal and deep-sea sediments [82,83]. Three no-rank bacteria genera belonging to the phylum Cyanobacteriota (f_uncultured diatom, o_Chloroplast, and f_uncultured cyanobacterium) were only abundant in 2017 (Figure 3), and they are thought to play crucial roles in the nitrogen and carbon cycles in marine ecosystems [84]. Furthermore, Blastopirellula and Rhodopirellula were two highly abundant genera in 2020. The genus Rhodopirellula is a globally distributed bacterium that can be found attached to sediment particles and is associated with phytoplankton, sponges, and macroalgae [85,86,87,88,89,90]. The genus Blastopirellula, a member of Planctomycetota, is chemoheterotrophic and uses carbohydrates as its main carbon and energy sources [91].
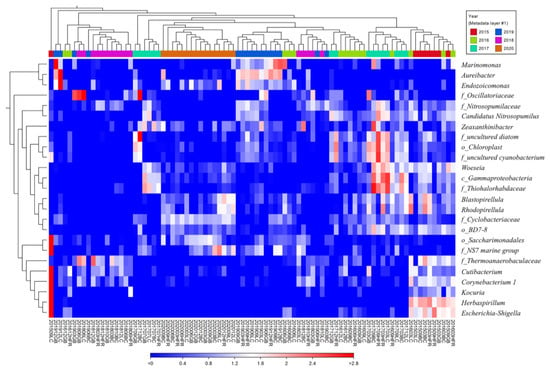
Figure 3.
Heat map of the relative abundance of the 25 genera with highest coefficients of variation. Clusters based on microbial community composition are above the heat map; clusters based on the genera are on the left of the heat map. The data were scaled per column. The values represent the relative abundance of genera in the sample.
In 2015, Cutibacterium, Corynebacterium 1, Kocuria, Herbaspirillum, and Escherichia-Shigella were dominant in the microbial community, especially in the sample 201509LC. However, these genera were relatively rare in other years. The genus Cutibacterium primarily resides on the human skin and is recognized as an important human pathogen [92,93,94,95,96]. Corynebacteria, found on the human skin and the upper respiratory system, is a human and animal pathogen [97,98]. The genus Kocuria, inhabiting human skin and various environments, is not only considered a pathogen but also antibiotic-resistant bacteria [99,100,101]. Herbaspirillum is a plant-associated N2-fixer, as well as an opportunistic pathogen and potential degrader of pollutants [102]. The Escherichia-Shigella genus, which comprises Escherichia and Shigella species, could induce human shigellosis and several foodborne diseases [103,104]. Escherichia coli, a member of the genus Escherichia-Shigella, is used as an indicator of pollution in food, water, and the environment [105]. All five aforementioned genera in 201509LC are human-associated bacteria. In short, our data showed that the microbial community of Liuqiu Island may exhibit different signature taxa each year. Most human-associated bacteria were found to be dominant in 2015, especially in the sample 201509LC. The above results were carried out based on the genera level annotation of the 16S amplicon; therefore, the accuracy issue may not be ignored. Further investigation on the species level may help to increase the resolution of the human-associated bacteria evaluation.
3.2. Temporal and Spatial Differences in Microbial Communities
Permutational multivariate analysis of variance (PERMANOVA) was performed to test statistical differences in the structure of microbial communities based on year, season, and location at the phylum and genus level. The results showed that the differences at both the phylum and genus level were significant across years, except between 2015 and 2016, but insignificant among seasons and locations (Table S4). The ANOSIM and NMDS analysis also showed that the temporal difference was significant but that the spatial difference was insignificant in the microbial communities of Liuqiu Island (Figure 4). The differences within sampling years were lower than the differences between sampling years based on ANOSIM analysis (Figure 4a). The within-group differences for the year 2020 were the lowest, followed by 2015 and 2016. This result was consistent with the NMDS analysis (Figure 4b). The NMDS plot also showed that the sample dots for years 2020, 2016, and 2015 were the most condensed, except for the sample dot 201509LC. Meanwhile, ANOSIM analysis showed that the differences between locations were similar to those within locations (Figure 4d). The NMDS plot also showed that the sample dots of the location groups were mixed together (Figure 4e). The above results indicate that the temporal difference was significant, and the spatial difference was insignificant in the microbial communities of Liuqiu Island. Thus, the microbial community structures of Liuqiu Island were further compared across different sampling years (Figure 4c) and locations (Figure 4f) at the genus level to reveal the signature genera for each sampling year and location. The temporal differences among the samples were significant in all of the top 25 genera (Figure 4c). For example, the abundances of genera Cutibacterium, Herbaspirillum, Corynebacterium 1, Escherichia-Shigella, and Kocuria in 2015 were significantly higher than those of other years. These five genera were abundant in sample 201509LC, with the relative abundances of 16.0%, 11.7%, 8.9%, 1.8%, and 0.7%, respectively. Furthermore, the genera Marinomonas, Aureibacter, and Endozoicomonas were the most abundant in 2019. The abundance of the genus Marinomonas was 1.5% in the sample 201912GB, and the abundance of genus Aureibacter and Endozoicomonas was 16.9% and 3.3%, respectively, in the sample 201912LC. The genus Candidatus Nitrosopumilus was the most abundant in 2017, and the most abundant sample was 201712LC with a relative abundance of 5.1%. The f_Thiohalorhabdaceae, Blastopirellula, Rhodopirellula, and Woeseia, were the most abundant genera from 2015 to 2020. The abundances of o_Chloroplast and uncultured cyanobacterium increased dramatically in 2017 but decreased in 2018 and 2020. In 2017, the abundances of o_Chloroplast, uncultured-cyanobacterium, and f_Thiohalorhabdaceae increased dramatically, while that of f_Thermoanaerobaculaceae decreased. In 2018 and 2020, o_Chloroplast, uncultured cyanobacterium, and CandidatusNitrosopumilus exhibited extra-low abundance. Furthermore, f_Thermoanaerobaculaceae and f_Nitrosopumilaceae also exhibited low abundance in 2020. Dynamic microbial community structures were exhibited at both the phylum and genus level. A study on marine micro-organisms showed that the increase or decrease of micro-organisms may reflect the conditions of the environment [52]. Therefore, the dynamic microbial communities of Liuqiu Island may reflect the changes of the environmental conditions.
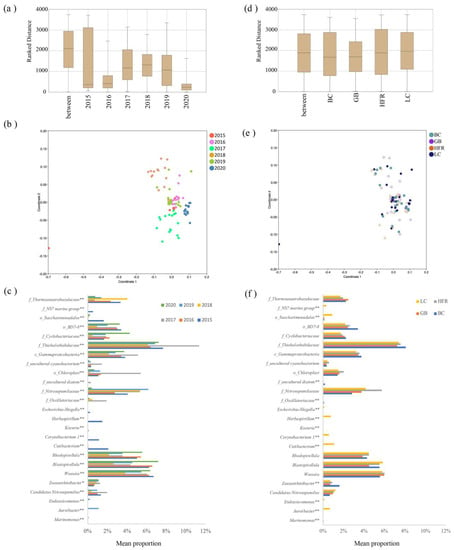
Figure 4.
Differences in microbial communities among different years and locations. (a,d) show the similarities within and between years and locations, respectively. ANOSIM analysis based on Bray-Curtis distance of bacterial community structure at the genus level. (b,e) show that NMDS analysis based on Bray-Curtis distance of bacterial community structure at the genus level among all samples, (b) based on year and (e) based on location. (c,f) exhibit the differences in bacterial abundance across different years and locations, respectively. The differences were calculated using the likelihood ratio test in the CLC Microbial Genomics Module. The p-value was checked using FDR. * p < 0.05; ** p < 0.01. The BC, GB, HFR, and LC indicate the sites Beauty Cave, Geban Bay, Houshi Fringing Reef, and Lobster Cave, accordingly.
In addition to the temporal difference, the spatial differences in genus abundances were also calculated. Only 13 genera exhibited differences among the sampling locations (Figure 4f). The genera Aureibacter, Endozoicomonas, Cutibacterium, Herbaspirillum, Corynebacterium 1, Escherichia-Shigella, and Kocuria were statistically more abundant in the Lobster Cave site than in the other three sites, while the genus Zeaxanthinibacter was significantly more abundant in Beauty Cave. The relative abundances of the genera Aureibacter and Endozoicomonas were 16.9% and 3.3%, respectively, in 201912LC, while those of Cutibacterium, Herbaspirillum, Corynebacterium 1, Escherichia-Shigella, and Kocuria were 16.0%, 11.7%, 8.9%, 1.8%, and 0.7%, respectively, in 201509LC. The genus Zeaxanthinibacter was 4.5% and 3.3% in 201812BC and 201709BC, respectively. The human-associated bacteriagenera, Cutibacterium, Herbaspirillum, Corynebacterium 1, Escherichia-Shigella, and Kocuria, were abundant in LC, while the oil-spill-related genus Zeaxanthinibacter was abundant in BC. The BC and LC were the closest sites from the mouth of Gaoping River and may be affected by the plume of Gaoping River easier than the other sites. The above results may suggest the northern Liuqiu Island might be affected by human activity easily.
Although the reef check showed the benthic communities were different among the four sites [40] where we collected samples, the differences among their microbial communities were insignificant. This may indicate that the main forces shaping the microbial communities in the coral reef sediments of Liuqiu Island were not local environmental factors but large-scale or global events. On the other hand, the significant temporal differences in microbial communities of Liuqiu Island may suggest that the influential factors may change annually or periodically. The near-shore environment of Liuqiu Island is affected not only by local coastal currents but also by large-scale events, such as the Kuroshio Branch Current (KBC), the monsoon-induced South China Sea Surface Current (SCSSC), and the internal-wave-induced upwelling [106,107,108,109]. The SCSSC brings warm and low-salinity water into the sea area surrounding Liuqiu Island in the summer, while the KBC carries the warm and high-salinity water into the sea area surrounding the island in winter. Furthermore, the upwelling provides Liuqiu Island with an extra flux of nutrient-rich water [109,110]. The aforementioned events could shape the near-shore environments of Liuqiu Island and are affected by El Niño–Southern Oscillation [106,107].
3.3. Putative Pathogen and Human-Related Bacteria
Many bacteria genera were reported as indicators of pathogenicity and pollution [111,112,113,114,115,116,117]. For example, in a study of the South China Sea, the genera Vibrio, Pseudoalteromonas, Escherichia-Shigella, Psychrobacter, and Pseudomonas were used as environmental indicators to evaluate the impact of human activity [34]. Another study in Hong Kong further showed the positive relationship between four genera of Desulfobacteraceae and the degree of pollution level [33]. Therefore, based on the taxonomic profiles from the literature [111,112,113,114,115,116,117], 20 opportunistic pathogen and human-related genera were identified from the microbial communities of all samples (Figure 5 and Table S5). In general, Vibrio was the most abundant (0.01%~1.06%) genus in most of the samples. Two typical opportunistic pathogenic species—V. vulnificus and V. harveyi—were found in all the samples. V. harveyi was the dominant species and was recognized as a significant pathogen of marine vertebrates/invertebrates [117]. The genera Cutibacterium and Corynebacterium 1 appeared in most samples, and both of them were pathogens of human skin [93,95,96,118]. Three desulfurized genera, Desulfatitalea, Desulfobacter, and Desulfococcus, that correlated with the degree of pollution level [33] were also identified in most samples. The accumulated abundances of the remaining 20 putative pathogens and human-related bacteria were relatively low (<1.3%) across all samples, except in samples 201509LC and 201707HFR. The relative abundance of all putative pathogens and human-related bacteria in most samples was not high in Liuqiu Island. However, their appearances may indicate that human activities have affected the marine environment of Liuqiu Island, even in the coral reef sediments, 10 m deep. On the other hand, human activities have had a very serious impact on Liuqiu Island over the past decade. The number of tourists grew from 2.3 million in 2015 to 2.9 million in 2019 (MOTC 2022), bringing a lot of pollutants such as garbage and wastewater. The excessive tourists may have contributed to the emergence of these possible pathogens. However, our data show that the abundance of the putative anthropogenic bacteria in the coral reef sediments remained at a low ratio. The abundance of several landmark species, such as E. coli, is even lower than that of coral reef islands in the South China Sea [34,119]. Our findings imply that the sediments around Liuqiu Island might be less prone to accumulating environmental pollutants. Rather than factors related to human activities, other physical and chemical factors could be the main forces that shape the microbial communities of the coral reefs in the Liuqiu Island. Further studies are required to reveal the key forces—such as currents, topography, and tides—that affect the sedimented microbial communities of Liuqiu Island.

Figure 5.
Heat map of the putative pathogen and human-related genera in all samples. The scale is based on relative abundances ranging from 0 to 1%. The BC, GB, HFR, and LC indicate the sites Beauty Cave, Geban Bay, Houshi Fringing Reef, and Lobster Cave, accordingly.
Although most samples exhibited a low abundance of putative pathogens, two remarkable samples, 201509LC and 201707HFR, exhibited an extremely high abundance of the putative pathogens. In 201509LC, the accumulated abundance of these 20 putative pathogens and human-related genera was extremely high, accounting for 27.6% of the entire microbial community. The most accumulated abundant genus was Cutibacterium (16.0%), followed by Corynebacterium 1 (8.94%), Escherichia-Shigella (1.79%), and Kocuria (0.75%). The first genus, Cutibacterium, is associated with human disease [93,95,96], and the dominant OTU in this study was the pathogenic Cutibacterium ance (92.6%). The second genus, Corynebacterium 1, was dominated by a potential multi-drug-resistant bacterium (94.6%), Corynebacterium tuberculostearicum [120], in our analysis. In the third genus, Escherichia-Shigella, the dominant species was identified as E. coli, a pollution indicator [105]. The fourth genus, Kocuria, is an opportunist pathogen of humans and aquaculture [94,99], and Kocuriaarsenatis sp., associated with arsenic pollution [121], was the most abundant OTU (98%). In the sample 201707HFR, the total reads of the putative human-related genera accounted for 2.75%, and Desulfococcus was the most abundant genus (2.60%). Desulfococcus is usually found in anaerobic or polluted environments [33,35,122,123]. The unusually abundant putative pathogen and human-related bacteria in samples 201509LC and 201707HFR may imply a sudden outburst of ecological events occurred in 2015 and 2017. The above results were carried out based on the genera level annotation of 16S amplicon; therefore, the pathogenic prediction of micro-organisms may be misleading due to the accuracy issue. Further investigation based on genomic level may increase the resolution of the putative pathogenic analysis.
3.4. Prospect of Long-Term Monitoring
Many bacteria genera were reported as indicators of pathogenicity and pollution. The results of this study show that the microbial community structure of Liuqiu Island from 2015 to 2020 was dynamic and imply that the benthic ecology in the Liuqiu coral reef ecosystem is relatively unsteady in southwest Taiwan. Of all our samples taken from the changing microbial community of Liuqiu Island, those that showed unordinary microbial community structures may best reflect the massive alteration of habitat environment. For example, the sample with the greatest difference was 201509LC, in which the abundance of phyla Actinomycetota and Patescibacteria increased significantly (Figure 2). Figure 3 shows seven genera that were significantly abundant in 201509LC. They are Cutibacterium (class Actinomycetes), Herbaspirillum (class Betaproteobacteria), Corynebacterium 1 (class Actinomycetes), Escherichia-Shigella (class Gammaproteobacteria), Kocuria (class Actinomycetes), and two no-rank genera (class Saccharibacteria). Saccharibacteria (class Patescibacteria) are human-associated bacteria isolated from the oral microbiome and can be found in various environments, such as activated sludges [124,125], water treatment plant sludge [126], and rainforest soil [127]. These micro-organisms associated with human activities were only abundant in 201509LC and were very rare in the other samples/sites. Meanwhile, the Shannon entropy and Simpson’s index from the LC site dropped to 6.902 and 0.962, respectively, in September 2015. However, the two values later rose to 8.89 and 0.992 in March 2016 and 9.93 and 0.997 in June 2016 (Table S1). These dynamic changes imply that an unknown disturbance occurred in LC between March and September 2015. The disturbance in LC may have been influenced by the adjacent environments and resulted in these unusual changes in the microbial community. The microbiota was back to its normal rarity in site LC about six to twelve months after March 2016. The recovery of the microbial community may have been due to the resilience of the coral reef system [21] in Liuqiu Island.
In addition to the KBC, SCSSC, and upwelling, the plume of the Gaoping River induced by a typhoon or heavy rainfall also affected the near-shore environment of Liuqiu Island [38,39,109]. The plume brought abundant nutrients, sediments, freshwater, and pollutants from the Gaoping River Basin into Liuqiu Island and shaped its near-shore environment suddenly. In August 2015, Typhoon SOUDELOR passed near Liuqiu Island and brought high rainfall, causing the plume of the Gaoping River to surround Liuqiu Island. A month later, sample 201509LC was harvested for this study. This strong hurricane might be one of the factors that altered the microbial community of 201509LC. However, the local events, such as the heavy rain which induced the discharge of wastewater and sludge from Liuqiu Island, should not be excluded. This special case in the long-term monitoring study reveals that the coral reef of Liuqiu Island is resilient, not only in the fast recovery of the microbial community structure but also in the diversity index. In short, long-term monitoring of a microbial community not only establishes ecological background information and shows the dynamic changes in microbial indicators, but it reveals whether putative events might influence marine environments.
4. Conclusions
In this study, we sampled sediments seasonally from four sites around the island, Beauty Cave, Geban Bay, Houshi Fringing Reef, and Lobster Cave, from 2015–2020. The V5–V6 hypervariable region of 16S rRNA was amplified and sequenced using the Illumina MiSeq platform to identify the microbial communities. In total, 11,438 OTUs were classified into 65 phyla based on the SILVA 132 database. Through the analysis of the microbial community structures, several conclusions could be drawn. First, the dominant phylum was Pseudomonadota, followed by Planctomycetota, Bacteroidota, Nitrososphaerota, and Acidobacteriota. The high abundance of Pseudomonadota, Planctomycetota, and Bacteroidota might reflect the eutrophic environments of the sediments in Liuqiu Island. Second, the identification of putative pathogens and human-related genera suggested the human activities affected the marine environment of Liuqiu Island. Third, the insignificant spatial differences suggested the local environmental factors may not be the main force shaping microbial communities of Liuqiu Island; however, the differential abundance analysis suggested that the northern Liuqiu Island might be affected by human activity easily. On the other hand, the significant temporal differences in microbial communities in Liuqiu Island indicate that annual or periodical events, such as the KBC and SCSSC, could shape the microbial communities of Liuqiu Island. Finally, the dramatic alteration of the microbial community of 201509LC may have been induced by a strong climate event, such as a typhoon or heavy rainfall. This study has showed that long-term monitoring of a microbial community not only establishes ecological background information and shows the dynamic changes in microbial indicators but reveals whether putative events might influence marine environments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jmse11010085/s1, Table S1: Summary of reads, operational taxonomic unit (OTU) numbers, Shannon entropy, and Simpson’s index of microbial communities from sites of Liuqiu island, Taiwan; Table S2: One-way ANOVA test of the Shannon entropy and Simpson’s index; Table S3: The microbial community structure of each sample at OTUs level; Table S4: PERMANOVA analysis (Bray–Curtis) of microbial communities of Liuqiu sediment samples; Table S5: The relative abundance of the putative pathogen and human-related genera in all samples.
Author Contributions
Conceptualization, F.-Y.W. and M.-Y.L.; methodology, F.-Y.W.; formal analysis, F.-Y.W.; investigation, F.-Y.W.; resources, F.-Y.W. and M.-Y.L.; data curation, F.-Y.W. and M.-Y.L.; writing—original draft preparation, F.-Y.W.; writing—review and editing, F.-Y.W. and M.-Y.L.; visualization, F.-Y.W.; supervision, F.-Y.W.; project administration, F.-Y.W.; funding acquisition, F.-Y.W. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Taiwan Ocean Research Institute and the Ministry of Science and Technology, Taiwan (MOST 109-2119-M-001-011- and MOST 110-2119-M-001-007-).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We appreciate the suggestions of Li-Lian Liu and Tzu-Hsuan Tu, the administrative support of Yi Yang, Lei Feng, and Chau-Chang Wang.
Conflicts of Interest
The authors declare no conflict of interest and the funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Ainsworth, T.D.; Renzi, J.J.; Silliman, B.R. Positive Interactions in the Coral Macro and Microbiome. Trends Microbiol. 2020, 28, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Bellwood, D.R.; Hughes, T.P.; Folke, C.; Nyström, M. Confronting the coral reef crisis. Nature 2004, 429, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Capone, D.G.; Dunham, S.E.; Horrigan, S.G.; Duguay, L.E. Microbial nitrogen transformations in unconsolidated coral reef sediments. Mar. Ecol. Prog. Ser. 1992, 80, 75–88. [Google Scholar] [CrossRef]
- Sakka, A.; Legendre, L.; Gosselin, M.; Niquil, N.; Delesalle, B. Carbon budget of the planktonic food web in an atoll lagoon (Takapoto, French Polynesia). J. Plankton Res. 2002, 24, 301–320. [Google Scholar] [CrossRef]
- Werner, U.; Blazejak, A.; Bird, P.; Eickert, G.; Schoon, R.; Abed, R.M.M.; Bissett, A.; de Beer, D. Microbial photosynthesis in coral reef sediments (Heron Reef, Australia). Estuar. Coast. Shelf Sci. 2008, 76, 876–888. [Google Scholar] [CrossRef]
- Mydlarz, L.D.; McGinty, E.S.; Harvell, C.D. What are the physiological and immunological responses of coral to climate warming and disease? J. Exp. Biol. 2010, 213, 934–945. [Google Scholar] [CrossRef]
- Bulan, D.E.; Wilantho, A.; Tongsima, S.; Viyakarn, V.; Chavanich, S.; Somboonna, N. Microbial and small eukaryotes associated with reefs in the upper Gulf of Thailand. Front. Mar. Sci. 2018, 5, 436. [Google Scholar] [CrossRef]
- Lima, L.F.; Alker, A.T.; Papudeshi, B.; Morris, M.M.; Edwards, R.A.; De Putron, S.J.; Dinsdale, E.A. Coral and seawater metagenomes reveal key microbial functions to coral health and ecosystem functioning shaped at reef scale. Microb. Ecol. 2022, 1–16. [Google Scholar] [CrossRef]
- Rusch, A.; Hannides, A.K.; Gaidos, E. Diverse communities of active Bacteria and Archaea along oxygen gradients in coral reef sediments. Coral Reefs 2009, 28, 15–26. [Google Scholar] [CrossRef]
- Alongi, D.M.; Trott, L.A.; Pfitzner, J. Deposition, mineralization, and storage of carbon and nitrogen in sediments of the far northern and northern Great Barrier Reef shelf. Cont. Shelf Res. 2007, 27, 2595–2622. [Google Scholar] [CrossRef]
- Wild, C.; Huettel, M.; Klueter, A.; Kremb, S.G.; Rasheed, M.Y.; Jørgensen, B.B. Coral mucus functions as an energy carrier and particle trap in the reef ecosystem. Nature 2004, 428, 66–70. [Google Scholar] [CrossRef]
- Wild, C.; Rasheed, M.; Werner, U.; Franke, U.; Johnstone, R.; Huettel, M. Degradation and mineralization of coral mucus in reef environments. Mar. Ecol. Prog. Ser. 2004, 267, 159–171. [Google Scholar] [CrossRef]
- Nelson, C.E.; Wegley Kelly, L.; Haas, A.F. Microbial Interactions with Dissolved Organic Matter Are Central to Coral Reef Ecosystem Function and Resilience. Annu. Rev. Mar. Sci. 2022, 15, 1234. [Google Scholar] [CrossRef]
- Cardini, U.; Bednarz, V.N.; Foster, R.A.; Wild, C. Benthic N2 fixation in coral reefs and the potential effects of human-induced environmental change. Ecol. Evol. 2014, 4, 1706–1727. [Google Scholar] [CrossRef]
- D’Elia, C.F.; Wiebe, W. Biogeochemical nutrient cycles in coral-reef ecosystems. Ecosyst. World 1990, 25, 49–74. [Google Scholar] [CrossRef]
- Tribble, G.; Atkinson, M.; Sansone, F.; Smith, S. Reef metabolism and endo-upwelling in perspective. Coral Reefs 1994, 13, 199–201. [Google Scholar] [CrossRef]
- Schöttner, S.; Pfitzner, B.; Grünke, S.; Rasheed, M.; Wild, C.; Ramette, A. Drivers of bacterial diversity dynamics in permeable carbonate and silicate coral reef sands from the Red Sea. Environ. Microbiol. 2011, 13, 1815–1826. [Google Scholar] [CrossRef]
- Carlos, C.; Torres, T.T.; Ottoboni, L.M. Bacterial communities and species-specific associations with the mucus of Brazilian coral species. Sci. Rep. 2013, 3, 1624. [Google Scholar] [CrossRef]
- Glasl, B.; Herndl, G.J.; Frade, P.R. The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 2016, 10, 2280–2292. [Google Scholar] [CrossRef]
- Uthicke, S.; McGuire, K. Bacterial communities in Great Barrier Reef calcareous sediments: Contrasting 16S rDNA libraries from nearshore and outer shelf reefs. Estuar. Coast. Shelf Sci. 2007, 72, 188–200. [Google Scholar] [CrossRef]
- Glasl, B.; Bourne, D.G.; Frade, P.R.; Thomas, T.; Schaffelke, B.; Webster, N.S. Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome 2019, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, F.; Suzuki, M.; Takai, K.; Oida, H.; Sakamoto, T.; Aoki, K.; Nealson, K.H.; Horikoshi, K. Microbial communities associated with geological horizons in coastal subseafloor sediments from the sea of okhotsk. Appl. Environ. Microbiol. 2003, 69, 7224–7235. [Google Scholar] [CrossRef] [PubMed]
- Newberry, C.J.; Webster, G.; Cragg, B.A.; Parkes, R.J.; Weightman, A.J.; Fry, J.C. Diversity of prokaryotes and methanogenesis in deep subsurface sediments from the Nankai Trough, Ocean Drilling Program Leg 190. Environ. Microbiol. 2004, 6, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Rochelle, P.A.; Cragg, B.A.; Fry, J.C.; John Parkes, R.; Weightman, A.J. Effect of sample handling on estimation of bacterial diversity in marine sediments by 16S rRNA gene sequence analysis. FEMS Microbiol. Ecol. 1994, 15, 215–225. [Google Scholar] [CrossRef]
- Soliman, T.; Reimer, J.D.; Yang, S.Y.; Villar-Briones, A.; Roy, M.C.; Jenke-Kodama, H. Diversity of Microbial Communities and Quantitative Chemodiversity in Layers of Marine Sediment Cores from a Causeway (Kaichu-Doro) in Okinawa Island, Japan. Front. Microbiol. 2017, 8, 2451. [Google Scholar] [CrossRef]
- Webster, G.; Parkes, R.J.; Cragg, B.A.; Newberry, C.J.; Weightman, A.J.; Fry, J.C. Prokaryotic community composition and biogeochemical processes in deep subseafloor sediments from the Peru Margin. FEMS Microbiol. Ecol. 2006, 58, 65–85. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.; Huang, J.; Guo, X.; Zhang, Y.; Liu, D.; Wu, R.; He, H.; Wang, J. Diversity of the microbial community and cultivable protease-producing bacteria in the sediments of the Bohai Sea, Yellow Sea and South China Sea. PLoS ONE 2019, 14, e0215328. [Google Scholar] [CrossRef]
- Guan, X.; Zhu, L.; Li, Y.; Xie, Y.; Zhao, M.; Luo, X. Composition and variation of sediment bacterial and nirS-harboring bacterial communities at representative sites of the Bohai Gulf coastal zone, China. World J. Microbiol. Biotechnol. 2014, 30, 1291–1300. [Google Scholar] [CrossRef]
- Köchling, T.; Lara-Martín, P.; González-Mazo, E.; Amils, R.; Sanz, J.L. Microbial community composition of anoxic marine sediments in the Bay of Cádiz (Spain). Int. Microbiol. 2011, 14, 143–154. [Google Scholar] [CrossRef]
- Lu, X.M.; Chen, C.; Zheng, T.L.; Chen, J.J. Temporal-spatial variation of bacterial diversity in estuary sediments in the south of Zhejiang Province, China. Appl. Microbiol. Biotechnol. 2016, 100, 2817–2828. [Google Scholar] [CrossRef]
- Su, Z.; Dai, T.; Tang, Y.; Tao, Y.; Huang, B.; Mu, Q.; Wen, D. Sediment bacterial community structures and their predicted functions implied the impacts from natural processes and anthropogenic activities in coastal area. Mar. Pollut. Bull. 2018, 131, 481–495. [Google Scholar] [CrossRef]
- Wang, L.; Liu, L.; Zheng, B.; Zhu, Y.; Wang, X. Analysis of the bacterial community in the two typical intertidal sediments of Bohai Bay, China by pyrosequencing. Mar. Pollut. Bull. 2013, 72, 181–187. [Google Scholar] [CrossRef]
- Chen, J.; McIlroy, S.E.; Archana, A.; Baker, D.M.; Panagiotou, G. A pollution gradient contributes to the taxonomic, functional, and resistome diversity of microbial communities in marine sediments. Microbiome 2019, 7, 104. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Xiang, S.Z.; Yan, Y.; Yang, R.; Lin, M.P.; Wang, X.M.; Xue, Y.L.; Guan, X.Y. Sediment Microbial Communities and Their Potential Role as Environmental Pollution Indicators in Xuande Atoll, South China Sea. Front. Microbiol. 2020, 11, 1011. [Google Scholar] [CrossRef]
- Zhang, W.; Ki, J.-S.; Qian, P.-Y. Microbial diversity in polluted harbor sediments I: Bacterial community assessment based on four clone libraries of 16S rDNA. Estuar. Coast. Shelf Sci. 2008, 76, 668–681. [Google Scholar] [CrossRef]
- Quero, G.M.; Ape, F.; Manini, E.; Mirto, S.; Luna, G.M. Temporal changes in microbial communities beneath fish farm sediments are related to organic enrichment and fish biomass over a production cycle. Front. Mar. Sci. 2020, 7, 524. [Google Scholar] [CrossRef]
- Verhoeven, J.T.P.; Salvo, F.; Knight, R.; Hamoutene, D.; Dufour, S.C. Temporal Bacterial Surveillance of Salmon Aquaculture Sites Indicates a Long Lasting Benthic Impact With Minimal Recovery. Front. Microbiol. 2018, 9, 3054. [Google Scholar] [CrossRef]
- Chung, H.W.; Liu, C.C.; Chiu, Y.S.; Liu, J.T. Spatiotemporal variation of Gaoping River plume observed by Formosat-2 high resolution imagery. J. Mar. Syst. 2014, 132, 28–37. [Google Scholar] [CrossRef]
- Liao, J.-m.; Yang, Y.; Lai, J.-w.; Chen, S.-h. High resolution ocean current model in the Southwest Taiwan Sea. In Proceedings of the 2015 Conference on Weather Analysis and Forecasting, Chicago, IL, USA, 29 June–3 July 2015; p. A5-3. [Google Scholar]
- Chen, J.-P.; Wang, F.-Y.; Liu, M.-Y.; Ho, S.-H. The coral paradise—Liuqiu Island. Sci. Dev. 2017, 536, 36–43. (In Chinese) [Google Scholar]
- Guo, F.; Zhang, T. Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Appl. Microbiol. Biotechnol. 2013, 97, 4607–4616. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, P.Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Jost, L. Entropy and diversity. Oikos 2006, 113, 363–375. [Google Scholar] [CrossRef]
- Masisi, L.; Nelwamondo, V.; Marwala, T. The use of entropy to measure structural diversity. In Proceedings of the 2008 IEEE International Conference on Computational Cybernetics, Stara Lesna, Slovakia, 27–29 November 2008; pp. 41–45. [Google Scholar]
- Beals, E.W. Bray-Curtis Ordination: An Effective Strategy for Analysis of Multivariate Ecological Data. In Advances in Ecological Research; MacFadyen, A., Ford, E.D., Eds.; Academic Press: Cambridge, MA, USA, 1984; Volume 14, pp. 1–55. [Google Scholar]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar] [CrossRef]
- Ye, Q.; Wu, Y.; Zhu, Z.; Wang, X.; Li, Z.; Zhang, J. Bacterial diversity in the surface sediments of the hypoxic zone near the Changjiang Estuary and in the East China Sea. MicrobiologyOpen 2016, 5, 323–339. [Google Scholar] [CrossRef]
- Zhou, M.-Y.; Wang, G.-L.; Li, D.; Zhao, D.-L.; Qin, Q.-L.; Chen, X.-L.; Chen, B.; Zhou, B.-C.; Zhang, X.-Y.; Zhang, Y.-Z. Diversity of both the cultivable protease-producing bacteria and bacterial extracellular proteases in the coastal sediments of King George Island, Antarctica. PLoS ONE 2013, 8, e79668. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Han, X.X.; Chen, X.L.; Dang, H.Y.; Xie, B.B.; Qin, Q.L.; Shi, M.; Zhou, B.C.; Zhang, Y.Z. Diversity of cultivable protease-producing bacteria in sediments of Jiaozhou Bay, China. Front. Microbiol. 2015, 6, 1021. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Haldar, A.; Bhattacharyya, M.; Ghosh, A. Anthropogenic influence shapes the distribution of antibiotic resistant bacteria (ARB) in the sediment of Sundarban estuary in India. Sci. Total Environ. 2019, 647, 1626–1639. [Google Scholar] [CrossRef]
- Ramesh, S.; Rajesh, M.; Mathivanan, N. Characterization of a thermostable alkaline protease produced by marine Streptomyces fungicidicus MML1614. Bioprocess Biosyst. Eng. 2009, 32, 791–800. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Church, M.J. Resource control of bacterial dynamics in the sea. Microb. Ecol. Ocean. 2008, 1, 335–382. [Google Scholar] [CrossRef]
- Kim, H.-J.; Miller, A.J.; McGowan, J.; Carter, M.L. Coastal phytoplankton blooms in the Southern California Bight. Prog. Oceanogr. 2009, 82, 137–147. [Google Scholar] [CrossRef]
- Ploug, H.; Grossart, H.-P.; Azam, F.; Jørgensen, B.B. Photosynthesis, respiration, and carbon turnover in sinking marine snow from surface waters of Southern California Bight: Implications for the carbon cycle in the ocean. Mar. Ecol. Prog. Ser. 1999, 179, 1–11. [Google Scholar] [CrossRef]
- Rath, J.; Wu, K.Y.; Herndl, G.J.; DeLong, E.F. High phylogenetic diversity in a marine-snow-associated bacterial assemblage. Aquat. Microb. Ecol. 1998, 14, 261–269. [Google Scholar] [CrossRef]
- Goodfellow, M.; Haynes, J. Actinomycetes in Marine Sediments. Biological, Biochemical and Biomedical Aspects of Actinomycetes; Academic Press: New York, NY, USA, 1984; pp. 453–472. [Google Scholar]
- Das, S.; Lyla, P.; Khan, S.A. Marine microbial diversity and ecology: Importance and future perspectives. Curr. Sci. 2006, 90, 1325–1335. [Google Scholar] [CrossRef]
- Castelle, C.J.; Brown, C.T.; Thomas, B.C.; Williams, K.H.; Banfield, J.F. Unusual respiratory capacity and nitrogen metabolism in a Parcubacterium (OD1) of the Candidate Phyla Radiation. Sci. Rep. 2017, 7, 40101. [Google Scholar] [CrossRef]
- Rahlff, J.; Giebel, H.-A.; Stolle, C.; Wurl, O.; Probst, A.J.; Herlemann, D.P. Overlooked diversity of ultramicrobacterial minorities at the air-sea interface. Atmosphere 2020, 11, 1214. [Google Scholar] [CrossRef]
- Chimetto, L.A.; Cleenwerck, I.; Brocchi, M.; Willems, A.; De Vos, P.; Thompson, F.L. Marinomonas brasilensis sp. nov., isolated from the coral Mussismilia hispida, and reclassification of Marinomonas basaltis as a later heterotypic synonym of Marinomonas communis. Int. J. Syst. Evol. Microbiol. 2011, 61, 1170–1175. [Google Scholar] [CrossRef]
- Lucas-Elío, P.; Marco-Noales, E.; Espinosa, E.; Ordax, M.; López, M.M.; Garcías-Bonet, N.; Marba, N.; Duarte, C.M.; Sanchez-Amat, A. Marinomonas alcarazii sp. nov., M. rhizomae sp. nov., M. foliarum sp. nov., M. posidonica sp. nov. and M. aquiplantarum sp. nov., isolated from the microbiota of the seagrass Posidonia oceanica. Int. J. Syst. Evol. Microbiol. 2011, 61, 2191–2196. [Google Scholar] [CrossRef]
- Romanenko, L.A.; Tanaka, N.; Frolova, G.M. Marinomonas arenicola sp. nov., isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 2009, 59, 2834–2838. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Kang, S.-J.; Oh, T.-K. Marinomonas dokdonensis sp. nov., isolated from sea water. Int. J. Syst. Evol. Microbiol. 2005, 55, 2303–2307. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Nonstad, I.; Faksness, L.-G.; Brandvik, P.J. Responses of microbial communities in Arctic sea ice after contamination by crude petroleum oil. Microb. Ecol. 2008, 55, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Curson, A.R.; Todd, J.D.; Sullivan, M.J.; Johnston, A.W. Catabolism of dimethylsulphoniopropionate: Microorganisms, enzymes and genes. Nat. Rev. Microbiol. 2011, 9, 849–859. [Google Scholar] [CrossRef]
- Iwaki, H.; Nishimura, A.; Hasegawa, Y. Isolation and characterization of marine bacteria capable of utilizing phthalate. World J. Microbiol. Biotechnol. 2012, 28, 1321–1325. [Google Scholar] [CrossRef]
- Bartz, J.-O.; Blom, J.; Busse, H.-J.; Mvie, J.B.; Hardt, M.; Schubert, P.; Wilke, T.; Goessmann, A.; Wilharm, G.; Bender, J. Parendozoicomonas haliclonae gen. nov. sp. nov. isolated from a marine sponge of the genus Haliclona and description of the family Endozoicomonadaceae fam. nov. comprising the genera Endozoicomonas, Parendozoicomonas, and Kistimonas. Syst. Appl. Microbiol. 2018, 41, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.-Y.; Shiu, J.-H.; Chen, W.-M.; Chiang, Y.-R.; Tang, S.-L. Genomic insight into the host–endosymbiont relationship of Endozoicomonas montiporae CL-33T with its coral host. Front. Microbiol. 2016, 7, 251. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, M.; Yokota, A. Endozoicomonas elysicola gen. nov., sp. nov., a γ-proteobacterium isolated from the sea slug Elysia ornata. Syst. Appl. Microbiol. 2007, 30, 202–206. [Google Scholar] [CrossRef]
- Neave, M.J.; Apprill, A.; Ferrier-Pagès, C.; Voolstra, C.R. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl. Microbiol. Biotechnol. 2016, 100, 8315–8324. [Google Scholar] [CrossRef]
- Neave, M.J.; Michell, C.T.; Apprill, A.; Voolstra, C.R. Whole-genome sequences of three symbiotic Endozoicomonas strains. Genome Announc. 2014, 2, e00802-00814. [Google Scholar] [CrossRef]
- Neave, M.J.; Michell, C.T.; Apprill, A.; Voolstra, C.R. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci. Rep. 2017, 7, 40579. [Google Scholar] [CrossRef]
- Tandon, K.; Lu, C.-Y.; Chiang, P.-W.; Wada, N.; Yang, S.-H.; Chan, Y.-F.; Chen, P.-Y.; Chang, H.-Y.; Chiou, Y.-J.; Chou, M.-S. Comparative genomics: Dominant coral-bacterium Endozoicomonas acroporae metabolizes dimethylsulfoniopropionate (DMSP). ISME J. 2020, 14, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Camp, E.F.; Suggett, D.J.; Pogoreutz, C.; Nitschke, M.R.; Houlbreque, F.; Hume, B.C.C.; Gardner, S.G.; Zampighi, M.; Rodolfo-Metalpa, R.; Voolstra, C.R. Corals exhibit distinct patterns of microbial reorganisation to thrive in an extreme inshore environment. Coral Reefs 2020, 39, 701–716. [Google Scholar] [CrossRef]
- Yoon, J.; Adachi, K.; Park, S.; Kasai, H.; Yokota, A. Aureibacter tunicatorum gen. nov., sp. nov., a marine bacterium isolated from a coral reef sea squirt, and description of Flammeovirgaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2011, 61, 2342–2347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferguson, R.M.; Coulon, F.; Villa, R. Understanding microbial ecology can help improve biogas production in AD. Sci. Total Environ. 2018, 642, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Asker, D.; Beppu, T.; Ueda, K. Zeaxanthinibacter enoshimensis gen. nov., sp. nov., a novel zeaxanthin-producing marine bacterium of the family Flavobacteriaceae, isolated from seawater off Enoshima Island, Japan. Int. J. Syst. Evol. Microbiol. 2007, 57, 837–843. [Google Scholar] [CrossRef]
- Lee, Y.; Jeong, H.I.; Jeong, S.E.; Jeon, C.O. Zeaxanthinibacter aestuarii sp. nov., isolated from estuary sediment and emended description of the genus Zeaxanthinibacter Asker et al. 2007. Int. J. Syst. Evol. Microbiol. 2016, 66, 3264–3269. [Google Scholar] [CrossRef]
- Newton, R.J.; Huse, S.M.; Morrison, H.G.; Peake, C.S.; Sogin, M.L.; McLellan, S.L. Shifts in the microbial community composition of Gulf Coast beaches following beach oiling. PLoS ONE 2013, 8, e74265. [Google Scholar] [CrossRef]
- Hoffmann, K.; Bienhold, C.; Buttigieg, P.L.; Knittel, K.; Laso-Perez, R.; Rapp, J.Z.; Boetius, A.; Offre, P. Diversity and metabolism of Woeseiales bacteria, global members of marine sediment communities. ISME J. 2020, 14, 1042–1056. [Google Scholar] [CrossRef]
- Mußmann, M.; Pjevac, P.; Krüger, K.; Dyksma, S. Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J. 2017, 11, 1276–1281. [Google Scholar] [CrossRef]
- Klawonn, I.; Nahar, N.; Walve, J.; Andersson, B.; Olofsson, M.; Svedén, J.; Littmann, S.; Whitehouse, M.J.; Kuypers, M.; Ploug, H. Cell-specific nitrogen-and carbon-fixation of cyanobacteria in a temperate marine system (Baltic Sea). Environ. Microbiol. 2016, 18, 4596–4609. [Google Scholar] [CrossRef]
- Bengtsson, M.M.; Øvreås, L. Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol. 2010, 10, 261. [Google Scholar] [CrossRef]
- DeLong, E.F.; Franks, D.G.; Alldredge, A.L. Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 1993, 38, 924–934. [Google Scholar] [CrossRef]
- Fuerst, J.A.; Gwilliam, H.G.; Lindsay, M.; Lichanska, A.; Belcher, C.; Vickers, J.E.; Hugenholtz, P. Isolation and molecular identification of planctomycete bacteria from postlarvae of the giant tiger prawn, Penaeus monodon. Appl. Environ. Microbiol. 1997, 63, 254–262. [Google Scholar] [CrossRef]
- Pimentel-Elardo, S.; Wehrl, M.; Friedrich, A.B.; Jensen, P.R.; Hentschel, U. Isolation of planctomycetes from Aplysina sponges. Aquat. Microb. Ecol. 2003, 33, 239–245. [Google Scholar] [CrossRef]
- Winkelmann, N.; Harder, J. An improved isolation method for attached-living Planctomycetes of the genus Rhodopirellula. J. Microbiol. Methods 2009, 77, 276–284. [Google Scholar] [CrossRef]
- Žure, M.; Fernandez-Guerra, A.; Munn, C.B.; Harder, J. Geographic distribution at subspecies resolution level: Closely related Rhodopirellula species in European coastal sediments. ISME J. 2017, 11, 478–489. [Google Scholar] [CrossRef]
- Schlesner, H.; Rensmann, C.; Tindall, B.J.; Gade, D.; Rabus, R.; Pfeiffer, S.; Hirsch, P. Taxonomic heterogeneity within the Planctomycetales as derived by DNA–DNA hybridization, description of Rhodopirellula baltica gen. nov., sp. nov., transfer of Pirellula marina to the genus Blastopirellula gen. nov. as Blastopirellula marina comb. nov. and emended description of the genus Pirellula. Int. J. Syst. Evol. Microbiol. 2004, 54, 1567–1580. [Google Scholar] [CrossRef]
- Dekio, I.; Asahina, A.; Shah, H.N. Unravelling the eco-specificity and pathophysiological properties of Cutibacterium species in the light of recent taxonomic changes. Anaerobe 2021, 71, 102411. [Google Scholar] [CrossRef]
- McDowell, A.; Patrick, S.; Eishi, Y.; Lambert, P.; Eady, A. Propionibacterium acnes in human health and disease. BioMed Res. Int. 2013, 2013, 493564. [Google Scholar] [CrossRef]
- Pękala, A.; Paździor, E.; Antychowicz, J.; Bernad, A.; Głowacka, H.; Więcek, B.; Niemczuk, W. Kocuria rhizophila and Micrococcus luteus as emerging opportunist pathogens in brown trout (Salmo trutta Linnaeus, 1758) and rainbow trout ( Oncorhynchus mykiss Walbaum, 1792). Aquaculture 2018, 486, 285–289. [Google Scholar] [CrossRef]
- Platsidaki, E.; Dessinioti, C. Recent advances in understanding Propionibacterium acnes (Cutibacterium acnes) in acne. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.F.; Kilian, M. The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus Propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, S.J.; Knoop, F.C. Corynebacterium infections. In xPharm: The Comprehensive Pharmacology Reference; Elsevier Inc.: Amsterdam, The Netherlands, 2007; pp. 1–5. [Google Scholar]
- Schaechter, M. Encyclopedia of Microbiology; Academic Press: Cambridge, MA, USA, 2009. [Google Scholar]
- Kandi, V.; Palange, P.; Vaish, R.; Bhatti, A.B.; Kale, V.; Kandi, M.R.; Bhoomagiri, M.R. Emerging Bacterial Infection: Identification and Clinical Significance of Kocuria Species. Cureus 2016, 8, e731. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Kim, M.S.; Roh, S.W.; Jung, M.J.; Bae, J.W. Kocuria atrinae sp. nov., isolated from traditional Korean fermented seafood. Int. J. Syst. Evol. Microbiol. 2010, 60, 914–918. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stackebrandt, E.; Koch, C.; Gvozdiak, O.; Schumann, P. Taxonomic Dissection of the Genus Micrococcus: Kocuria gen. nov., Nesterenkonia gen. nov., Kytococcus gen. nov., Dermacoccus gen. nov., and Micrococcus Cohn 1872 gen. emend. Int. J. Syst. Evol. Microbiol. 1995, 45, 682–692. [Google Scholar] [CrossRef]
- Matteoli, F.P.; Olivares, F.L.; Venancio, T.M.; da Rocha, L.O.; da Silva Irineu, L.E.S.; Canellas, L.P. Herbaspirillum. In Beneficial Microbes in Agro-Ecology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 493–508. [Google Scholar]
- Lampel, K.A. Shigella; ASM Press: Washington, DC, USA, 2009; pp. 131–145. [Google Scholar]
- Pond, K.; World Health Organization; United States Environmental Protection Agency. Water Recreation and Disease: Plausibility of Associated Infections: Acute Effects, Sequelae and Mortality/K. Pond; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Brunette, G.W.; Kozarsky, P.E.; Cohen, N.J. CDC Health Information for International Travel 2016: The Yellow Book; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Kuo, N.-J.; Ho, C.-R. ENSO effect on the sea surface wind and sea surface temperature in the Taiwan Strait. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef]
- Huang, T.-H.; Chen, C.-T.A.; Zhang, W.-Z.; Zhuang, X.-F. Varying intensity of Kuroshio intrusion into Southeast Taiwan Strait during ENSO events. Cont. Shelf Res. 2015, 103, 79–87. [Google Scholar] [CrossRef]
- Keshavmurthy, S.; Kuo, C.-Y.; Huang, Y.-Y.; Carballo-Bolaños, R.; Meng, P.-J.; Wang, J.-T.; Chen, C.A. Coral reef resilience in Taiwan: Lessons from long-term ecological research on the Coral Reefs of Kenting National Park (Taiwan). J. Mar. Sci. Eng. 2019, 7, 388. [Google Scholar] [CrossRef]
- Hsieh, H.-Y.; Meng, P.-J.; Chang, Y.-C.; Lo, W.-T. Temporal and spatial occurrence of mesopelagic fish larvae during epipelagic drift associated with hydrographic features in the Gaoping coastal waters off southwestern Taiwan. Mar. Coast. Fish. 2017, 9, 244–259. [Google Scholar] [CrossRef]
- Liu, J.T.; Hsu, R.T.; Hung, J.-J.; Chang, Y.-P.; Wang, Y.-H.; Rendle-Bühring, R.H.; Lee, C.-L.; Huh, C.-A.; Yang, R.J. From the highest to the deepest: The Gaoping River–Gaoping Submarine Canyon dispersal system. Earth-Sci. Rev. 2016, 153, 274–300. [Google Scholar] [CrossRef]
- Hornick, K.M.; Buschmann, A.H. Insights into the diversity and metabolic function of bacterial communities in sediments from Chilean salmon aquaculture sites. Ann. Microbiol. 2018, 68, 63–77. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost microbiomes: The state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 2014, 5, 207. [Google Scholar] [CrossRef] [PubMed]
- Moncada, C.; Hassenrück, C.; Gärdes, A.; Conaco, C. Microbial community composition of sediments influenced by intensive mariculture activity. FEMS Microbiol. Ecol. 2019, 95, fiz006. [Google Scholar] [CrossRef] [PubMed]
- Nogales, B.; Lanfranconi, M.P.; Piña-Villalonga, J.M.; Bosch, R. Anthropogenic perturbations in marine microbial communities. FEMS Microbiol. Rev. 2011, 35, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.M.; Crosswell, J.; Metcalfe, S.S.; Carlin, G.; Morrison, P.D.; Karpe, A.V.; Palombo, E.A.; Steven, A.D.L.; Beale, D.J. Influence of Human Activities on Broad-Scale Estuarine-Marine Habitats Using Omics-Based Approaches Applied to Marine Sediments. Microorganisms 2019, 7, 419. [Google Scholar] [CrossRef]
- Ziegler, M.; Roik, A.; Porter, A.; Zubier, K.; Mudarris, M.S.; Ormond, R.; Voolstra, C.R. Coral microbial community dynamics in response to anthropogenic impacts near a major city in the central Red Sea. Mar. Pollut. Bull. 2016, 105, 629–640. [Google Scholar] [CrossRef]
- Austin, B.; Zhang, X.H. Vibrio harveyi: A significant pathogen of marine vertebrates and invertebrates. Lett. Appl. Microbiol. 2006, 43, 119–124. [Google Scholar] [CrossRef]
- Altonsy, M.O.; Kurwa, H.A.; Lauzon, G.J.; Amrein, M.; Gerber, A.N.; Almishri, W.; Mydlarski, P.R. Corynebacterium tuberculostearicum, a human skin colonizer, induces the canonical nuclear factor-kappaB inflammatory signaling pathway in human skin cells. Immun. Inflamm. Dis. 2020, 8, 62–79. [Google Scholar] [CrossRef]
- Dong, X.; Lan, H.; Huang, L.; Zhang, H.; Lin, X.; Weng, S.; Peng, Y.; Lin, J.; Wang, J.H.; Peng, J.; et al. Metagenomic Views of Microbial Communities in Sand Sediments Associated with Coral Reefs. Microb. Ecol. 2022, 1–13. [Google Scholar] [CrossRef]
- Hinić, V.; Lang, C.; Weisser, M.; Straub, C.; Frei, R.; Goldenberger, D. Corynebacterium tuberculostearicum: A potentially misidentified and multiresistant Corynebacterium species isolated from clinical specimens. J. Clin. Microbiol. 2012, 50, 2561–2567. [Google Scholar] [CrossRef]
- Roman-Ponce, B.; Wang, D.; Vásquez-Murrieta, M.S.; Chen, W.F.; Estrada-de Los Santos, P.; Sui, X.H.; Wang, E.T. Kocuria arsenatis sp. nov., an arsenic-resistant endophytic actinobacterium associated with Prosopis laegivata grown on high-arsenic-polluted mine tailing. Int. J. Syst. Evol. Microbiol. 2016, 66, 1027–1033. [Google Scholar] [CrossRef]
- Banchi, E.; Del Negro, P.; Celussi, M.; Malfatti, F. Sediment Features and Human Activities Structure the Surface Microbial Communities of the Venice Lagoon. Front. Mar. Sci. 2021, 8, 762292. [Google Scholar] [CrossRef]
- Guo, F.; Li, B.; Yang, Y.; Deng, Y.; Qiu, J.-W.; Li, X.; Leung, K.M.; Zhang, T. Impacts of human activities on distribution of sulfate-reducing prokaryotes and antibiotic resistance genes in marine coastal sediments of Hong Kong. FEMS Microbiol. Ecol. 2016, 92, fiw128. [Google Scholar] [CrossRef]
- Bond, P.L.; Hugenholtz, P.; Keller, J.; Blackall, L.L. Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl. Environ. Microbiol. 1995, 61, 1910–1916. [Google Scholar] [CrossRef]
- Godon, J.-J.; Zumstein, E.; Dabert, P.; Habouzit, F.; Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 1997, 63, 2802–2813. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Tyson, G.W.; Webb, R.I.; Wagner, A.M.; Blackall, L.L. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl. Environ. Microbiol. 2001, 67, 411–419. [Google Scholar] [CrossRef]
- Borneman, J.; Triplett, E.W. Molecular microbial diversity in soils from eastern Amazonia: Evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl. Environ. Microbiol. 1997, 63, 2647–2653. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).