




Isolation and Detection of Exosomal Mir210 Using Carbon Nanomaterial-Coated Magnetic Beads




Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Instrumentation
2.3. Exosome Isolation, Quantification, and Particle Size Determination
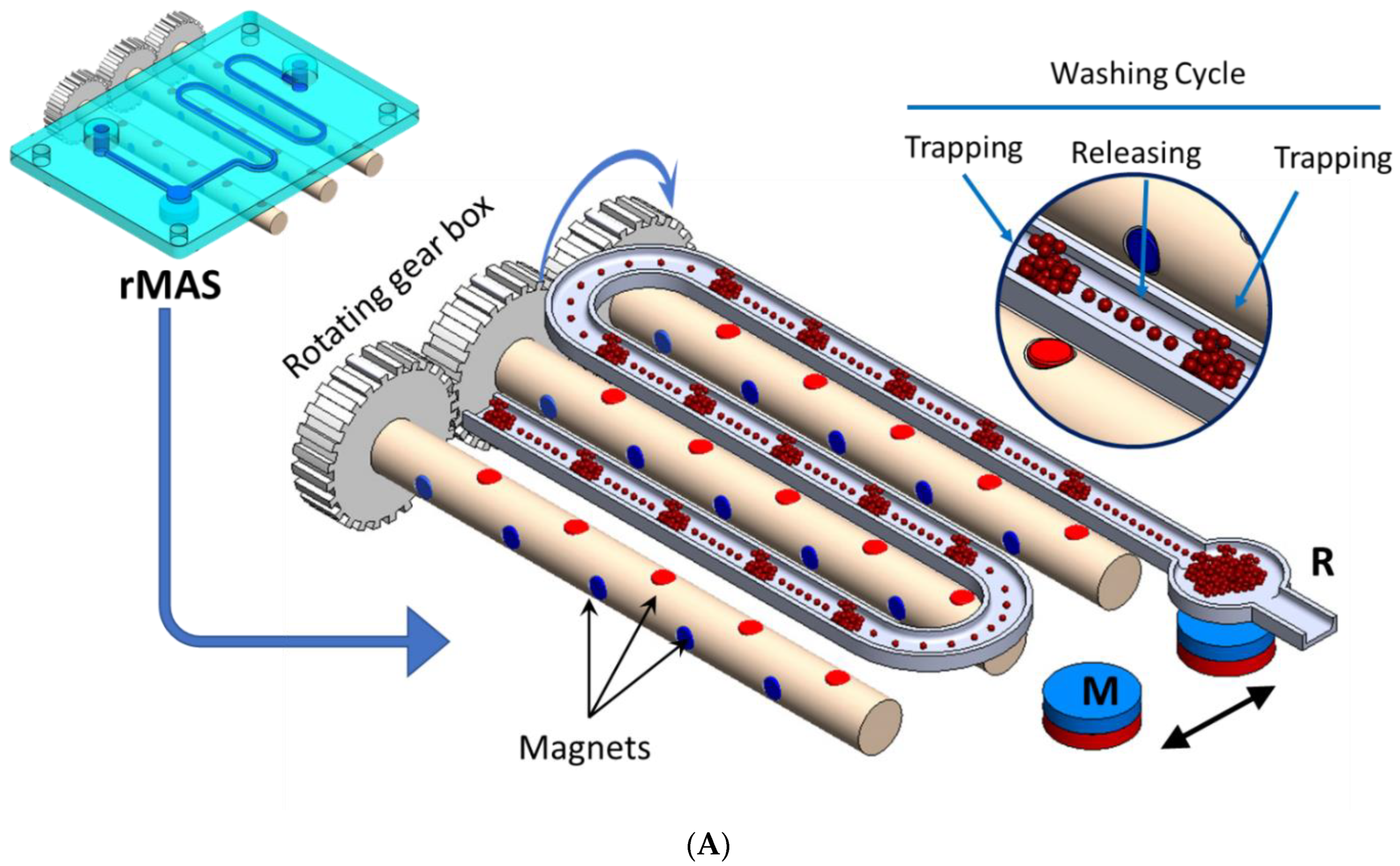
2.4. Magnetic Bead-Based Isolation of Exosomes (Apta-Magnetic Separation System)
2.5. RNA Isolation and Quantification
2.6. Reverse Transcription and RT-qPCR
2.7. Magnetic Nanobeads Fluorescence Sensor Platform for Mir210 Detection
2.8. Specificity
3. Results and Discussions
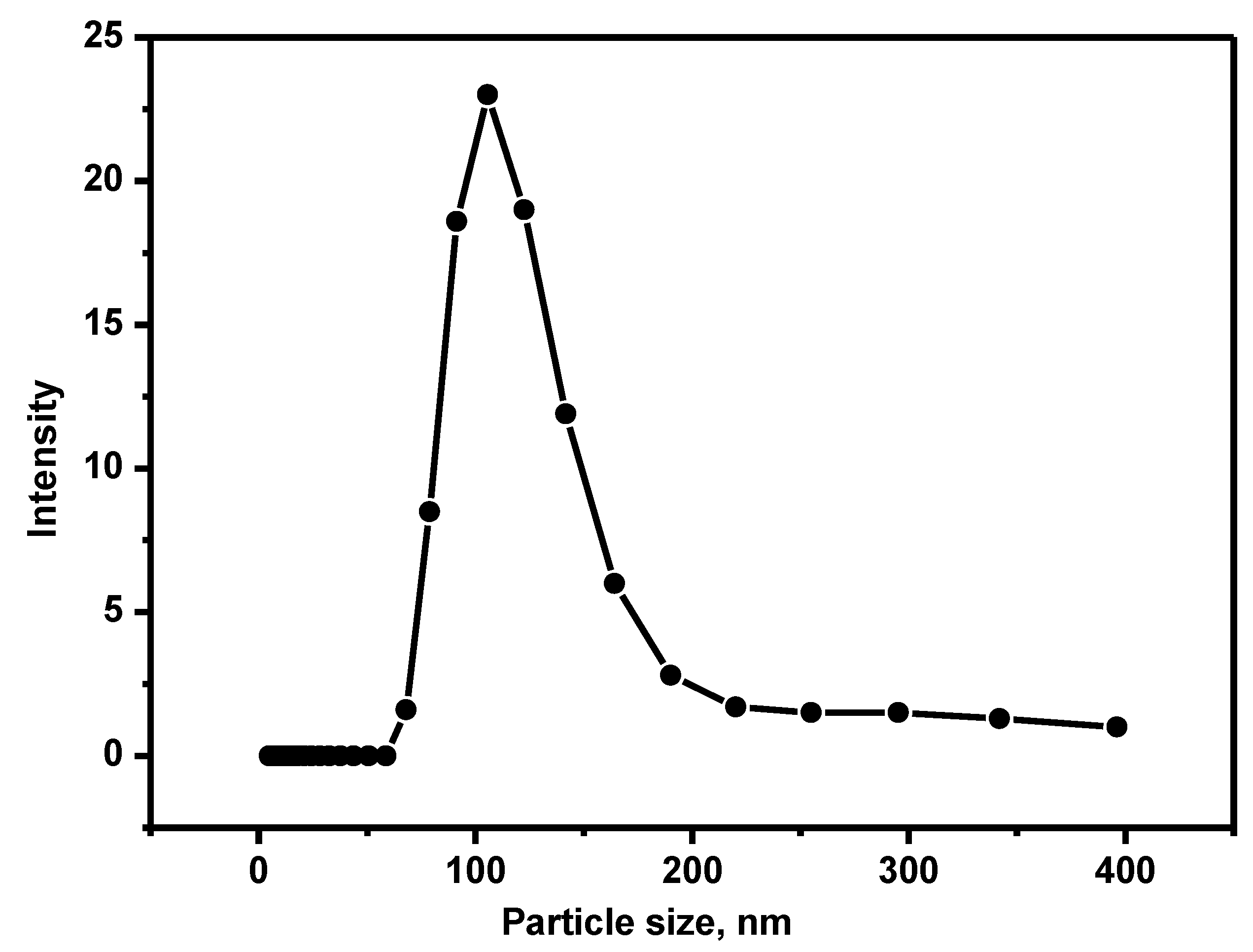
3.1. Apta-Magnetic Separation (AMS) and Particle Size Determination of Exosomes
3.2. Isolation and Measurement of Particle Size of Exosome
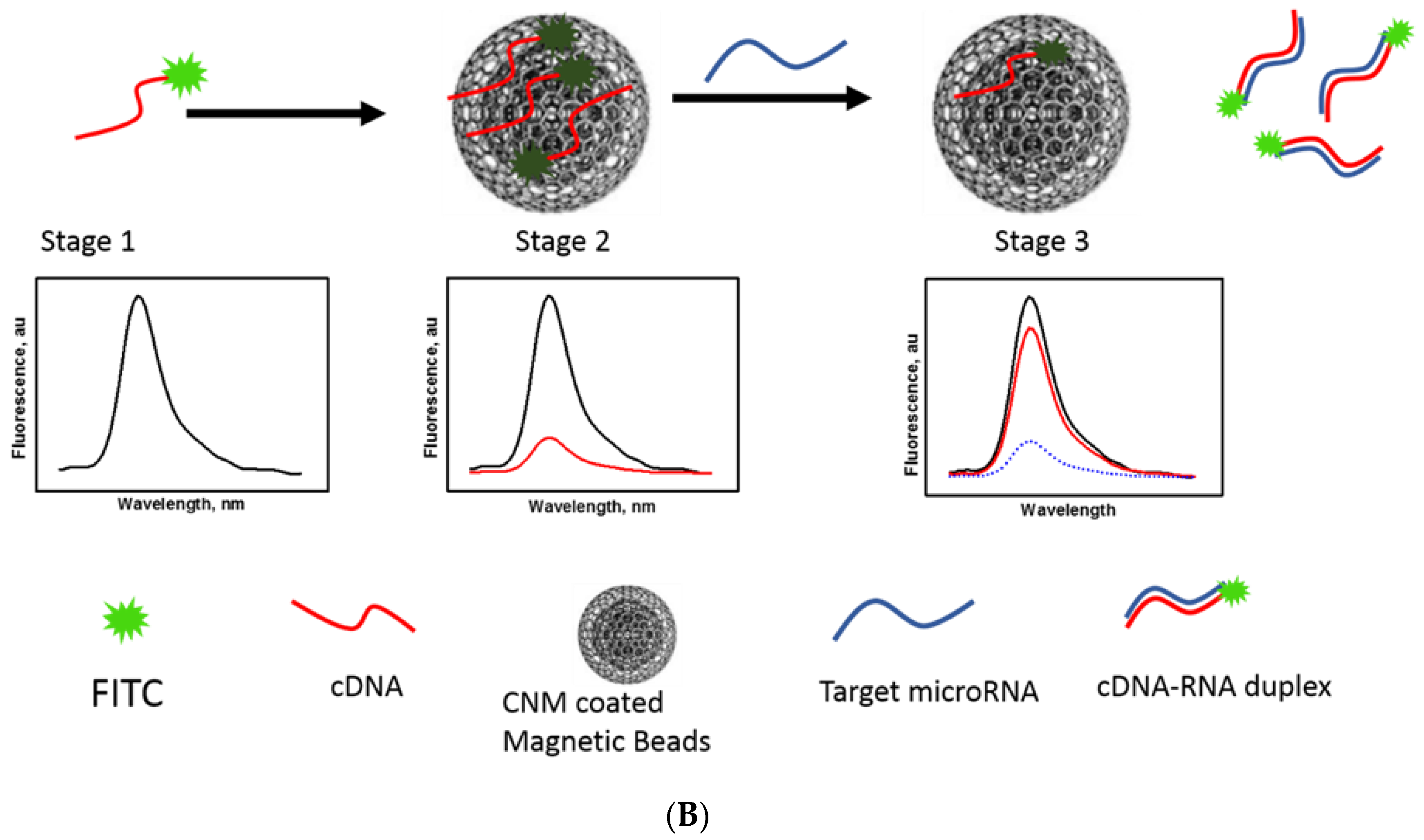
3.3. CCM Beads as a Fluorescence Sensing Substrate
3.4. Optimization of CNM/FAM-DNA Ratio (Fluorescence off State)
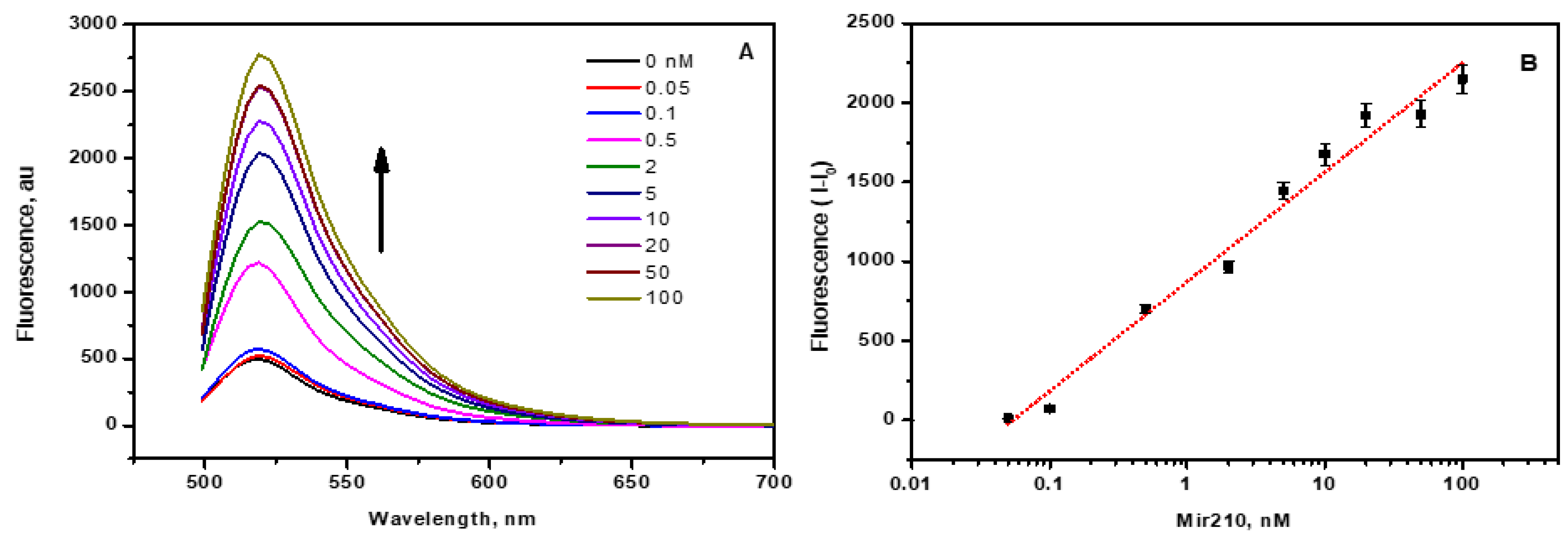
3.5. Mir210 Detection Using CCM Beads Fluorescence Assay
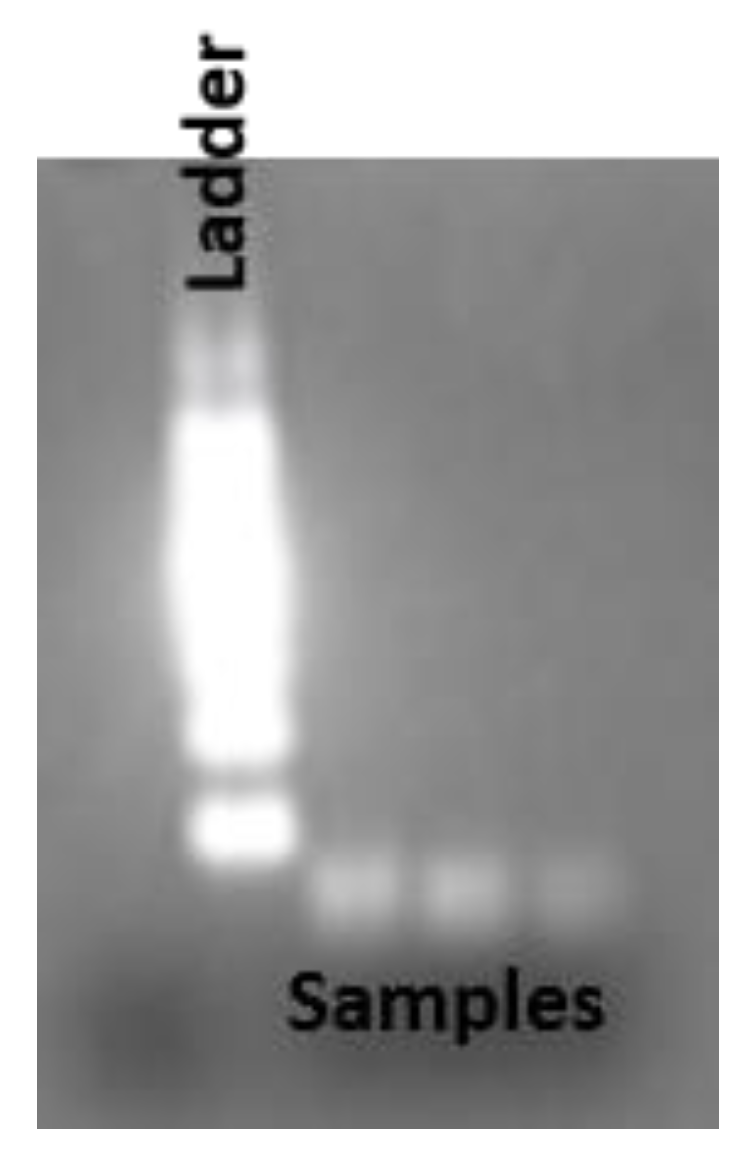
3.6. Reverse Transcription and RT-qPCR
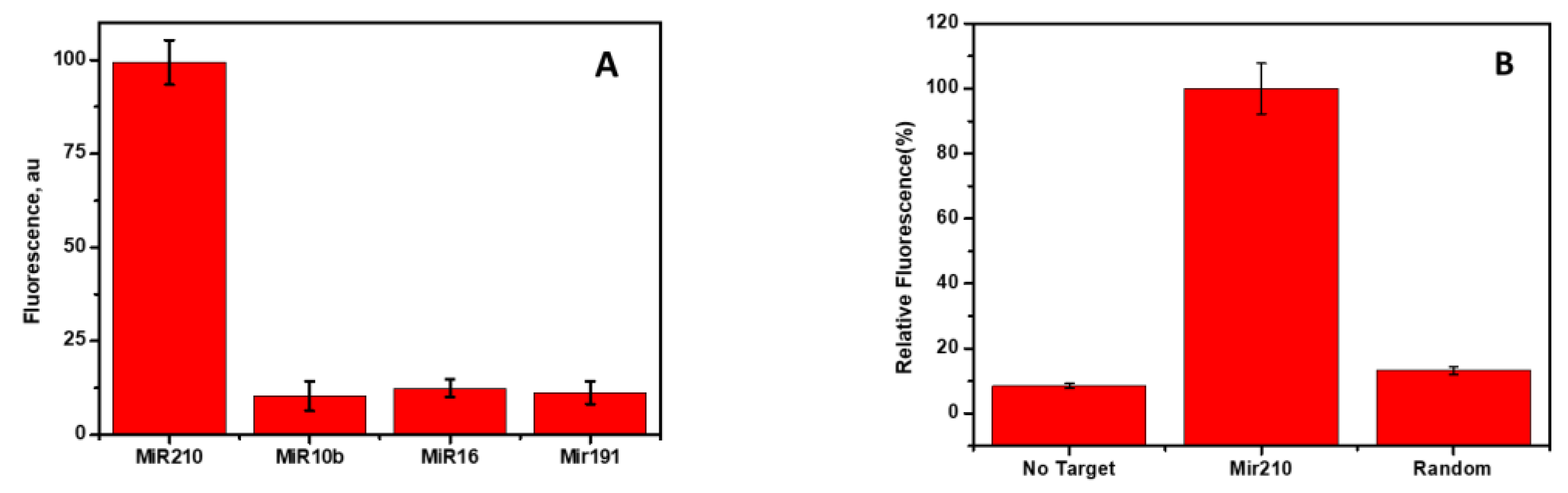
3.7. Cross-Reactivity and Specificity
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer. 2020. Available online: https://gco.iarc.fr/today (accessed on 11 February 2021).
- Vaidyanathan, R.; Soon, R.H.; Zhang, P.; Jiang, K.; Lim, C.T. Cancer diagnosis: From tumor to liquid biopsy and beyond. Lab Chip 2019, 19, 11–34. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Meng, J.; Zhu, L.; Peng, Y. Exosomal noncoding RNAs in Glioma: Biological functions and potential clinical applications. Mol. Cancer 2020, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M.; Sharifi, M. Exosomal miRNAs as novel cancer biomarkers: Challenges and opportunities. J. Cell. Physiol. 2018, 233, 6370–6380. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Honma, K.; Saji, S.; Moriwaki, H.; Ochiya, T.; Hoffman, R.M. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 2013, 65, 383–390. [Google Scholar] [CrossRef]
- Nahand, J.S.; Moghoofei, M.; Salmaninejad, A.; Bahmanpour, Z.; Karimzadeh, M.; Nasiri, M.; Mirzaei, H.R.; Pourhanifeh, M.H.; Bokharaei-Salim, F.; Mirzaei, H. Pathogenic role of exosomes and microRNAs in HPV-mediated inflammation and cervical cancer: A review. Int. J. Cancer 2020, 146, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.W.; Fletcher, S.; Wilton, D.S. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol. Life Sci. 2012, 69, 3613–3634. [Google Scholar] [CrossRef]
- Bottani, M.; Banfi, G.; Lombardi, G. Circulating miRNAs as diagnostic and prognostic biomarkers in common solid tumors: Focus on lung, breast, prostate cancers, and osteosarcoma. J. Clin. Med. 2019, 8, 1661. [Google Scholar] [CrossRef]
- Bayraktar, R.; Van Roosbroeck, K.; Calin, G.A. Cell-to-cell communication: MicroRNAs as hormones. Mol. Oncol. 2017, 11, 1673–1686. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids—The mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Javidi, M.A.; Ahmadi, A.H.; Bakhshinejad, B.; Nouraee, N.; Babashah, S.; Sadeghizadeh, M. Cell-free microRNAs as cancer biomarkers: The odyssey of miRNAs through body fluids. Med. Oncol. 2014, 31, 1–11. [Google Scholar] [CrossRef]
- Chen, J.; Li, P.; Zhang, T.; Xu, Z.; Huang, X.; Wang, R.; Du, L. Review on strategies and technologies for exosome isolation and purification. Front. Bioeng. Biotechnol. 2022, 9, 811971. [Google Scholar] [CrossRef] [PubMed]
- Sidhom, K.; Obi, P.O.; Saleem, A. A review of exosomal isolation methods: Is size exclusion chromatography the best option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.M.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 5335. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Mao, J.; Barrero, R.A.; Wang, P.; Zhang, F.; Wang, T. Development of a CD63 aptamer for efficient cancer immunochemistry and immunoaffinity-based exosome isolation. Molecules 2020, 25, 5585. [Google Scholar] [CrossRef] [PubMed]
- Alomran, N.; Chinnappan, R.; Alsolaiss, J.; Casewell, N.R.; Zourob, M. Exploring the Utility of ssDNA Aptamers Directed against Snake Venom Toxins as New Therapeutics for Snakebite Envenoming. Toxins 2022, 14, 469. [Google Scholar] [CrossRef]
- Grass, R.N.; Stark, W.J. Gas phase synthesis of fcc-cobalt nanoparticles. J. Mater. Chem. 2006, 16, 1825–1830. [Google Scholar] [CrossRef]
- Al Faraj, A.; Shaik, A.S.; Al Sayed, B. Preferential magnetic targeting of carbon nanotubes to cancer sites: Noninvasive tracking using MRI in a murine breast cancer model. Nanomedicine 2015, 10, 931–948. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Monguió-Tortajada, M.; Carreras-Planella, L.; Franquesa, M.; Beyer, K.; Borràs, F.E. Size-Exclusion Chromatography-based isolation minimally alters Extracellular Vesicles’ characteristics compared to precipitating agents. Sci. Rep. 2016, 6, 33641. [Google Scholar] [CrossRef]
- Ramadan, Q.; Christophe, L.; Teo, W.; Li, S.J.; Hua, F.H. Flow-through immunomagnetic separation system for waterborne pathogen isolation and detection: Application to Giardia and Cryptosporidium cell isolation. Anal. Chim. Acta 2010, 673, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.E.; Sin, S.H.; Ozgur, S.; Henry, D.H.; Menezes, P.; Griffith, J.; Eron, J.J.; Damania, B.; Dittmer, D.P. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog. 2013, 9, e1003484. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Ferracin, M.; Trerè, D.; Milazzo, M.; Marinelli, S.; Galassi, M.; Venerandi, L.; Pollutri, D.; Patrizi, C.; Borghi, A. Circulating microRNAs, miR-939, miR-595, miR-519d and miR-494, identify cirrhotic patients with HCC. PLoS ONE 2015, 10, e0141448. [Google Scholar] [CrossRef]
- Köberle, V.; Pleli, T.; Schmithals, C.; Augusto Alonso, E.; Haupenthal, J.; Bönig, H.; Peveling-Oberhag, J.; Biondi, R.M.; Zeuzem Kronenberger, S.B. Differential stability of cell-free circulating microRNAs: Implications for their utilization as biomarkers. PLoS ONE 2013, 8, e75184. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Kryukov, F.; Slaby, O.; Dementyeva, E.; Jarkovsky, J.; Nekvindova, J.; Radova, L.; Greslikova, H.; Kuglik, P.; Vetesnikova, E. Circulating serum microRNAs as novel diagnostic and prognostic biomarkers for multiple myeloma and monoclonal gammopathy of undetermined significance. Haematologica 2014, 99, 511. [Google Scholar] [CrossRef]
- Zhang, K.; Yue, Y.; Wu, S.; Liu, W.; Shi, J.; Zhang, Z. Rapid Capture and Nondestructive Release of Extracellular Vesicles Using Aptamer-Based Magnetic Isolation. ACS Sens. 2019, 4, 1245–1251. [Google Scholar] [CrossRef]
- Palmieri, V.; Lucchetti, D.; Gatto, I.; Maiorana, A.; Marcantoni, M.; Maulucci, G.; Papi, M.; Pola, R.; De Spirito, M.; Sgambato, A. Dynamic light scattering for the characterization and counting of extracellular vesicles: A powerful noninvasive tool. J. Nanoparticle Res. 2014, 16, 2583. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Wang, L.; Tian, J.; Sun, X. Nucleic acid detection using carbon nanoparticles as a fluorescent sensing platform. Chem. Commun. 2011, 47, 961–963. [Google Scholar] [CrossRef]
- Chinnappan, R.; Rahamn, A.A.; AlZabn, R.; Kamath, S.; Lopata, A.L.; Abu-Salah, K.M.; Zourob, M. Aptameric biosensor for the sensitive detection of major shrimp allergen, tropomyosin. Food Chem. 2020, 314, 126133. [Google Scholar] [CrossRef]
- Chinnappan, R.; AlZabn, R.; Fataftah, A.K.; Alhoshani, A.; Zourob, M. Probing high-affinity aptamer binding region and development of aptasensor platform for the detection of cylindrospermopsin. Anal. Bioanal. Chem. 2020, 412, 4691–4701. [Google Scholar] [CrossRef]
- Ai, X.; Zhao, H.; Hu, T.; Yan, Y.; He, H.; Ma, C. A signal-on fluorescence-based strategy for detection of microRNA-21 based on graphene oxide and λ exonuclease-based signal amplification. Anal. Methods 2021, 13, 2107–2113. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, C.; Chen, H.; Li, X.; Li, Y.; Fan, X. A simple G-quadruplex molecular beacon-based biosensor for highly selective detection of microRNA. Biosens. Bioelectron. 2017, 87, 552–557. [Google Scholar] [CrossRef]
- Wang, W.; Kong, T.; Zhang, D.; Zhang, J.; Cheng, G. Label-free microRNA detection based on fluorescence quenching of gold nanoparticles with a competitive hybridization. Anal. Chem. 2015, 87, 10822–10829. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yang, F.; Zhang, Y.; Ning, Y.; Yao, Q.; Zhang, G.J. Amplified fluorescence sensing of miRNA by combination of graphene oxide with duplex-specific nuclease. Anal. Methods 2014, 6, 3598–3603. [Google Scholar] [CrossRef]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamer | Oligonucleotides (5′-3′) Used |
|---|---|
| Mir210 | CUGUGCGUGUGACAGCGGCUGA |
| Mir210 cDNA | GACACGCACACTGTCGCCGACT |
| Reverse complement | FAM-TCAGCCGCTGTCACACGCACAG |
| Anti-CEA Aptamer | H2NTCGCGCGAGTCGTCTGGGGAACCATCGAGTTACACCGACCTTCTATGTGCGGCCCCCCGCATCGTCCTCCC |
| Reverse complementary of CEA aptamer | GGGAGGACGATGCGGGGGGCCGCACATAGAAGGTCGGTGTAACTCGATGGTTCCCCAGACGACTCGCGCGA |
| Primers | |
|---|---|
| Reverse transcription primer | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGCC |
| Forward primer | GTATACCTGTGCGTGTGACAG |
| Reverse primer | GTGCAGGGTCCGAGGT |
| Final product | 5′-GTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGCCGCTGTCACACGCACAGGTATAC-3′ 3′-CACGTCCCAGGCTCCATAAGCGTGACCTATGCTGAGTCGGCGACAGTGTGCGTGTCCATATG-5′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinnappan, R.; Ramadan, Q.; Zourob, M. Isolation and Detection of Exosomal Mir210 Using Carbon Nanomaterial-Coated Magnetic Beads. J. Funct. Biomater. 2023, 14, 441. https://doi.org/10.3390/jfb14090441
Chinnappan R, Ramadan Q, Zourob M. Isolation and Detection of Exosomal Mir210 Using Carbon Nanomaterial-Coated Magnetic Beads. Journal of Functional Biomaterials. 2023; 14(9):441. https://doi.org/10.3390/jfb14090441
Chicago/Turabian StyleChinnappan, Raja, Qasem Ramadan, and Mohammed Zourob. 2023. "Isolation and Detection of Exosomal Mir210 Using Carbon Nanomaterial-Coated Magnetic Beads" Journal of Functional Biomaterials 14, no. 9: 441. https://doi.org/10.3390/jfb14090441
APA StyleChinnappan, R., Ramadan, Q., & Zourob, M. (2023). Isolation and Detection of Exosomal Mir210 Using Carbon Nanomaterial-Coated Magnetic Beads. Journal of Functional Biomaterials, 14(9), 441. https://doi.org/10.3390/jfb14090441