
Exploring the Role of Fibrin Gels in Enhancing Cell Migration for Vasculature Formation

 , , ,
, , , 
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Type | Overview | Cells | Matrix/Design | ES | Ref. |
|---|---|---|---|---|---|
| Inserts | Test of topographical and biochemical cues in endothelial barrier | HUVECs | Collagen-coated PET-transwell membrane electrospinning with PLC nanofibers | - | [9] |
| Organoid | Organoid formation for tumor cell Migration studies | Primary EC and A549 lung cancer cells in 3:1 ratio in methylcellulose | Dispersed fibroblasts in fibrin | - | [10] |
| (Bio)scaffolds | Test diffusion limits in a thick vascularized model | Fibrin gel populated with dispersed fibroblasts and beads coated with HUVECs | - | [11] | |
| Test MSC role in angiogenesis | Fibrin gel populated with dispersed MSC and beads coated with HUVECs | - | [12] | ||
| Normal and tumor vasculature formation | HUVECs, smooth muscle cells and fibroblasts, MSC, HepG2 cell line | starPEG-heparin hydrogels | - | [13] | |
| Organ-on-a-chip | PDMS device for vasculature formation studies | Primary EC and fibroblasts | PDMS device coated with fibrin | M | [14] |
| Study of angiogenesis and thrombosis in a microvessel | HUVECs pericytes | PDMS reservoir filled with collagen with dispersed pericytes. | M | [15] | |
| 3D bioprinting | 3D lattice structure resembling vasculature network | hMSC HUVECs | Vasculature ink: Pluronic F-127 and thrombinCell ink: gelatin, fibrinogen, and cells | M | [16] |
2. Materials and Methods
2.1. Part 1: 2D Cell Characterization
2.1.1. Cell Culture
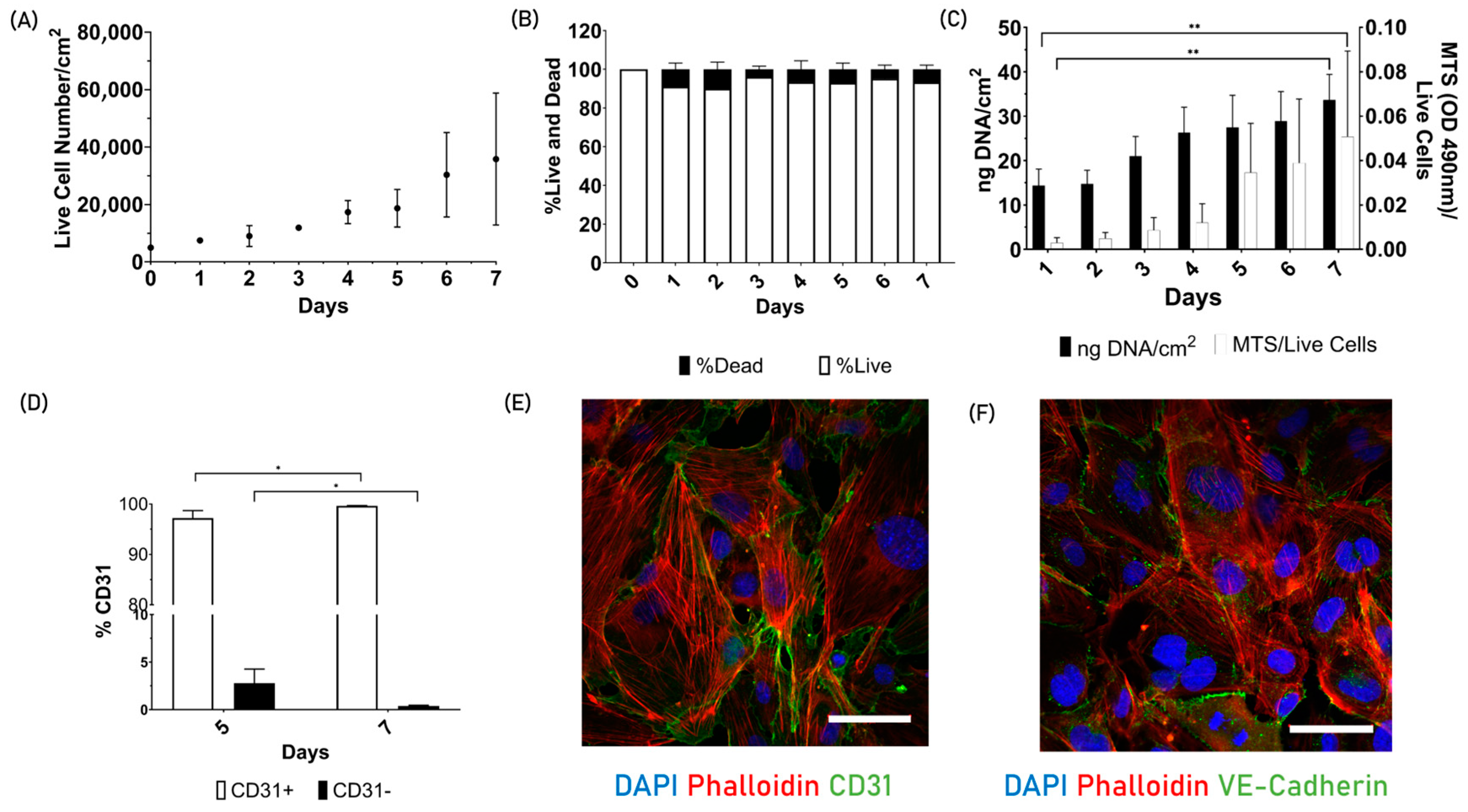
2.1.2. Endothelial Cells 2D Cell Characterization
- Cellular viability
- Metabolic activity
- Cell proliferation
- CD31 surface marker
- Cell morphology
2.2. Part 2: Acellular Fibrin Gel Characterization
2.2.1. Rheologic Characterization
2.2.2. Confocal Microscopy
2.2.3. Scanning Electron Microscopy
2.3. Part 3: Endothelial Cell Migration
2.3.1. Spheroids Formation
2.3.2. Spheroids Fibrin Encapsulation
Hoechst and Propidium Iodine
2.4. Data Presentation and Statistical Analysis
3. Results
3.1. Part 1: 2D Cell Characterization
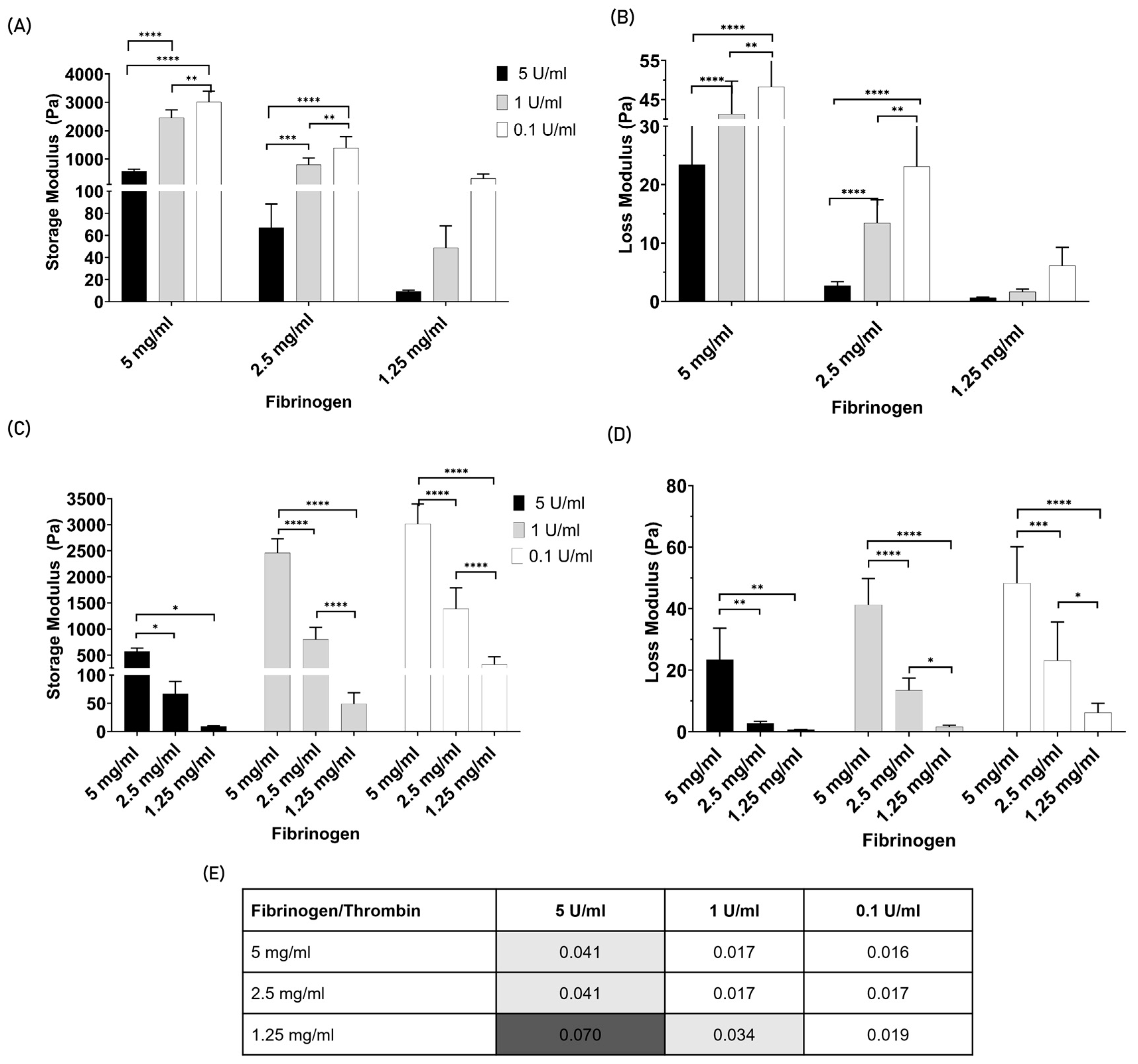
3.2. Part 2: Acellular Fibrin Gel Characterization
3.2.1. Rheologic Characterization
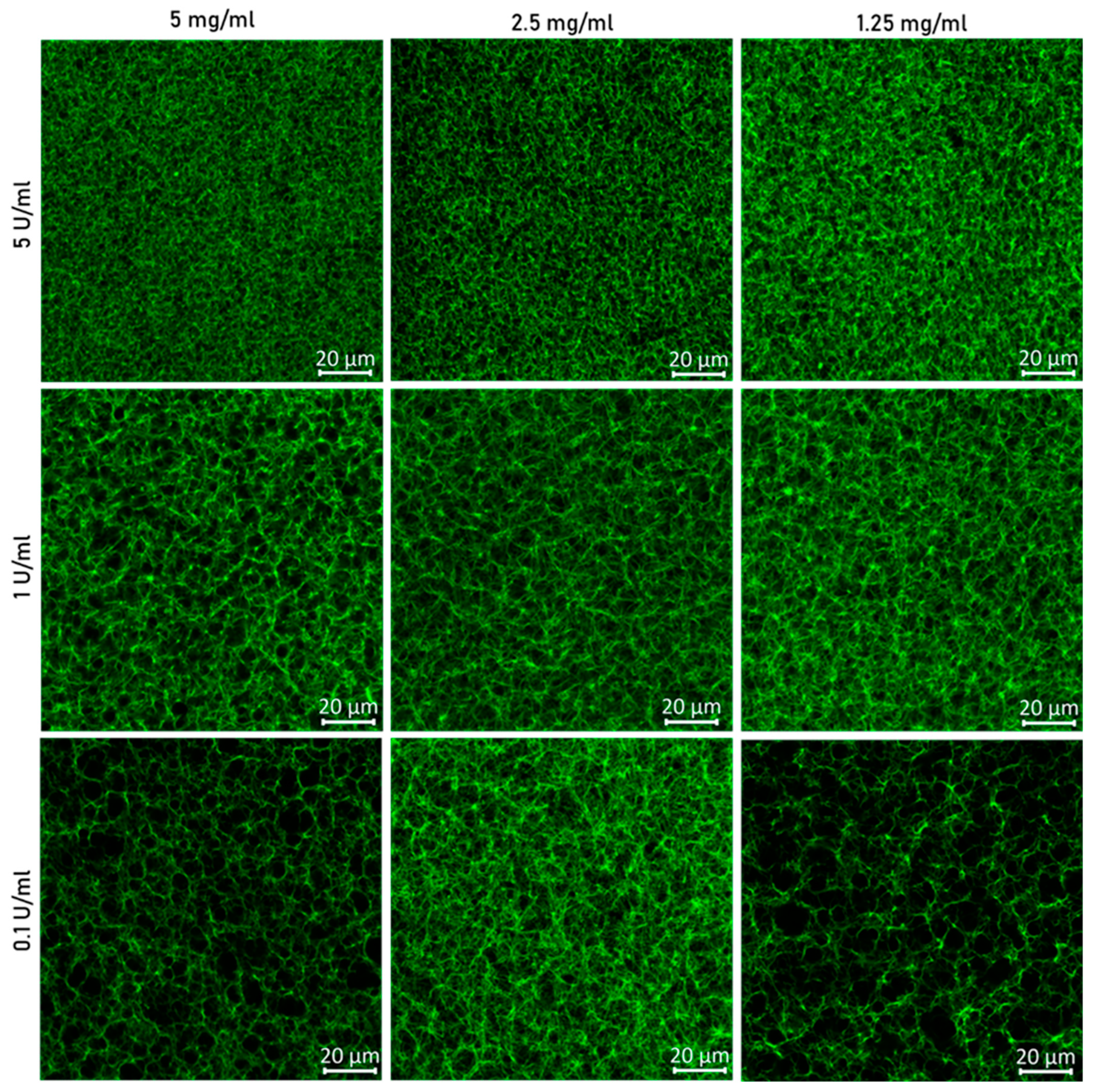
3.2.2. Confocal Microscopy
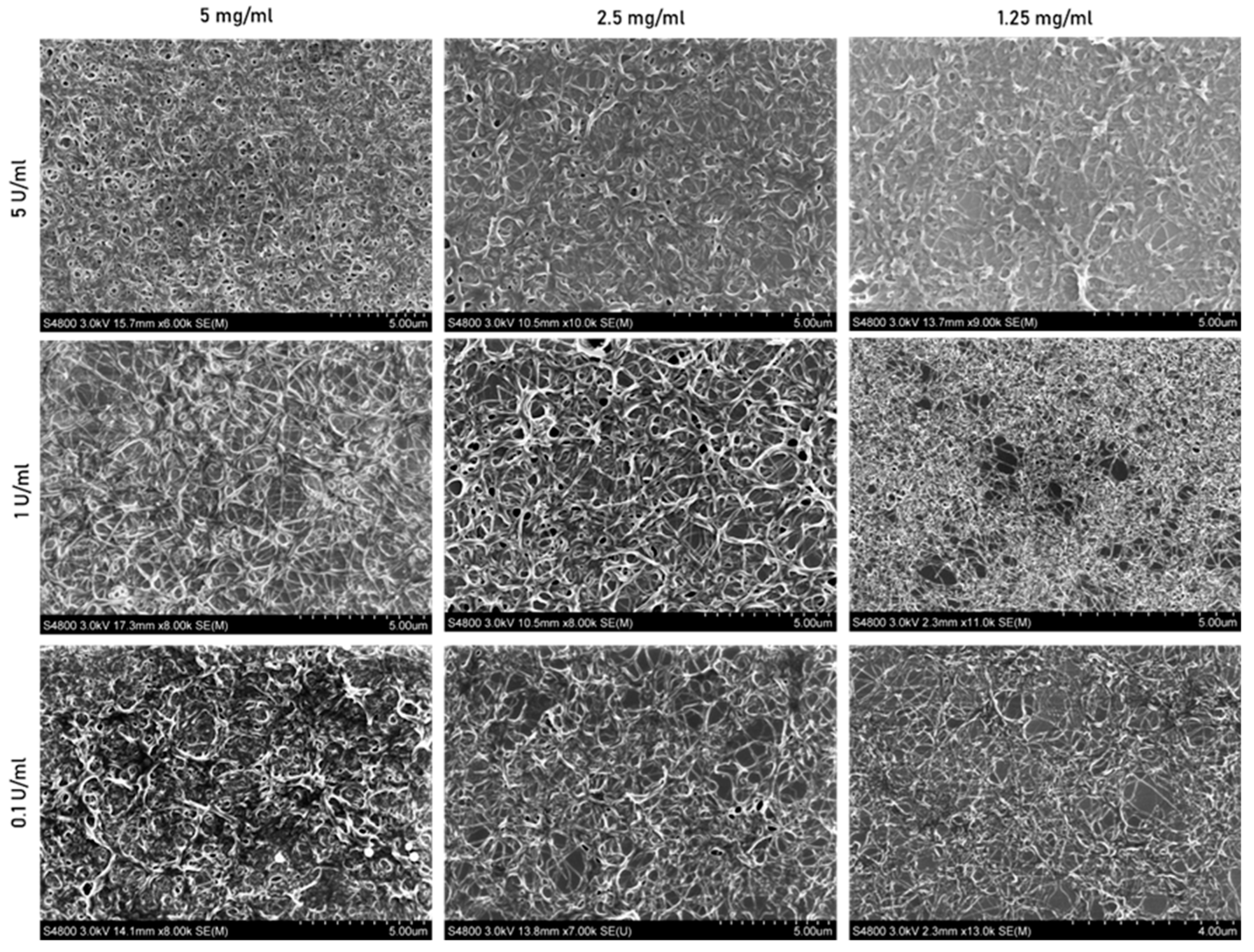
3.2.3. Scanning Electron Microscopy
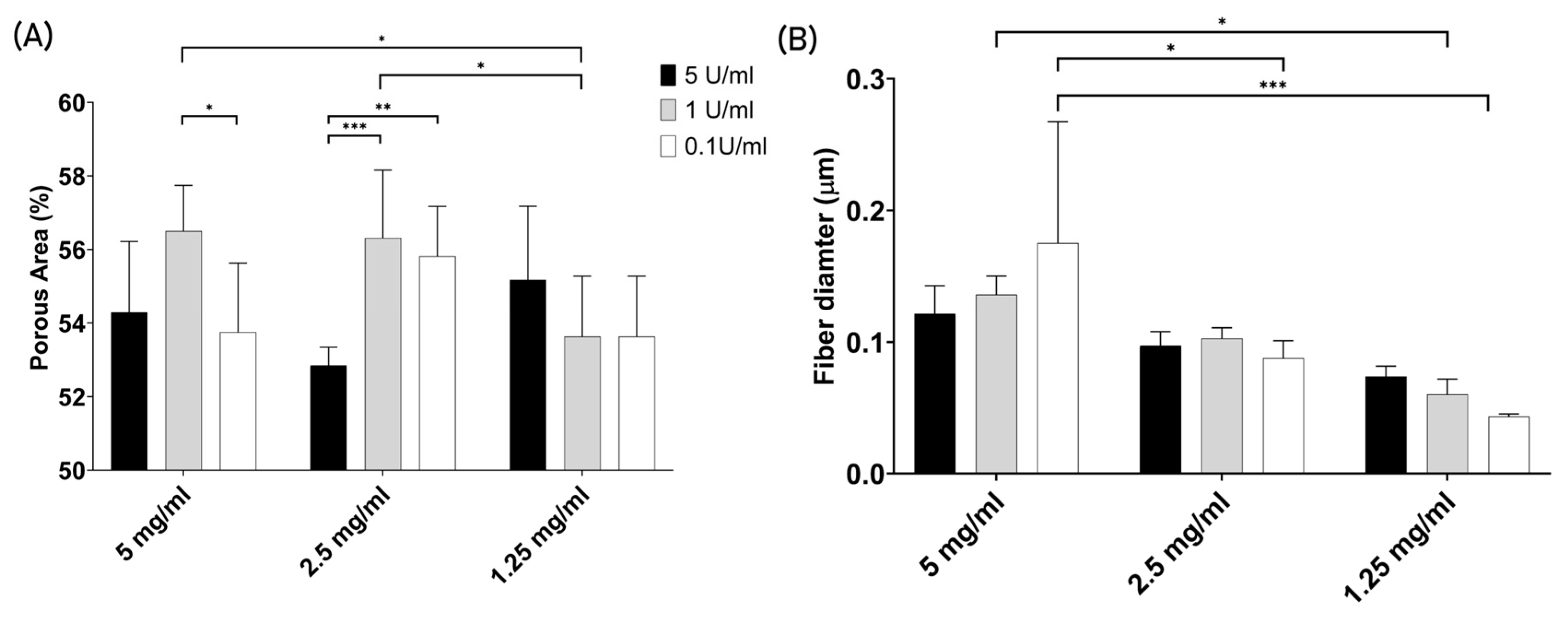
3.2.4. Porosity and Fiber Thickness
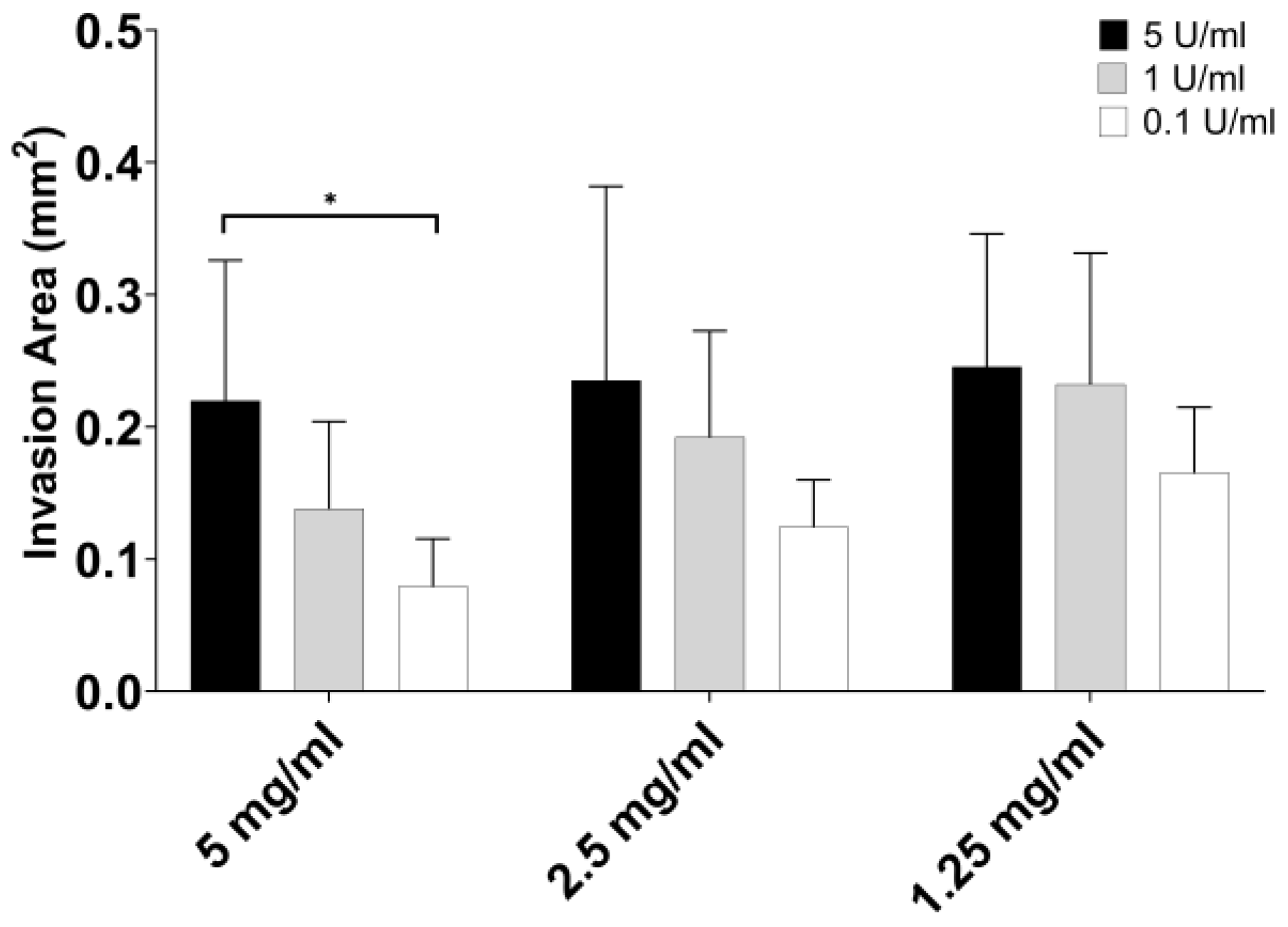
3.3. Part 3: Endothelial Cell Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolte, D.; McClung, J.A.; Aronow, W.S. Vasculogenesis and Angiogenesis. In Translational Research in Coronary Artery Disease; Elsevier: Amsterdam, The Netherlands, 2016; pp. 49–65. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free. Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef]
- Morin, K.T.; Tranquillo, R.T. In vitro models of angiogenesis and vasculogenesis in fibrin gel. Exp. Cell Res. 2013, 319, 2409–2417. [Google Scholar] [CrossRef]
- Bouis, D.; Kusumanto, Y.; Meijer, C.; Mulder, N.; Hospers, G. A review on pro- and anti-angiogenic factors as targets of clinical intervention. Pharmacol. Res. 2006, 53, 89–103. [Google Scholar] [CrossRef]
- Dvorak, H.F. Angiogenesis: Update 2005. J. Thromb. Haemost. 2005, 3, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Sottile, J. Regulation of angiogenesis by extracellular matrix. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2004, 1654, 13–22. [Google Scholar] [CrossRef]
- Stéphanou, A.; Meskaoui, G.; Vailhé, B.; Tracqui, P. The rigidity in fibrin gels as a contributing factor to the dynamics of in vitro vascular cord formation. Microvasc. Res. 2007, 73, 182–190. [Google Scholar] [CrossRef]
- Kim, D.; Eom, S.; Park, S.M.; Hong, H.; Kim, D.S. A collagen gel-coated, aligned nanofiber membrane for enhanced endothelial barrier function. Sci. Rep. 2019, 9, 14915. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, S.M.; Welch-Reardon, K.M.; Waterman, M.L.; Hughes, C.C.W.; George, S.C. A three-dimensional in vitro model of tumor cell intravasation. Integr. Biol. 2014, 6, 603. [Google Scholar] [CrossRef]
- Griffith, C.K.; Miller, C.; Sainson, R.C.; Calvert, J.W.; Jeon, N.L.; Hughes, C.C.; George, S.C. Diffusion Limits of an in Vitro Thick Prevascularized Tissue. Tissue Eng. 2005, 11, 257–266. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Blevins, K.S.; Hughes, C.C.W.; George, S.C.; Putnam, A.J. Mesenchymal Stem Cells Enhance Angiogenesis in Mechanically Viable Prevascularized Tissues via Early Matrix Metalloproteinase Upregulation. Tissue Eng. 2006, 12, 2875–2888. [Google Scholar] [CrossRef] [PubMed]
- Chwalek, K.; Tsurkan, M.V.; Freudenberg, U.; Werner, C. Glycosaminoglycan-based hydrogels to modulate heterocellular communication in in vitro angiogenesis models. Sci. Rep. 2014, 4, 4414. [Google Scholar] [CrossRef] [PubMed]
- Moya, M.L.; Hsu, Y.-H.; Lee, A.P.; Hughes, C.C.W.; George, S.C. In Vitro Perfused Human Capillary Networks. Tissue Eng. Part. C Methods 2013, 19, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, J.; Craven, M.; Choi, N.W.; Totorica, S.; Diaz-Santana, A.; Kermani, P.; Hempstead, B.; Fischbach-Teschl, C.; López, J.A.; et al. In vitro microvessels for the study of angiogenesis and thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9342–9347. [Google Scholar] [CrossRef] [PubMed]
- Kolesky, D.B.; Homan, K.A.; Skylar-Scott, M.A.; Lewis, J.A. Three-dimensional bioprinting of thick vascularized tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 3179–3184. [Google Scholar] [CrossRef]
- Levy, J.H.; Welsby, I.; Goodnough, L.T. Fibrinogen as a therapeutic target for bleeding: A review of critical levels and replacement therapy. Transfusion 2014, 54, 1389–1405. [Google Scholar] [CrossRef]
- Mann, K.G.; Brummel, K.; Butenas, S. What is all that thrombin for? J. Thromb. Haemost. 2003, 1, 1504–1514. [Google Scholar] [CrossRef]
- Vinci, M.; Box, C.; Eccles, S.A. Three-Dimensional (3D) Tumor Spheroid Invasion Assay. J. Vis. Exp. 2015, 99, e52686. [Google Scholar] [CrossRef]
- Parigoris, E.; Lee, S.; Mertz, D.; Turner, M.; Liu, A.Y.; Sentosa, J.; Djomehri, S.; Chang, H.C.; Luker, K.; Luker, G.; et al. Cancer Cell Invasion of Mammary Organoids with Basal-In Phenotype. Adv. Healthc. Mater. 2021, 10, 2000810. [Google Scholar] [CrossRef]
- Kam, Y.; Guess, C.; Estrada, L.; Weidow, B.; Quaranta, V. A novel circular invasion assay mimics in vivo invasive behavior of cancer cell lines and distinguishes single-cell motility in vitro. BMC Cancer 2008, 8, 198. [Google Scholar] [CrossRef]
- Barosova, H.; Meldrum, K.; Karakocak, B.B.; Balog, S.; Doak, S.H.; Petri-Fink, A.; Clift, M.J.D.; Rothen-Rutishauser, B. Inter-laboratory variability of A549 epithelial cells grown under submerged and air-liquid interface conditions. Toxicol. Vitr. 2021, 75, 105178. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.A.; Mockros, L.F.; Weisel, J.W.; Lorand, L. Structural origins of fibrin clot rheology. Biophys. J. 1999, 77, 2813–2826. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Nagaswami, C. Computer modeling of fibrin polymerization kinetics correlated with electron microscope and turbidity observations: Clot structure and assembly are kinetically controlled. Biophys. J. 1992, 63, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W. The mechanical properties of fibrin for basic scientists and clinicians. Biophys. Chem. 2004, 112, 267–276. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moura, J.A.; Barlow, H.J.; Doak, S.H.; Hawkins, K.; Muller, I.; Clift, M.J.D. Exploring the Role of Fibrin Gels in Enhancing Cell Migration for Vasculature Formation. J. Funct. Biomater. 2024, 15, 265. https://doi.org/10.3390/jfb15090265
Moura JA, Barlow HJ, Doak SH, Hawkins K, Muller I, Clift MJD. Exploring the Role of Fibrin Gels in Enhancing Cell Migration for Vasculature Formation. Journal of Functional Biomaterials. 2024; 15(9):265. https://doi.org/10.3390/jfb15090265
Chicago/Turabian StyleMoura, Joana A., Hugh J. Barlow, Shareen H. Doak, Karl Hawkins, Iris Muller, and Martin J. D. Clift. 2024. "Exploring the Role of Fibrin Gels in Enhancing Cell Migration for Vasculature Formation" Journal of Functional Biomaterials 15, no. 9: 265. https://doi.org/10.3390/jfb15090265