

Sorafenib—Drug Delivery Strategies in Primary Liver Cancer
 , , and
, , and
Abstract
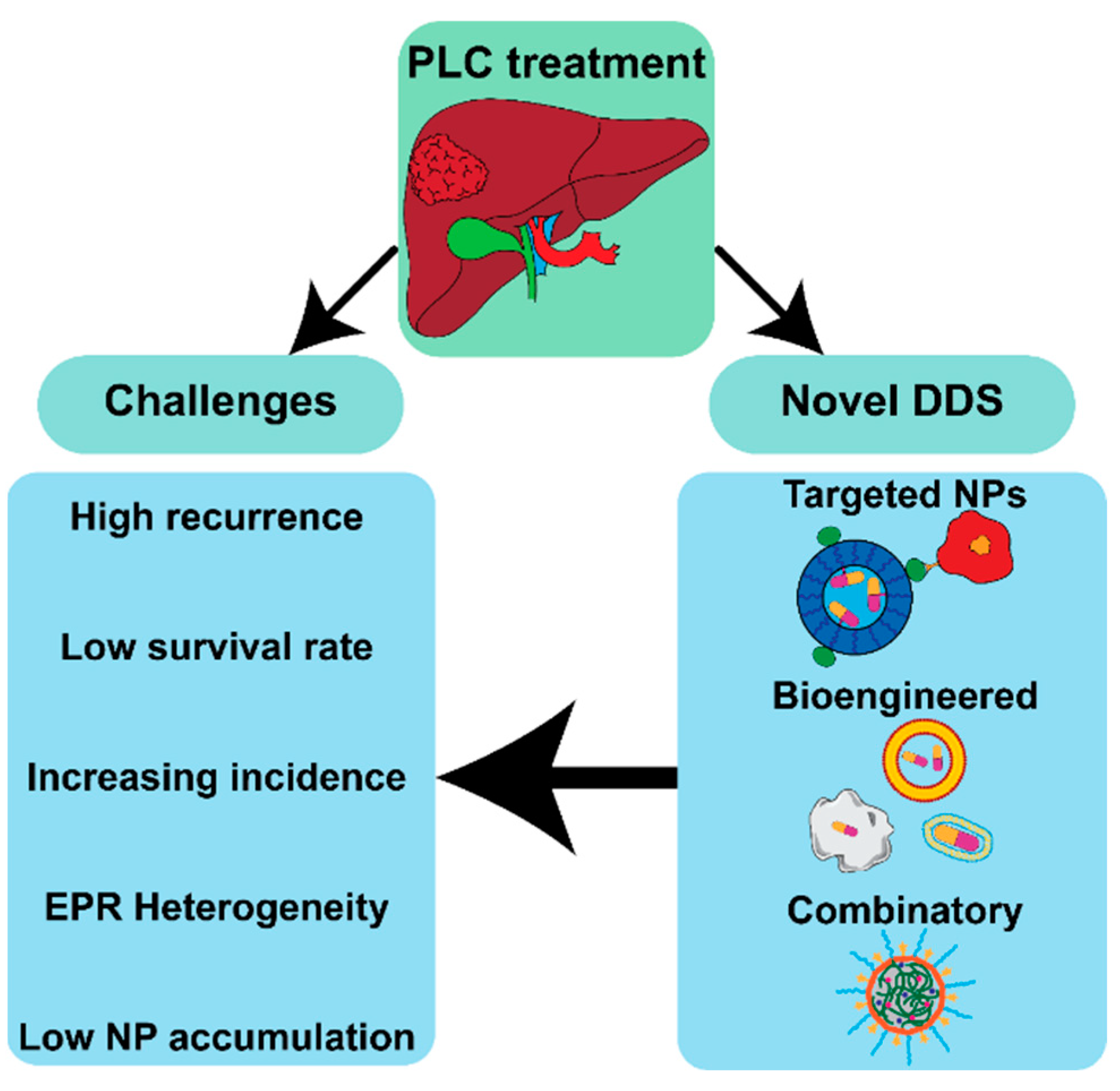
:1. Introduction
2. Liver Cancer Treatment
2.1. Current Approach
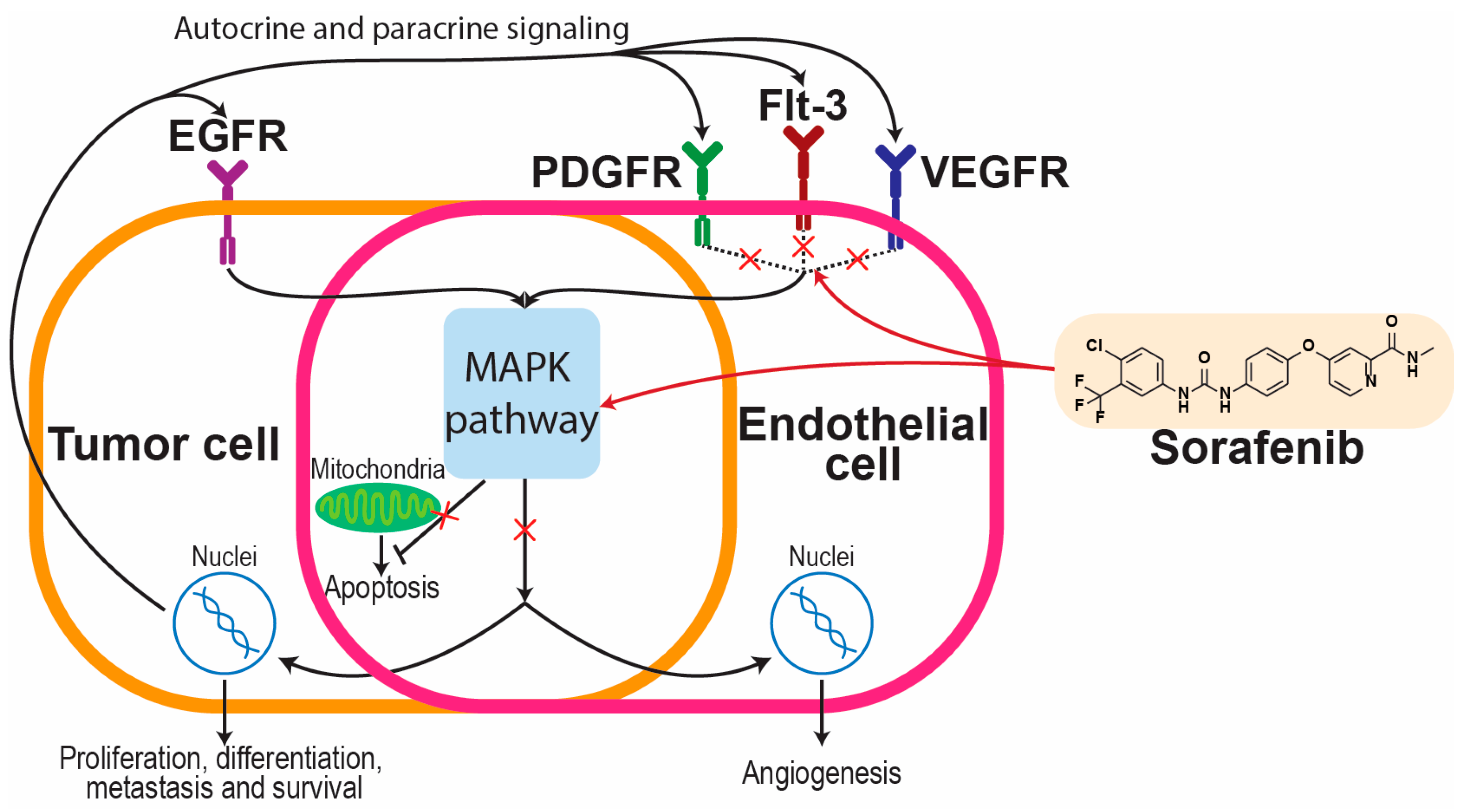
2.2. Sorafenib in Liver Cancers
3. Primary Liver Cancer Targeting
3.1. Passive Accumulation
3.2. Receptor-Targeted Accumulation
3.2.1. Glypican-3
3.2.2. CXCR4
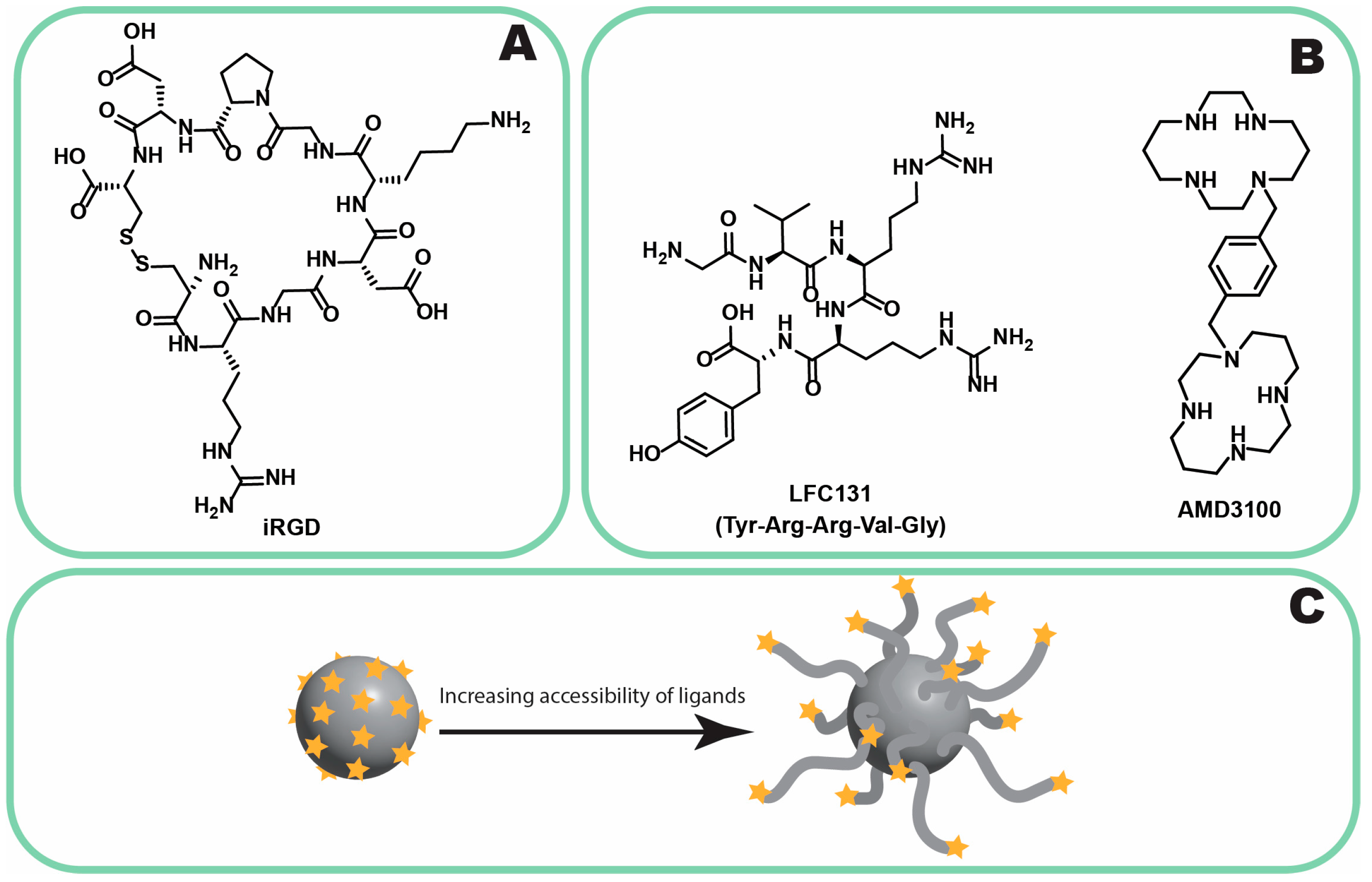
3.2.3. Neuropilin-1
3.2.4. GRP78
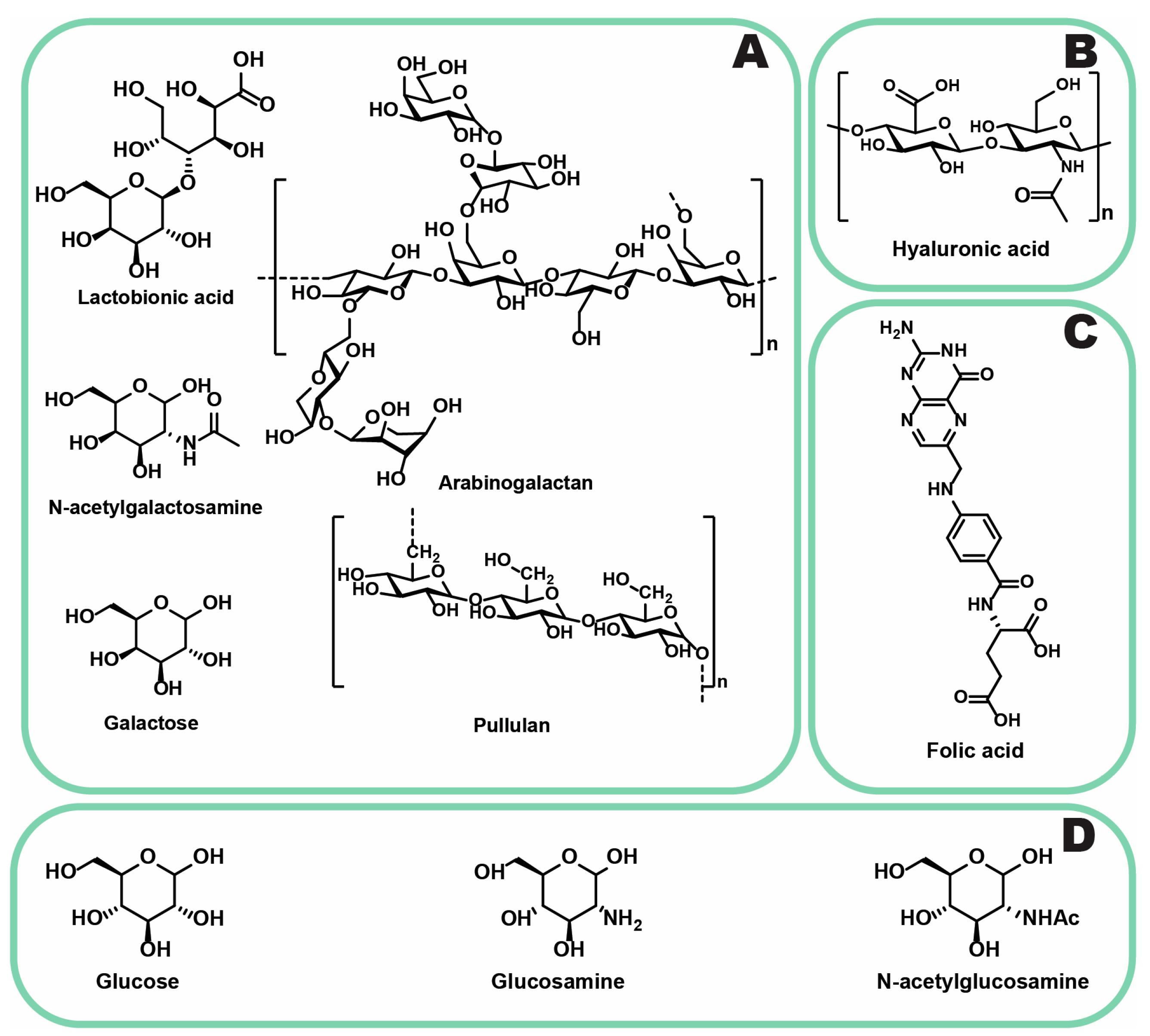
3.2.5. ASGPR
3.2.6. GLUT-1
3.2.7. LDL Receptor
3.2.8. CD44
3.2.9. Folate Receptor
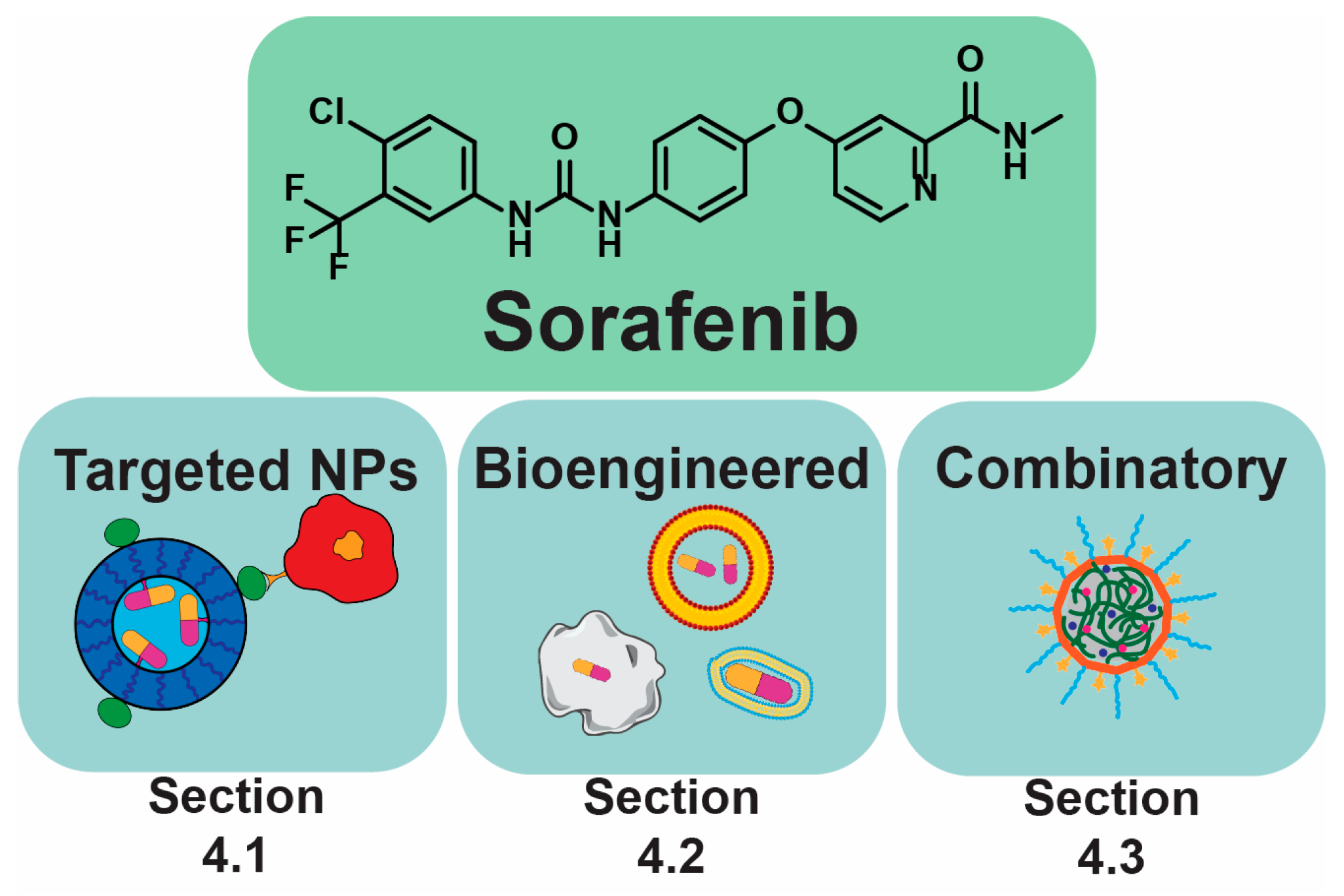
4. Sorafenib and Its Derivatives in PLC
4.1. Targeting
4.1.1. Antibodies
4.1.2. ASGPR
4.1.3. Apolipoproteins
4.1.4. GLUT and CD44
4.1.5. Folate Receptor
4.1.6. Vitamin E
4.1.7. CXCR4
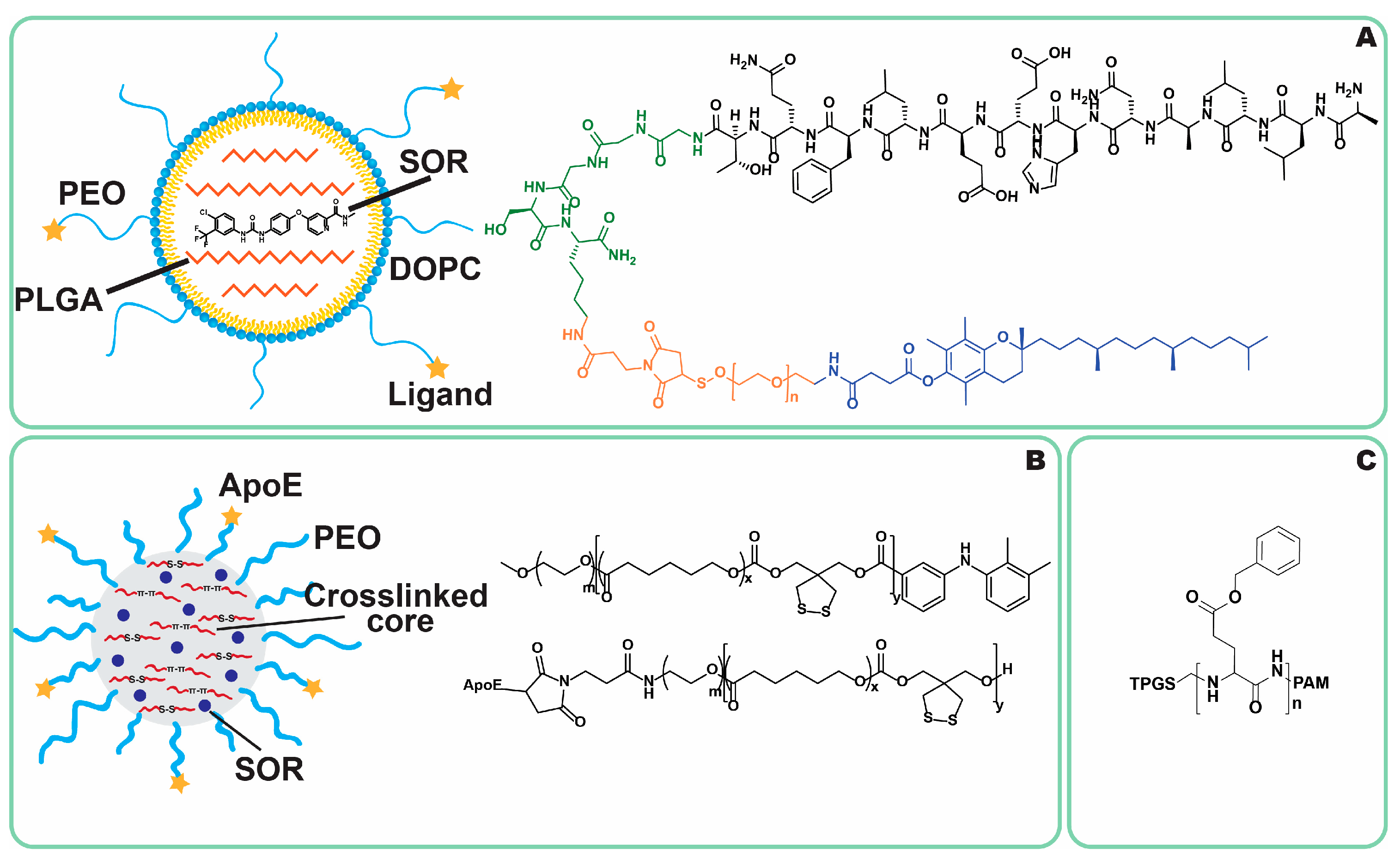
4.2. Bioengineering Approaches
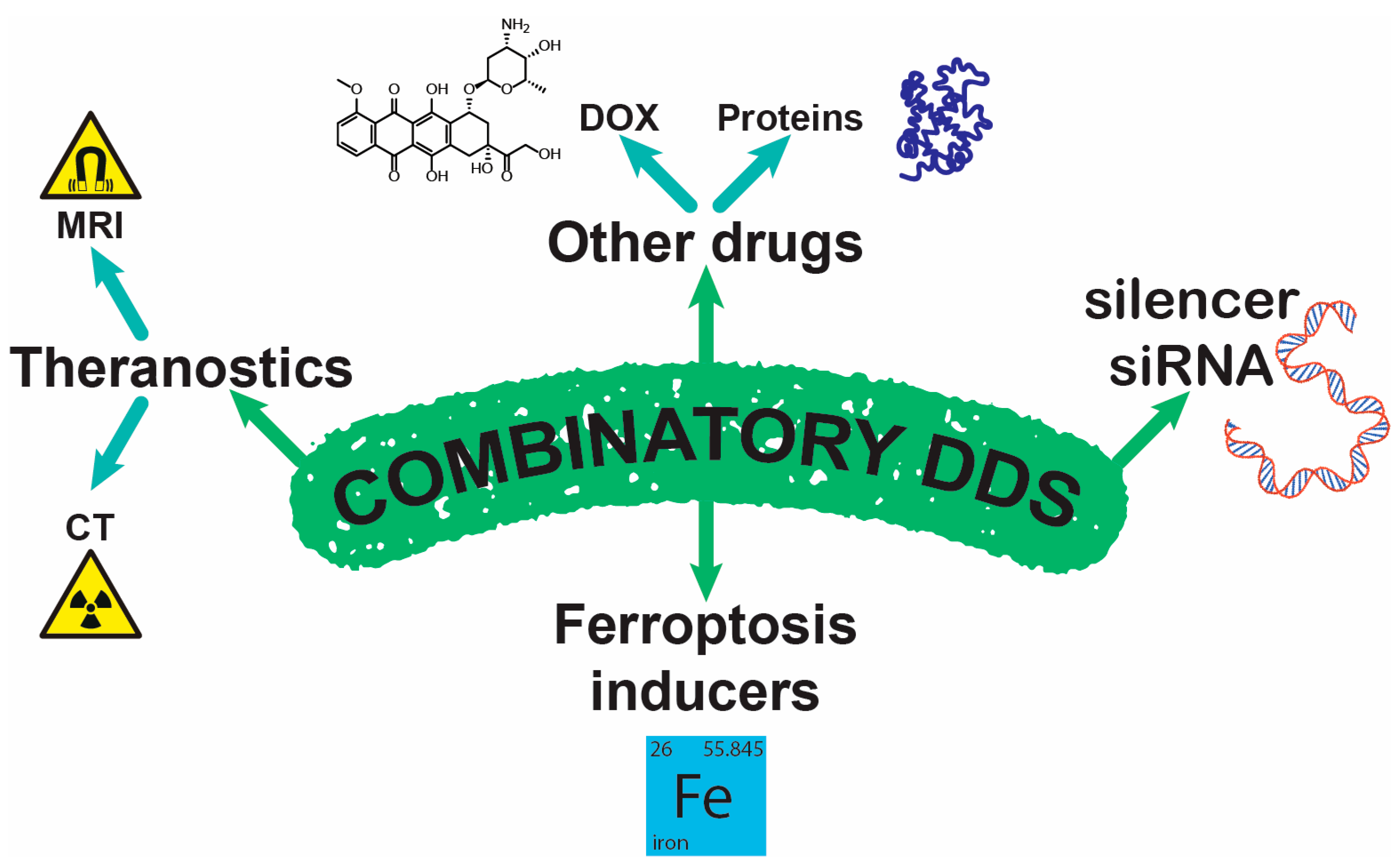
4.3. Combinatory NPs
4.3.1. Theranostics
4.3.2. Iron-Containing
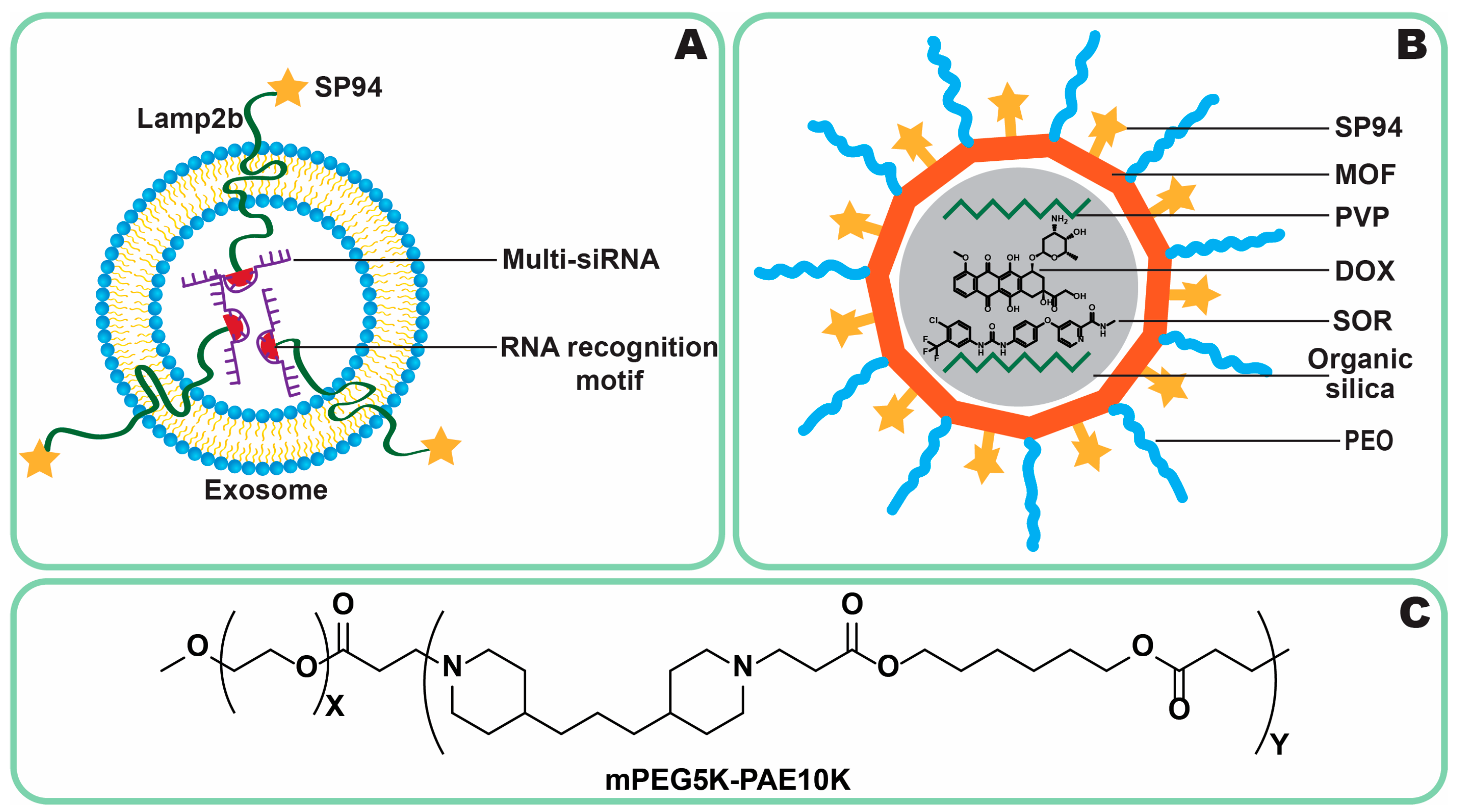
4.3.3. Co-Delivery
4.3.4. siRNA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Apos | Apolipoproteins |
| ASGPR1 | Asialoglycoprotein receptor 1 |
| BBB | Blood–brain barrier |
| BSA | Bovine serum albumin |
| CD44 | A cell-surface glycoprotein involved in cell–cell interactions |
| CT | Computer tomography |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| DDS | Drug delivery system |
| FR | Folate receptor |
| GLUT-1 | Glucose transporter 1 |
| GPC-3 | Glypican-3 |
| GRP78 | 78 kDa glucose-regulated protein, heat shock protein |
| HA | Hyaluronic acid |
| HCC | Hepatocellular carcinoma |
| iRGD | Cyclic peptide, ligand of neuropilin-1 |
| LDLR | Low-density lipoprotein receptor |
| MRI | Magnetic resonance imaging |
| NP | Nanoparticle |
| NP-1 | Neuropilin-1 |
| PCL | Polycaprolactone |
| PEO | Poly(ethylene oxide) |
| PLC | Primary liver cancer |
| PLGA | Poly(lactic-co-glycolic) acid |
| PVA | Poly(vinyl alcohol) |
| RGD | Arginine–glycine–aspartic |
| ROS | Reactive oxygen species |
| SI | Selectivity index |
| SOR | Sorafenib |
| SP94 | Peptide targeting GRP78 |
| TACE | Transarterial chemoembolization |
| TAE | Transarterial embolization |
| TPGS | Tocopherol polyethylene glycol succinate |
References
- Ananthakrishnan, A.; Gogineni, V.; Saeian, K. Epidemiology of Primary and Secondary Liver Cancers. Semin Intervent. Radiol. 2006, 23, 047–063. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Ribes, J.; Borràs, J. Epidemiology of Primary Liver Cancer. Semin. Liver. Dis. 1999, 19, 271–285. [Google Scholar] [CrossRef]
- Ferrante, N.D.; Pillai, A.; Singal, A.G. Update on the Diagnosis and Treatment of Hepatocellular Carcinoma. Gastroenterol. Hepatol. 2020, 16, 506–516. [Google Scholar]
- Wu, L.-J.; Pan, Y.-D.; Pei, X.-Y.; Chen, H.; Nguyen, S.; Kashyap, A.; Liu, J.; Wu, J. Capturing Circulating Tumor Cells of Hepatocellular Carcinoma. Cancer Lett. 2012, 326, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Da, X.; Mo, J.; Li, Q.; Cao, B.; Huang, J.; Lu, Y.; Lu, L.; Fan, M.; Lu, H. Targeted Co-Delivery of PD-L1 Monoclonal Antibody and Sorafenib to Circulating Tumor Cells via Platelet-Functionalized Nanocarriers. Biochem. Biophys. Res. Commun. 2023, 671, 335–342. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Sig. Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Ferri, L.E. Neutrophils Promote Liver Metastasis via Mac-1–Mediated Interactions with Circulating Tumor Cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef]
- Espejo-Cruz, M.L.; González-Rubio, S.; Zamora-Olaya, J.; Amado-Torres, V.; Alejandre, R.; Sánchez-Frías, M.; Ciria, R.; De La Mata, M.; Rodríguez-Perálvarez, M.; Ferrín, G. Circulating Tumor Cells in Hepatocellular Carcinoma: A Comprehensive Review and Critical Appraisal. Int. J. Mol. Sci. 2021, 22, 13073. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Xu, J.; Zhang, A.; Wang, X.; Tang, R.; Zhang, X.; Yin, H.; Liu, M.; Wang, D.D.; et al. Quantified Postsurgical Small Cell Size CTCs and EpCAM+ Circulating Tumor Stem Cells with Cytogenetic Abnormalities in Hepatocellular Carcinoma Patients Determine Cancer Relapse. Cancer Lett. 2018, 412, 99–107. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC Strategy for Prognosis Prediction and Treatment Recommendation: The 2022 Update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Ming, Y.; Gong, Y.; Fu, X.; Ouyang, X.; Peng, Y.; Pu, W. Small-Molecule-Based Targeted Therapy in Liver Cancer. Mol. Ther. 2024, 32, 3260–3287. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Tian, H.; Cheng, J.; Zou, J.; Luan, F.; Qiao, J.; Zhang, D.; Tian, Y.; Zhai, B.; Guo, D. Research Progress of Sorafenib Drug Delivery System in the Treatment of Hepatocellular Carcinoma: An Update. Biomed. Pharmacother. 2024, 177, 117118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, W.; Wang, Y.; Zhu, J.; Zhou, M.; Peng, C.; He, Z.; Sun, J.; Li, Z.; Gui, S. Emerging Nanotaxanes for Cancer Therapy. Biomaterials 2021, 272, 120790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, J.; Yuan, Z. Strategies and Challenges to Improve the Performance of Tumor-Associated Active Targeting. J. Mater. Chem. B 2020, 8, 3959–3971. [Google Scholar] [CrossRef]
- Chainoglou, E.; Hadjipavlou-Litina, D. Curcumin Analogues and Derivatives with Anti-Proliferative and Anti-Inflammatory Activity: Structural Characteristics and Molecular Targets. Expert Opin. Drug Discov. 2019, 14, 821–842. [Google Scholar] [CrossRef]
- Dziubańska-Kusibab, P.J.; Nevedomskaya, E.; Haendler, B. Preclinical Anticipation of On- and Off-Target Resistance Mechanisms to Anti-Cancer Drugs: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 705. [Google Scholar] [CrossRef]
- Li, Z.; Gao, J.; Zheng, S.; Wang, Y.; Xiang, X.; Cheng, Q.; Zhu, J. Therapeutic Efficacy of Sorafenib in Patients with Hepatocellular Carcinoma Recurrence After Liver Transplantation: A Systematic Review and Meta-Analysis. Turk. J. Gastroenterol. 2021, 32, 30–41. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Ran, X.; Tang, H.; Cao, D. Sorafenib-Based Drug Delivery Systems: Applications and Perspectives. Polymers 2023, 15, 2638. [Google Scholar] [CrossRef]
- Alavi, M.; Hamidi, M. Passive and Active Targeting in Cancer Therapy by Liposomes and Lipid Nanoparticles. Drug Metab. Pers. Ther. 2019, 34, 20180032. [Google Scholar] [CrossRef]
- Shreya, A.B.; Raut, S.Y.; Managuli, R.S.; Udupa, N.; Mutalik, S. Active Targeting of Drugs and Bioactive Molecules via Oral Administration by Ligand-Conjugated Lipidic Nanocarriers: Recent Advances. AAPS PharmSciTech 2019, 20, 15. [Google Scholar] [CrossRef]
- Huang, M.; Zhai, B.-T.; Fan, Y.; Sun, J.; Shi, Y.-J.; Zhang, X.-F.; Zou, J.-B.; Wang, J.-W.; Guo, D.-Y. Targeted Drug Delivery Systems for Curcumin in Breast Cancer Therapy. Int. J. Nanomed. 2023, 18, 4275–4311. [Google Scholar] [CrossRef]
- Ma, Z.; Fan, Y.; Wu, Y.; Kebebe, D.; Zhang, B.; Lu, P.; Pi, J.; Liu, Z. Traditional Chinese Medicine-Combination Therapies Utilizing Nanotechnology-Based Targeted Delivery Systems: A New Strategy for Antitumor Treatment. Int. J. Nanomed. 2019, 14, 2029–2053. [Google Scholar] [CrossRef] [PubMed]
- Liver Cancer Treatment—NCI. Available online: https://www.cancer.gov/types/liver/what-is-liver-cancer/treatment (accessed on 10 December 2024).
- Knavel, E.M.; Brace, C.L. Tumor Ablation: Common Modalities and General Practices. Tech. Vasc. Interv. Radiol. 2013, 16, 192–200. [Google Scholar] [CrossRef]
- Bannangkoon, K.; Hongsakul, K.; Tubtawee, T. Lipiodol Accumulation Patterns and Their Impact on Survival Outcomes in Transarterial Chemoembolization for Hepatocellular Carcinoma: A Single Institution Retrospective Analysis. Sci. Rep. 2024, 14, 18979. [Google Scholar] [CrossRef] [PubMed]
- Miszczuk, M.A.; Chapiro, J.; Geschwind, J.-F.H.; Thakur, V.; Nezami, N.; Laage-Gaupp, F.; Kulon, M.; Van Breugel, J.M.M.; Fereydooni, A.; Lin, M.; et al. Lipiodol as an Imaging Biomarker of Tumor Response After Conventional Transarterial Chemoembolization: Prospective Clinical Validation in Patients with Primary and Secondary Liver Cancer. Transl. Oncol. 2020, 13, 100742. [Google Scholar] [CrossRef]
- Schwarz, L.; Bubenheim, M.; Gardin, I.; Huet, E.; Riachi, G.; Clavier, E.; Goria, O.; Vera, P.; Scotté, M. Adjuvant I-131 Lipiodol After Resection or Radiofrequency Ablation for Hepatocellular Carcinoma. World J. Surg. 2016, 40, 1941–1950. [Google Scholar] [CrossRef]
- Kim, S.J.; Choi, M.S.; Kang, J.Y.; Choi, D.I.; Park, C.K.; Gwak, G.Y.; Lee, J.H.; Koh, K.C.; Paik, S.W.; Yoo, B.C. Prediction of Complete Necrosis of Hepatocellular Carcinoma Treated with Transarterial Chemoembolization Prior to Liver Transplantation. Gut Liver 2009, 3, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Idée, J.-M.; Guiu, B. Use of Lipiodol as a Drug-Delivery System for Transcatheter Arterial Chemoembolization of Hepatocellular Carcinoma: A Review. Crit. Rev. Oncol. Hematol. 2013, 88, 530–549. [Google Scholar] [CrossRef]
- Shukla, S.K.; Goyal, M.; Kanabar, D.D.; Ayehunie, S.; Deore, B.; Sanhueza, C.A.; Muth, A.; Gupta, V. Exploring the Enhanced Stability and Therapeutic Efficacy of Sorafenib-Cyclodextrin Inclusion Complex. J. Mol. Liq. 2024, 401, 124701. [Google Scholar] [CrossRef]
- Khan, M.A.; Raza, A.; Ovais, M.; Sohail, M.F.; Ali, S. Current State and Prospects of Nano-Delivery Systems for Sorafenib. J. Polym. Mater. Polym. Biomater. 2018, 67, 1105–1115. [Google Scholar] [CrossRef]
- Zhang, Z.; Niu, B.; Chen, J.; He, X.; Bao, X.; Zhu, J.; Yu, H.; Li, Y. The Use of Lipid-Coated Nanodiamond to Improve Bioavailability and Efficacy of Sorafenib in Resisting Metastasis of Gastric Cancer. Biomaterials 2014, 35, 4565–4572. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, Z.; Chen, Y.; Lu, J.; Li, Y.; Wang, S.; Wu, G.; Qian, F. Improving Oral Bioavailability of Sorafenib by Optimizing the “Spring” and “Parachute” Based on Molecular Interaction Mechanisms. Mol. Pharmaceutics 2016, 13, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Wiergowska, G.; Stasiłowicz, A.; Miklaszewski, A.; Lewandowska, K.; Cielecka-Piontek, J. Structural Polymorphism of Sorafenib Tosylate as a Key Factor in Its Solubility Differentiation. Pharmaceutics 2021, 13, 384. [Google Scholar] [CrossRef]
- Kong, F.-H.; Ye, Q.-F.; Miao, X.-Y.; Liu, X.; Huang, S.-Q.; Xiong, L.; Wen, Y.; Zhang, Z.-J. Current Status of Sorafenib Nanoparticle Delivery Systems in the Treatment of Hepatocellular Carcinoma. Theranostics 2021, 11, 5464–5490. [Google Scholar] [CrossRef] [PubMed]
- Izadiyan, Z.; Basri, M.; Fard Masoumi, H.R.; Abedi Karjiban, R.; Salim, N.; Shameli, K. Modeling and Optimization of Nanoemulsion Containing Sorafenib for Cancer Treatment by Response Surface Methodology. Chem. Cent. J. 2017, 11, 21. [Google Scholar] [CrossRef]
- Bruix, J.; Sherman, M. Management of Hepatocellular Carcinoma: An Update Δσ. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef]
- Li, D.; Wan, S.; Li, W.; Cheng, C.; Xu, L.; Gu, P. Sorafenib Exhibits Lower Toxicity and Comparable Efficacy to Sunitinib as a First-Line Treatment for Metastatic Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Medicine 2023, 102, e34983. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal Cell Carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef]
- Ramadori, G.; Cameron, S. Effects of Systemic Chemotherapy on the Liver. Ann. Hepatol. 2010, 9, 133–143. [Google Scholar] [CrossRef]
- White, P.T.; Cohen, M.S. The Discovery and Development of Sorafenib for the Treatment of Thyroid Cancer. Expert Opin. Drug Discov. 2015, 10, 427–439. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, C.; Zhang, A.; Li, Y.; Li, J.; Li, Z.; Yang, X.; Hou, Y. ATF4 Protects against Sorafenib-Induced Cardiotoxicity by Suppressing Ferroptosis. Biomed. Pharmacother. 2022, 153, 113280. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yang, X.-R.; Chung, W.-Y.; Dennison, A.R.; Zhou, J. Targeted Therapy for Hepatocellular Carcinoma. Sig. Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- Strumberg, D.; Richly, H.; Hilger, R.A.; Schleucher, N.; Korfee, S.; Tewes, M.; Faghih, M.; Brendel, E.; Voliotis, D.; Haase, C.G.; et al. Phase I Clinical and Pharmacokinetic Study of the Novel Raf Kinase and Vascular Endothelial Growth Factor Receptor Inhibitor BAY 43-9006 in Patients With Advanced Refractory Solid Tumors. J. Clin. Oncol. 2005, 23, 965–972. [Google Scholar] [CrossRef]
- Zheng, L.; Guo, C.-Y.; Chen, C.-S.; Xiao, J.-C.; Hu, H.-T.; Cheng, H.-T.; Zong, D.-W.; Jiang, L.; Li, H.-L. Sorafenib Improves Lipiodol Deposition in Transarterial Chemoembolization of Chinese Patients with Hepatocellular Carcinoma: A Long-Term, Retrospective Study. Oncotarget 2017, 8, 97613–97622. [Google Scholar] [CrossRef]
- Asmis, T.; Balaa, F.; Scully, L.; Papadatos, D.; Marginean, C.; Fasih, N.; Shaw–Stiffel, T.; Goel, R. Diagnosis and Management of Hepatocellular Carcinoma: Results of a Consensus Meeting of The Ottawa Hospital Cancer Centre. Curr. Oncol. 2010, 17, 6–12. [Google Scholar] [CrossRef]
- Mikhail, A.S.; Negussie, A.H.; Mauda-Havakuk, M.; Owen, J.W.; Pritchard, W.F.; Lewis, A.L.; Wood, B.J. Drug-Eluting Embolic Microspheres: State-of-the-Art and Emerging Clinical Applications. Expert Opin. Drug Deliv. 2021, 18, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Abdelgalil, A.A.; Alkahtani, H.M.; Al-Jenoobi, F.I. Sorafenib. Profiles Drug Subst. Excip. Relat. Methodol. 2019, 44, 239–266. [Google Scholar] [CrossRef]
- Ammar, U.M.; Abdel-Maksoud, M.S.; Oh, C.-H. Recent Advances of RAF (Rapidly Accelerated Fibrosarcoma) Inhibitors as Anti-Cancer Agents. Eur. J. Med. Chem. 2018, 158, 144–166. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Sig. Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Masłowska, K.; Halik, P.K.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The Role of VEGF Receptors as Molecular Target in Nuclear Medicine for Cancer Diagnosis and Combination Therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical Overview of Sorafenib, a Multikinase Inhibitor That Targets Both Raf and VEGF and PDGF Receptor Tyrosine Kinase Signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Hendrixson, M.; Gladkiy, Y.; Thyagarajan, A.; Sahu, R.P. Efficacy of Sorafenib-Based Therapies for Non-Small Cell Lung Cancer. Med. Sci. 2024, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Leifheit, M.E.; Johnson, G.; Kuzel, T.M.; Schneider, J.R.; Barker, E.; Yun, H.D.; Ustun, C.; Goldufsky, J.W.; Gupta, K.; Marzo, A.L. Enhancing Therapeutic Efficacy of FLT3 Inhibitors with Combination Therapy for Treatment of Acute Myeloid Leukemia. Int. J. Mol. Sci. 2024, 25, 9448. [Google Scholar] [CrossRef] [PubMed]
- Massarwa, M.; Benson, A.; Khoury, T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef]
- Gong, L.; Giacomini, M.M.; Giacomini, C.; Maitland, M.L.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Sorafenib Pathways. Pharm. Genom. 2017, 27, 240–246. [Google Scholar] [CrossRef]
- Pang, Y.; Eresen, A.; Zhang, Z.; Hou, Q.; Wang, Y.; Yaghmai, V.; Zhang, Z. Adverse Events of Sorafenib in Hepatocellular Carcinoma Treatment. Am. J. Cancer Res. 2022, 12, 2770–2782. [Google Scholar]
- Field, K.M.; Michael, M. Part II: Liver Function in Oncology: Towards Safer Chemotherapy Use. Lancet Oncol. 2008, 9, 1181–1190. [Google Scholar] [CrossRef]
- Li, J.; Zhang, L.; Ge, T.; Liu, J.; Wang, C.; Yu, Q. Understanding Sorafenib-Induced Cardiovascular Toxicity: Mechanisms and Treatment Implications. Drug Des. Dev. Ther. 2024, Volume 18, 829–843. [Google Scholar] [CrossRef]
- He, X.; Sun, H.; Jiang, Q.; Chai, Y.; Li, X.; Wang, Z.; Zhu, B.; You, S.; Li, B.; Hao, J.; et al. Hsa-miR-4277 Decelerates the Metabolism or Clearance of Sorafenib in HCC Cells and Enhances the Sensitivity of HCC Cells to Sorafenib by Targeting cyp3a4. Front. Oncol. 2021, 11, 735447. [Google Scholar] [CrossRef] [PubMed]
- Bæk Møller, N.; Budolfsen, C.; Grimm, D.; Krüger, M.; Infanger, M.; Wehland, M.; Magnusson, N.E. Drug-Induced Hypertension Caused by Multikinase Inhibitors (Sorafenib, Sunitinib, Lenvatinib and Axitinib) in Renal Cell Carcinoma Treatment. Int. J. Mol. Sci. 2019, 20, 4712. [Google Scholar] [CrossRef]
- Lathia, C.; Lettieri, J.; Cihon, F.; Gallentine, M.; Radtke, M.; Sundaresan, P. Lack of Effect of Ketoconazole-Mediated CYP3A Inhibition on Sorafenib Clinical Pharmacokinetics. Cancer Chemother. Pharmacol. 2006, 57, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, S.; Zhu, Y.; Liang, X.; Meng, H.; Chen, J.; Zhang, D.; Guo, H.; Shi, B. Incidence and Risk of Sorafenib-Induced Hypertension: A Systematic Review and Meta-Analysis. J. Clin. Hypertens. 2014, 16, 177–185. [Google Scholar] [CrossRef]
- Hsiao, W.-D.; Peng, C.-Y.; Chuang, P.-H.; Lai, H.-C.; Cheng, K.-S.; Chou, J.-W.; Chen, Y.-Y.; Yu, C.-J.; Feng, C.-L.; Su, W.-P.; et al. Evaluation of Dose-Efficacy of Sorafenib and Effect of Transarterial Chemoembolization in Hepatocellular Carcinoma Patients: A Retrospective Study. BMC Gastroenterol. 2016, 16, 50. [Google Scholar] [CrossRef]
- Geschwind, J.-F.H.; Gholam, P.M.; Goldenberg, A.; Mantry, P.; Martin, R.C.G.; Piperdi, B.; Zigmont, E.; Imperial, J.; Babajanyan, S.; Foreman, P.K.; et al. Use of Transarterial Chemoembolization (TACE) and Sorafenib in Patients with Unresectable Hepatocellular Carcinoma: US Regional Analysis of the GIDEON Registry. Liver Cancer 2016, 5, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, J.-F.; Kudo, M.; Marrero, J.A.; Venook, A.P.; Chen, X.-P.; Bronowicki, J.-P.; Dagher, L.; Furuse, J.; Ladrón De Guevara, L.; Papandreou, C.; et al. TACE Treatment in Patients with Sorafenib-Treated Unresectable Hepatocellular Carcinoma in Clinical Practice: Final Analysis of GIDEON. Radiology 2016, 279, 630–640. [Google Scholar] [CrossRef]
- Chung, Y.; Han, G.; Yoon, J.; Yang, J.; Wang, J.; Shao, G.; Kim, B.I.; Lee, T.; Chao, Y. Interim Analysis of START: Study in Asia of the Combination of TACE (Transcatheter Arterial Chemoembolization) with Sorafenib in Patients with Hepatocellular Carcinoma Trial. Int. J. Cancer 2013, 132, 2448–2458. [Google Scholar] [CrossRef]
- Yao, X.; Yan, D.; Zeng, H.; Liu, D.; Li, H. Concurrent Sorafenib Therapy Extends the Interval to Subsequent TACE for Patients with Unresectable Hepatocellular Carcinoma. J. Surg. Oncol. 2016, 113, 672–677. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled Drug Delivery Vehicles for Cancer Treatment and Their Performance. Sig. Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef]
- Dang, Y.; Guan, J. Nanoparticle-Based Drug Delivery Systems for Cancer Therapy. Smart Mater. Med. 2020, 1, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Shi, Y. Mechanisms of Targeted Drug Delivery for Liver Cancer: Active, Passive, and Subcellular Strategies. J. Biosci. Med. 2025, 13, 369–384. [Google Scholar] [CrossRef]
- Gavas, S.; Quazi, S.; Karpiński, T.M. Nanoparticles for Cancer Therapy: Current Progress and Challenges. Nanoscale Res. Lett. 2021, 16, 173. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; Achy, S.E.; Hussein, W.; Refaat, T. Passive Targeting of Nanoparticles to Cancer: A Comprehensive Review of the Literature. Mol. Clin. Oncol. 2014, 2, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, B.; Hojjat-Farsangi, M.; Mohammadi, H.; Anvari, E.; Ghalamfarsa, G.; Yousefi, M.; Jadidi-Niaragh, F. Nanoparticles and Targeted Drug Delivery in Cancer Therapy. Immunol. Lett. 2017, 190, 64–83. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, Q.; Zhao, R.; Guo, X.; Liu, C.; Zhang, K.; Zhang, W.; Liu, J.; Yu, J.; Wang, S.; et al. Designer Exosomes for Targeted Delivery of a Novel Therapeutic Cargo to Enhance Sorafenib-Mediated Ferroptosis in Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 898156. [Google Scholar] [CrossRef]
- Fang, Z.; Song, M.; Lai, K.; Cui, M.; Yin, M.; Liu, K. Kiwi-Derived Extracellular Vesicles for Oral Delivery of Sorafenib. Eur. J. Pharm. Sci. 2023, 191, 106604. [Google Scholar] [CrossRef]
- Hu, J.; Hu, J.; Wu, W.; Qin, Y.; Fu, J.; Zhou, J.; Liu, C.; Yin, J. N-Acetyl-Galactosamine Modified Metal-Organic Frameworks to Inhibit the Growth and Pulmonary Metastasis of Liver Cancer Stem Cells through Targeted Chemotherapy and Starvation Therapy. Acta Biomater. 2022, 151, 588–599. [Google Scholar] [CrossRef]
- Li, Y.; Wei, J.; Wei, Y.; Cheng, L.; Guo, B.; Meng, F.; Li, F.; Zhong, Z. Apolipoprotein E Peptide-Guided Disulfide-Cross-Linked Micelles for Targeted Delivery of Sorafenib to Hepatocellular Carcinoma. Biomacromolecules 2020, 21, 716–724. [Google Scholar] [CrossRef]
- Ma, G.; Du, X.; Zhu, J.; Xu, F.; Yu, H.; Li, J. Multi-Functionalized Dendrimers for Targeted Co-Delivery of Sorafenib and Paclitaxel in Liver Cancers. J. Drug Deliv. Sci. Technol. 2021, 63, 102493. [Google Scholar] [CrossRef]
- Shu, M.; Tang, J.; Chen, L.; Zeng, Q.; Li, C.; Xiao, S.; Jiang, Z.; Liu, J. Tumor Microenvironment Triple-Responsive Nanoparticles Enable Enhanced Tumor Penetration and Synergetic Chemo-Photodynamic Therapy. Biomaterials 2021, 268, 120574. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, S.; Wu, Q.; Qu, H.; Shi, X.; Wang, K.; Tang, C.; Yin, C. In Situ Customized Apolipoprotein B48-Enriched Protein Corona Enhances Oral Gene Delivery of Chitosan-Based Nanoparticles. Biomaterials 2024, 311, 122704. [Google Scholar] [CrossRef]
- Digiacomo, L.; Cardarelli, F.; Pozzi, D.; Palchetti, S.; Digman, M.A.; Gratton, E.; Capriotti, A.L.; Mahmoudi, M.; Caracciolo, G. An Apolipoprotein-Enriched Biomolecular Corona Switches the Cellular Uptake Mechanism and Trafficking Pathway of Lipid Nanoparticles. Nanoscale 2017, 9, 17254–17262. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, F.; Yanez Arteta, M.; Lerche, M.; Porcar, L.; Lang, C.; Bragg, R.A.; Elmore, C.S.; Krishnamurthy, V.R.; Russell, R.A.; Darwish, T.; et al. Apolipoprotein E Binding Drives Structural and Compositional Rearrangement of mRNA-Containing Lipid Nanoparticles. ACS Nano 2021, 15, 6709–6722. [Google Scholar] [CrossRef]
- Samuelsson, E.; Shen, H.; Blanco, E.; Ferrari, M.; Wolfram, J. Contribution of Kupffer Cells to Liposome Accumulation in the Liver. Colloids Surf. B Biointerfaces 2017, 158, 356–362. [Google Scholar] [CrossRef]
- Wang, H.; Thorling, C.A.; Liang, X.; Bridle, K.R.; Grice, J.E.; Zhu, Y.; Crawford, D.H.G.; Xu, Z.P.; Liu, X.; Roberts, M.S. Diagnostic Imaging and Therapeutic Application of Nanoparticles Targeting the Liver. J. Mater. Chem. B 2015, 3, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle–Liver Interactions: Cellular Uptake and Hepatobiliary Elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef]
- Park, J.; Choi, Y.; Chang, H.; Um, W.; Ryu, J.H.; Kwon, I.C. Alliance with EPR Effect: Combined Strategies to Improve the EPR Effect in the Tumor Microenvironment. Theranostics 2019, 9, 8073–8090. [Google Scholar] [CrossRef]
- Stylianopoulos, T. EPR-Effect: Utilizing Size-Dependent Nanoparticle Delivery to Solid Tumors. Ther. Deliv. 2013, 4, 421–423. [Google Scholar] [CrossRef]
- Kang, H.; Rho, S.; Stiles, W.R.; Hu, S.; Baek, Y.; Hwang, D.W.; Kashiwagi, S.; Kim, M.S.; Choi, H.S. Size-Dependent EPR Effect of Polymeric Nanoparticles on Tumor Targeting. Adv. Healthc. Mater. 2020, 9, 1901223. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and Fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Danhier, F. To Exploit the Tumor Microenvironment: Since the EPR Effect Fails in the Clinic, What Is the Future of Nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 1–12. [Google Scholar] [CrossRef]
- Ladd, A.D.; Duarte, S.; Sahin, I.; Zarrinpar, A. Mechanisms of Drug Resistance in HCC. Hepatology 2024, 79, 926–940. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Macias, R.I.R.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 24 December 2024).
- Zhang, G.; Liu, J.; Yu, X.; Deng, Y.; Sun, Y.; Liu, T.; Dong, L.; Zhu, C.; Shen, X.; Zhu, J.; et al. Bismuth-Based Mesoporous Nanoball Carrying Sorafenib for Computed Tomography Imaging and Synergetic Chemoradiotherapy of Hepatocellular Carcinoma. Adv. Healthc. Mater. 2020, 9, 2000650. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, S.; Zhang, Y.; Wang, J.; Zhang, S.; Yao, X.; Chen, L.; Gao, Z.; Xie, B. Improved Drug Targeting to Liver Tumor by Sorafenib-Loaded Folate-Decorated Bovine Serum Albumin Nanoparticles. Drug Deliv. 2019, 26, 89–97. [Google Scholar] [CrossRef]
- Qian, J.; Quan, F.; Zhao, F.; Wu, C.; Wang, Z.; Zhou, L. Aconitic Acid Derived Carbon Dots: Conjugated Interaction for the Detection of Folic Acid and Fluorescence Targeted Imaging of Folate Receptor Overexpressed Cancer Cells. Sens. Actuators B Chem. 2018, 262, 444–451. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein Receptor Mediated Hepatocyte Targeting — Strategies and Applications. J. Control. Release 2015, 203, 126–139. [Google Scholar] [CrossRef]
- Yamauchi, N.; Watanabe, A.; Hishinuma, M.; Ohashi, K.-I.; Midorikawa, Y.; Morishita, Y.; Niki, T.; Shibahara, J.; Mori, M.; Makuuchi, M.; et al. The Glypican 3 Oncofetal Protein Is a Promising Diagnostic Marker for Hepatocellular Carcinoma. Mod. Pathol. 2005, 18, 1591–1598. [Google Scholar] [CrossRef]
- Expression of GPC3 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000147257-GPC3/cancer (accessed on 25 November 2024).
- Zhang, X.; Chen, J.; Yin, Y.; Xiao, S.; Zhang, R.; Guo, H.; Yang, T.; Zhou, T.; Zhang, S.; Yang, Y.; et al. Design and Characterization of Glypican-3 Targeted Liposomes with Cantharidin Encapsulation for Hepatocellular Carcinoma Treatment. J. Drug Deliv. Sci. Technol. 2024, 99, 105934. [Google Scholar] [CrossRef]
- Lee, Y.L.; Ahn, B.-C.; Lee, Y.; Lee, S.-W.; Cho, J.-Y.; Lee, J. Targeting of Hepatocellular Carcinoma with Glypican-3-Targeting Peptide Ligand. J. Pept. Sci. 2011, 17, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zhou, J.; Li, Z.; Appelman, H.D.; Zhao, L.; Zhu, J.; Wang, T.D. Sorafenib Encapsulated in Nanocarrier Functionalized with Glypican-3 Specific Peptide for Targeted Therapy of Hepatocellular Carcinoma. Colloids Surf. B Biointerfaces 2019, 184, 110498. [Google Scholar] [CrossRef] [PubMed]
- Grega, S.D.; Zheng, D.X.; Zheng, Q.-H. Imaging Ligands Targeting Glypican-3 Receptor Expression in Hepatocellular Carcinoma. Am. J. Nucl. Med. Mol. Imaging 2022, 12, 113. [Google Scholar] [CrossRef]
- Tissue Expression of CXCR4—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000121966-CXCR4/tissue (accessed on 25 November 2024).
- Liu, J.-Y.; Chiang, T.; Liu, C.-H.; Chern, G.-G.; Lin, T.-T.; Gao, D.-Y.; Chen, Y. Delivery of siRNA Using CXCR4-Targeted Nanoparticles Modulates Tumor Microenvironment and Achieves a Potent Antitumor Response in Liver Cancer. Mol. Ther. 2015, 23, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.-Y.; Lin, T.-T.; Sung, Y.-C.; Liu, Y.C.; Chiang, W.-H.; Chang, C.-C.; Liu, J.-Y.; Chen, Y. CXCR4-Targeted Lipid-Coated PLGA Nanoparticles Deliver Sorafenib and Overcome Acquired Drug Resistance in Liver Cancer. Biomaterials 2015, 67, 194–203. [Google Scholar] [CrossRef]
- Zheng, N.; Liu, W.; Li, B.; Nie, H.; Liu, J.; Cheng, Y.; Wang, J.; Dong, H.; Jia, L. Co-Delivery of Sorafenib and Metapristone Encapsulated by CXCR4-Targeted PLGA-PEG Nanoparticles Overcomes Hepatocellular Carcinoma Resistance to Sorafenib. J. Exp. Clin. Cancer Res. 2019, 38, 232. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, W.; Dai, G.; Huang, Y. Cordycepin Suppresses the Migration and Invasion of Human Liver Cancer Cells by Downregulating the Expression of CXCR4. Int. J. Mol. Med. 2020, 45, 141–150. [Google Scholar] [CrossRef]
- Miao, H.Q.; Lee, P.; Lin, H.; Soker, S.; Klagsbrun, M. Neuropilin-1 Expression by Tumor Cells Promotes Tumor Angiogenesis and Progression. FASEB J. 2000, 14, 2532–2539. [Google Scholar] [CrossRef]
- Nikitovic, D.; Kukovyakina, E.; Berdiaki, A.; Tzanakakis, A.; Luss, A.; Vlaskina, E.; Yagolovich, A.; Tsatsakis, A.; Kuskov, A. Enhancing Tumor Targeted Therapy: The Role of iRGD Peptide in Advanced Drug Delivery Systems. Cancers 2024, 16, 3768. [Google Scholar] [CrossRef]
- Expression of NRP1 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000099250-NRP1/cancer (accessed on 20 November 2024).
- Liu, X.; Zhu, X.; Qi, X.; Meng, X.; Xu, K. Co-Administration of iRGD with Sorafenib-Loaded Iron-Based Metal-Organic Framework as a Targeted Ferroptosis Agent for Liver Cancer Therapy. Int. J. Nanomed. 2021, Volume 16, 1037–1050. [Google Scholar] [CrossRef]
- Mao, X.; Liu, J.; Gong, Z.; Zhang, H.; Lu, Y.; Zou, H.; Yu, Y.; Chen, Y.; Sun, Z.; Li, W.; et al. iRGD-Conjugated DSPE-PEG2000 Nanomicelles for Targeted Delivery of Salinomycin for Treatment of Both Liver Cancer Cells Cancer Stem Cells. Nanomedicine 2015, 10, 2677–2695. [Google Scholar] [CrossRef]
- Schmithals, C.; Köberle, V.; Korkusuz, H.; Pleli, T.; Kakoschky, B.; Augusto, E.A.; Ibrahim, A.A.; Arencibia, J.M.; Vafaizadeh, V.; Groner, B.; et al. Improving Drug Penetrability with iRGD Leverages the Therapeutic Response to Sorafenib and Doxorubicin in Hepatocellular Carcinoma. Cancer Res. 2015, 75, 3147–3154. [Google Scholar] [CrossRef]
- Qilu Pharmaceutical Co., Ltd. Phase 1b/2 Clinical Study on Safety, Pharmacokinetics, and Preliminary Efficacy of CEND-1 for Injection in Patients with Advanced Metastatic Pancreatic Ductal Adenocarcinoma; Qilu Pharmaceutical Co., Ltd.: Jinan, China, 2024.
- Qilu Pharmaceutical Co., Ltd. A Phase II, Randomized, Double-Blind, Multi-Center, Placebo-Controlled Study of the Efficacy and Safety of CEND-1 in Combination with Chemotherapy as First-Line Therapy in Patients with Locally Advanced Unresectable or Metastatic Pancreatic Ductal Adenocarcinoma; Qilu Pharmaceutical Co., Ltd.: Jinan, China, 2024.
- AustralasianGastro-Intestinal Trials Group. Australasian Gastro-Intestinal Trials Group A Randomised, Double-Blinded Phase II Study of Gemcitabine and Nab-Paclitaxel with LSTA1 (Certepetide) or Placebo in Patients with Untreated Metastatic Pancreatic Ductal Adenocarcinoma; AustralasianGastro-Intestinal Trials Group: Sydney, Australia, 2024.
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER Stress Sensor, Glucose Regulatory Protein 78 (GRP78) Regulates Redox Status in Pancreatic Cancer Thereby Maintaining “Stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Mofed, D.; Wahba, M.A.; Salem, T.Z. Genetically Engineered Hepatitis C Virus-like Particles (HCV-LPs) Tagged with SP94 Peptide to Acquire Selectivity to Liver Cancer Cells via Grp78. Curr. Issues Mol. Biol. 2022, 44, 3746–3756. [Google Scholar] [CrossRef]
- Medina, S.H.; Tiruchinapally, G.; Chevliakov, M.V.; Durmaz, Y.Y.; Stender, R.N.; Ensminger, W.D.; Shewach, D.S.; ElSayed, M.E.H. Targeting Hepatic Cancer Cells with PEGylated Dendrimers Displaying N-Acetylgalactosamine and SP94 Peptide Ligands. Adv. Healthc. Mater. 2013, 2, 1337–1350. [Google Scholar] [CrossRef] [PubMed]
- Zierke, M.A.; Rangger, C.; Samadikhah, K.; Panzer, M.; Dichtl, S.; Hörmann, N.; Wilflingseder, D.; Schmid, A.M.; Haubner, R. [68Ga]Ga-NODAGA-TriGalactan, a Low Molecular Weight Tracer for the Non-Invasive Imaging of the Functional Liver Reserve. EJNMMI Radiopharm. Chem. 2024, 9, 41. [Google Scholar] [CrossRef]
- Kim, J.T.; McPherson, A.K.; Rodriguez, A.A.; Chen, L.; Lu, Y.F.; Moskalev, N.; Upadhyay, S.K.; Green, C.; Whetsell, M.; Xiang-Rong, J.; et al. Development of an Optimized Process for the Liver-Targeted Triantennary N-Acetylgalactosamine Ligand. Org. Process Res. Dev. 2024, 28, 188–209. [Google Scholar] [CrossRef]
- Chirayil, T.J.; Kumar, G.S.V. Sorafenib-Entrapped, Self-Assembled Pullulan–Stearic Acid Biopolymer-Derived Drug Delivery System to PLC/PRF/5 Hepatocellular Carcinoma Model. Int. J. Nanomed. 2022, 17, 5099–5116. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, L.; Hou, J.; Xia, X.; Wang, J.; Ning, Q.; Jiang, S. Promising Positive Liver Targeting Delivery System Based on Arabinogalactan-Anchored Polymeric Micelles of Norcantharidin. Artif. Cells Nanomed. Biotechnol. 2018, 46, S630–S640. [Google Scholar] [CrossRef]
- Shi, B.; Abrams, M.; Sepp-Lorenzino, L. Expression of Asialoglycoprotein Receptor 1 in Human Hepatocellular Carcinoma. J. Histochem. Cytochem. 2013, 61, 901–909. [Google Scholar] [CrossRef]
- Okcu, O.; Sen, B.; Ozturk, C.; Guvendi, G.F.; Bedir, R. GLUT-1 Expression in Breast Cancer. Turk. J. Pathol. 2022, 38, 114. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Hu, Q.; Gu, J. Expressions of Carbohydrate Response Element Binding Protein and Glucose Transporters in Liver Cancer and Clinical Significance. Pathol. Oncol. Res. 2020, 26, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Lachaal, M.; Rampal, A.L.; Lee, W.; Shi, Y.; Jung, C.Y. GLUT1 Transmembrane Glucose Pathway. Affinity Labeling with a Transportable D-Glucose Diazirine. J. Biol. Chem. 1996, 271, 5225–5230. [Google Scholar] [CrossRef]
- Meng, L.; Liu, F.; Du, C.; Zhu, J.; Xiong, Q.; Li, J.; Sun, W. Glucosamine-Modified Reduction-Responsive Polymeric Micelles for Liver Cancer Therapy. Molecules 2023, 28, 3824. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Paknikar, K.M.; Gajbhiye, V. A Robust pH-Sensitive Unimolecular Dendritic Nanocarrier That Enables Targeted Anti-Cancer Drug Delivery via GLUT Transporters. Colloids Surf. B Biointerfaces 2018, 171, 437–444. [Google Scholar] [CrossRef]
- Amann, T.; Hellerbrand, C. GLUT1 as a Therapeutic Target in Hepatocellular Carcinoma. Expert Opin. Ther. Targets 2009, 13, 1411–1427. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, X.; Lv, Y.; Zhao, Y. Low Density Lipoprotein Receptor (LDLR)-Targeted Lipid Nanoparticles for the Delivery of Sorafenib and Dihydroartemisinin in Liver Cancers. Life Sci. 2019, 239, 117013. [Google Scholar] [CrossRef]
- Tissue Expression of LDLR—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000130164-LDLR/tissue (accessed on 16 November 2024).
- Matsui, M.; Sakurai, F.; Elbashir, S.; Foster, D.; Manoharan, M.; Corey, D.R. Activation of LDL Receptor (LDLR) Expression by Small RNAs Complementary to a Noncoding Transcript That Overlaps the LDLR Promoter. Chem. Biol. 2010, 17, 1344. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, J.; Wang, Y.; Zhou, M.; Li, D.; Zheng, S.; Yin, L.; Luo, C.; Zhang, H.; Zhong, L.; et al. In Situ Apolipoprotein E-Enriched Corona Guides Dihydroartemisinin-Decorating Nanoparticles towards LDLr-Mediated Tumor-Homing Chemotherapy. Asian J. Pharm. Sci. 2020, 15, 482–491. [Google Scholar] [CrossRef]
- Neves, A.R.; Queiroz, J.F.; Reis, S. Brain-Targeted Delivery of Resveratrol Using Solid Lipid Nanoparticles Functionalized with Apolipoprotein E. J. Nanobiotechnol. 2016, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Rozeik, M.S.; Hammam, O.A.; Ali, A.I.; Magdy, M.; Khalil, H.; Anas, A.; el Hassan, A.A.A.; Rahim, A.A.; El-Shabasy, A.I. Evaluation of CD44 and CD133 as Markers of Liver Cancer Stem Cells in Egyptian Patients with HCV-Induced Chronic Liver Diseases versus Hepatocellular Carcinoma. Electron. Physician 2017, 9, 4708. [Google Scholar] [CrossRef] [PubMed]
- Asai, R.; Tsuchiya, H.; Amisaki, M.; Makimoto, K.; Takenaga, A.; Sakabe, T.; Hoi, S.; Koyama, S.; Shiota, G. CD44 Standard Isoform Is Involved in Maintenance of Cancer Stem Cells of a Hepatocellular Carcinoma Cell Line. Cancer Med. 2019, 8, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Bao, S. Chemotherapy and Cancer Stem Cells. Cell Stem Cell 2007, 1, 353–355. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Cell Line—CD44—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000026508-CD44/cell+line (accessed on 2 March 2025).
- Expression of FOLR1 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000110195-FOLR1/cancer (accessed on 19 November 2024).
- Expression of FOLR2 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000165457-FOLR2/cancer (accessed on 19 November 2024).
- Wibowo, A.S.; Singh, M.; Reeder, K.M.; Carter, J.J.; Kovach, A.R.; Meng, W.; Ratnam, M.; Zhang, F.; Dann, C.E. Structures of Human Folate Receptors Reveal Biological Trafficking States and Diversity in Folate and Antifolate Recognition. Proc. Natl. Acad. Sci. USA 2013, 110, 15180–15188. [Google Scholar] [CrossRef]
- Grapp, M.; Wrede, A.; Schweizer, M.; Hüwel, S.; Galla, H.-J.; Snaidero, N.; Simons, M.; Bückers, J.; Low, P.S.; Urlaub, H.; et al. Choroid Plexus Transcytosis and Exosome Shuttling Deliver Folate into Brain Parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, X.; Lei, Y.; Wang, G.; Liu, M. Glypican-3: A Novel and Promising Target for the Treatment of Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 824208. [Google Scholar] [CrossRef]
- Tissue Expression of GPC3—Staining in Placenta—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000147257-GPC3/tissue/placenta (accessed on 25 November 2024).
- Filmus, J.; Capurro, M. Glypican-3 and Alphafetoprotein as Diagnostic Tests for Hepatocellular Carcinoma. CNS Drugs 2004, 8, 207–212. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, H.; Zheng, J.; Liu, Y. Glypican-3: A New Target for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Cancer 2020, 11, 2008–2021. [Google Scholar] [CrossRef]
- Su, D. The Transcatheter Arterial Chemoembolization Combined with Targeted Nanoparticle Delivering Sorafenib System for the Treatment of Microvascular Invasion of Hepatocellular Carcinoma. Bioengineered 2021, 12, 11124–11135. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cai, W.; Ma, Y.; Xu, R.; Huo, Z.; Song, L.; Qiu, X.; Zhang, Y.; Li, A.; Cao, W.; et al. hGC33-Modified and Sorafenib-Loaded Nanoparticles Have a Synergistic Anti-Hepatoma Effect by Inhibiting Wnt Signaling Pathway. Nanoscale Res. Lett. 2020, 15, 220. [Google Scholar] [CrossRef]
- Choi, W.-T.; Yang, Y.; Xu, Y.; An, J. Targeting Chemokine Receptor CXCR4 for Treatment of HIV-1 Infection, Tumor Progression, and Metastasis. Curr. Top. Med. Chem. 2014, 14, 1574–1589. [Google Scholar] [CrossRef]
- Xiang, Z.; Zeng, Z.; Tang, Z.; Fan, J.; Zhuang, P.; Liang, Y.; Tan, Y.; He, J. Chemokine Receptor CXCR4 Expression in Hepatocellular Carcinoma Patients Increases the Risk of Bone Metastases and Poor Survival. BMC Cancer 2009, 9, 176. [Google Scholar] [CrossRef]
- Chen, Y.; Ramjiawan, R.R.; Reiberger, T.; Ng, M.R.; Hato, T.; Huang, Y.; Ochiai, H.; Kitahara, S.; Unan, E.C.; Reddy, T.P.; et al. CXCR4 Inhibition in Tumor Microenvironment Facilitates Anti-Programmed Death Receptor-1 Immunotherapy in Sorafenib-Treated Hepatocellular Carcinoma in Mice. Hepatology 2015, 61, 1591–1602. [Google Scholar] [CrossRef]
- Caspar, B.; Cocchiara, P.; Melet, A.; Van Emelen, K.; Van der Aa, A.; Milligan, G.; Herbeuval, J.-P. CXCR4 as a Novel Target in Immunology: Moving Away from Typical Antagonists. Future Drug Discov. 2022, 4, FDD77. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Lin, C.-T.; Wu, H.-C. Hepatocellular Carcinoma Cell-Specific Peptide Ligand for Targeted Drug Delivery. Mol. Cancer Ther. 2008, 7, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Toita, R.; Murata, M.; Tabata, S.; Abe, K.; Narahara, S.; Piao, J.S.; Kang, J.-H.; Hashizume, M. Development of Human Hepatocellular Carcinoma Cell-Targeted Protein Cages. Bioconj. Chem. 2012, 23, 1494–1501. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Y.; Xiao, J.; Liu, G.; Li, X.; Zhao, Y.; Tan, H.; Shi, H.; Cheng, D. Investigation of SP94 Peptide as a Specific Probe for Hepatocellular Carcinoma Imaging and Therapy. Sci. Rep. 2016, 6, 33511. [Google Scholar] [CrossRef]
- Wu, C.-H.; Lan, C.-H.; Wu, K.-L.; Wu, Y.; Jane, W.-N.; Hsiao, M.; Wu, H.-C. Hepatocellular Carcinoma-Targeted Nanoparticles for Cancer Therapy. Int. J. Oncol. 2018, 52, 389–401. [Google Scholar] [CrossRef]
- Jin, Y.; Cheng, Z.; Yuan, Z.; Du, Y.; Tian, J.; Shao, B. Glucose-Regulated Protein 78 Targeting ICG and DOX Loaded Hollow Fe3O4 Nanoparticles for Hepatocellular Carcinoma Diagnosis and Therapy. Int. J. Nanomed. 2024, 19, 189. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Jiang, Y.; Yan, S.; Dang, H.; Yue, H.; Liu, K.; Kuang, L.; Liu, X.; Tang, H. A Novel pH- and Glutathione-Responsive Drug Delivery System Based on in Situ Growth of MOF199 on Mesoporous Organic Silica Nanoparticles Targeting the Hepatocellular Carcinoma Niche. Cancer Nanotechnol. 2022, 13, 32. [Google Scholar] [CrossRef]
- Raza, F.; Zheng, M.; Zhong, H.; Su, J.; He, B.; Yuan, W.-E.; Qiu, M. Engineered Tumor Microvesicles Modified by SP94 Peptide for Arsenic Trioxide Targeting Drug Delivery in Liver Cancer Therapy. Biomater. Adv. 2023, 155, 213683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Han, B.; Gao, C.; Liu, X.; Peng, Y.; Gong, C.; Hu, D.; Wang, N.; Tang, S.; Zhang, B.; et al. Integrated Platform of Oxygen Self-Enriched Nanovesicles: SP94 Peptide-Directed Chemo/Sonodynamic Therapy for Liver Cancer. Eur. J. Pharm. Biopharm. 2022, 179, 206–220. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Z.; Hao, H.; Luo, W.; Yang, J.; Li, M.; Yang, M.; Chen, Z.; Yan, R.; Li, H.; et al. Boron-Containing Mesoporous Silica Nanoparticles with Effective Delivery and Targeting of Liver Cancer Cells for Boron Neutron Capture Therapy. ACS Appl. Mater. Interfaces 2024, 16, 22934–22945. [Google Scholar] [CrossRef]
- Wu, M.; Xia, X.; Sun, D.; Zhong, C. Abstract 303: Surface Modification of Paclitaxel-Loaded Nanoparticles Based on Polydopamine with pH Sensitive Property for Targeted Therapy in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 303. [Google Scholar] [CrossRef]
- Wu, M.; Wang, Q.; Gong, Z.; Liu, Z.; Zheng, H.; Chen, X.; Zhong, C. Abstract 5736: Sonochemical Preparation of SP94 Peptide-Decorated pH-Responsive Biodegradable Biopolyester Nanoparticles for Targeted Therapy in Hepatocellular Carcinoma. Cancer Res. 2024, 84, 5736. [Google Scholar] [CrossRef]
- Kabil, M.F.; Gaber, S.A.A.; Hamzawy, M.A.; El-Sherbiny, I.M.; Nasr, M. Folic/Lactobionic Acid Dual-Targeted Polymeric Nanocapsules for Potential Treatment of Hepatocellular Carcinoma. Drug Deliv. Transl. Res. 2024, 14, 1338–1351. [Google Scholar] [CrossRef]
- Witzigmann, D.; Detampel, P.; Porta, F.; Huwyler, J. Isolation of Multiantennary N-Glycans from Glycoproteins for Hepatocyte Specific Targeting via the Asialoglycoprotein Receptor. RSC Adv. 2016, 6, 97636–97640. [Google Scholar] [CrossRef]
- Li, Y.; Huang, G.; Diakur, J.; Wiebe, L.I. Targeted Delivery of Macromolecular Drugs: Asialoglycoprotein Receptor (ASGPR) Expression by Selected Hepatoma Cell Lines Used in Antiviral Drug Development. Curr. Drug Deliv. 2008, 5, 299–302. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Wang, M.-D.; Zhou, X.; Qin, C.-J.; Fu, G.-B.; Tang, L.; Wu, H.; Huang, S.; Zhao, L.-H.; Zeng, M.; et al. Blocking Preferential Glucose Uptake Sensitizes Liver Tumor-Initiating Cells to Glucose Restriction and Sorafenib Treatment. Cancer Lett. 2017, 388, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Cho, H.-J.; Kim, D.-D. Poly((D,L)Lactic-Glycolic)Acid–Star Glucose Nanoparticles for Glucose Transporter and Hypoglycemia-Mediated Tumor Targeting. Int. J. Nanomed. 2017, 12, 7453–7467. [Google Scholar] [CrossRef]
- Ma, P.; Sun, Y.; Chen, J.; Li, H.; Zhu, H.; Gao, X.; Bi, X.; Zhang, Y. Enhanced Anti-Hepatocarcinoma Efficacy by GLUT1 Targeting and Cellular Microenvironment-Responsive PAMAM–Camptothecin Conjugate. Drug Deliv. 2018, 25, 153–165. [Google Scholar] [CrossRef]
- Mazumder, A.; Assawapanumat, W.; Dwivedi, A.; Reabroi, S.; Chairoungdua, A.; Nasongkla, N. Glucose Targeted Therapy against Liver Hepatocellular Carcinoma: In Vivo Study. J. Drug Deliv. Sci. Technol. 2019, 49, 502–512. [Google Scholar] [CrossRef]
- Dashty, M.; Motazacker, M.M.; Levels, J.; de Vries, M.; Mahmoudi, M.; Peppelenbosch, M.P.; Rezaee, F. Proteome of Human Plasma Very Low-Density Lipoprotein and Low-Density Lipoprotein Exhibits a Link with Coagulation and Lipid Metabolism. Thromb. Haemost. 2017, 112, 518–530. [Google Scholar] [CrossRef]
- Südhof, T.C.; Goldstein, J.L.; Brown, M.S.; Russell, D.W. The LDL Receptor Gene: A Mosaic of Exons Shared with Different Proteins. Science 1985, 228, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Dalhaimer, P.; Florey, B.; Isaac, S. Interactions of Apolipoproteins with Lipid-Based Nanoparticles. ACS Nano 2023. [Google Scholar] [CrossRef]
- Sneath, R.J.; Mangham, D.C. The Normal Structure and Function of CD44 and Its Role in Neoplasia. Mol. Pathol. 1998, 51, 191. [Google Scholar] [CrossRef]
- Spring, F.A.; Dalchau, R.; Daniels, G.L.; Mallinson, G.; Judson, P.A.; Parsons, S.F.; Fabre, J.W.; Anstee, D.J. The Ina and Inb Blood Group Antigens Are Located on a Glycoprotein of 80,000 MW (the CDw44 Glycoprotein) Whose Expression Is Influenced by the In(Lu) Gene. Immunology 1988, 64, 37. [Google Scholar]
- Xie, P.; Yan, J.; Wu, M.; Li, H.; Chen, Z.; Yu, M.; Zhang, B.; Chen, L.; Jin, L.; Zhou, B.; et al. CD44 Potentiates Hepatocellular Carcinoma Migration and Extrahepatic Metastases via the AKT/ERK Signaling CXCR4 Axis. Ann. Transl. Med. 2022, 10, 689. [Google Scholar] [CrossRef]
- Drug Delivery Systems (Definition). Available online: http://www.reference.md/files/D016/mD016503.html (accessed on 20 February 2025).
- Wu, F.-Q.; Fang, T.; Yu, L.-X.; Lv, G.-S.; Lv, H.-W.; Liang, D.; Li, T.; Wang, C.-Z.; Tan, Y.-X.; Ding, J.; et al. ADRB2 Signaling Promotes HCC Progression and Sorafenib Resistance by Inhibiting Autophagic Degradation of HIF1α. J. Hepatol. 2016, 65, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Vega-Vásquez, P.; Mosier, N.S.; Irudayaraj, J. Nanoscale Drug Delivery Systems: From Medicine to Agriculture. Front. Bioeng. Biotechnol. 2020, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, F.; Gan, P.; Li, B.; Li, S. Callispheres Drug-Eluting Bead Transhepatic Artery Chemoembolization with Oral Delivery of Sorafenib for the Treatment of Unresectable Liver Cancer. Front. Surg. 2022, 9, 981116. [Google Scholar] [CrossRef]
- Liu, S.; Liu, C.; Wang, Q.; Liu, Y.; Wang, D.; Zhao, G.; Yu, G. The Second-Line Treatment of Hepatocellular Carcinoma with CalliSpheres Drug-Eluting Bead Transarterial Chemoembolization Combined with Regorafenib: A Safety and Efficacy Analysis. Ir. J. Med. Sci. 2024, 193, 1215–1222. [Google Scholar] [CrossRef]
- Cho, S.; Min, N.G.; Park, W.; Kim, S.; Kim, D. Janus Microcarriers for Magnetic Field-Controlled Combination Chemotherapy of Hepatocellular Carcinoma. Adv. Funct. Mater. 2019, 29, 1901384. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Cho, S.; Ji, J.; Lewandowski, R.J.; Larson, A.C.; Kim, D.-H. Development and Validation of Sorafenib-Eluting Microspheres to Enhance Therapeutic Efficacy of Transcatheter Arterial Chemoembolization in a Rat Model of Hepatocellular Carcinoma. Radiol. Imaging Cancer 2021, 3, e200006. [Google Scholar] [CrossRef]
- Ahnfelt, E.; Degerstedt, O.; Lilienberg, E.; Sjögren, E.; Hansson, P.; Lennernäs, H. Lipiodol-Based Emulsions Used for Transarterial Chemoembolization and Drug Delivery: Effects of Composition on Stability and Product Quality. J. Drug Deliv. Sci. Technol. 2019, 53, 101143. [Google Scholar] [CrossRef]
- Li, X.; Yu, H.; Huang, Y.; Chen, Y.; Wang, J.; Xu, L.; Zhang, F.; Zhuge, Y.; Zou, X. Preparation of Microspheres Encapsulating Sorafenib and Catalase and Their Application in Rabbit VX2 Liver Tumor. Biomed. Pharmacother. 2020, 129, 110512. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Sun, J.; Sun, L.; Wan, J.; Wang, S.; Gu, D.; Yu, C.; Yang, C.; He, J.; et al. Self-Assembled and Self-Monitored Sorafenib/Indocyanine Green Nanodrug with Synergistic Antitumor Activity Mediated by Hyperthermia and Reactive Oxygen Species-Induced Apoptosis. ACS Appl. Mater. Interfaces 2019, 11, 43996–44006. [Google Scholar] [CrossRef]
- Tanaka, H.; Horioka, K.; Hasebe, T.; Sawada, K.; Nakajima, S.; Konishi, H.; Isozaki, S.; Goto, M.; Fujii, Y.; Kamikokura, Y.; et al. Treatment of Hepatocellular Carcinoma with Autologous Platelets Encapsulating Sorafenib or Lenvatinib: A Novel Therapy Exploiting Tumor-platelet Interactions. Int. J. Cancer 2022, 150, 1640–1653. [Google Scholar] [CrossRef]
- Li, Z.; Ye, L.; Liu, J.; Lian, D.; Li, X. Sorafenib-Loaded Nanoparticles Based on Biodegradable Dendritic Polymers for Enhanced Therapy of Hepatocellular Carcinoma. Int. J. Nanomed. 2020, 15, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Zhou, L.; Jin, H.; Chen, Y.; Cheng, D.; Jiang, Y. Sorafenib-Loaded Long-Circulating Nanoliposomes for Liver Cancer Therapy. BioMed Res. Int. 2020, 2020, 1351046. [Google Scholar] [CrossRef]
- Tunki, L.; Kulhari, H.; Vadithe, L.N.; Kuncha, M.; Bhargava, S.; Pooja, D.; Sistla, R. Modulating the Site-Specific Oral Delivery of Sorafenib Using Sugar-Grafted Nanoparticles for Hepatocellular Carcinoma Treatment. Eur. J. Pharm. Sci. 2019, 137, 104978. [Google Scholar] [CrossRef]
- Iacobazzi, R.M.; Vischio, F.; Arduino, I.; Canepa, F.; Laquintana, V.; Notarnicola, M.; Scavo, M.P.; Bianco, G.; Fanizza, E.; Lopedota, A.A.; et al. Magnetic Implants in Vivo Guiding Sorafenib Liver Delivery by Superparamagnetic Solid Lipid Nanoparticles. J. Colloid Interface Sci. 2022, 608, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Futschik, E.K.; Reyes-Ortega, F. Advantages and Disadvantages of Using Magnetic Nanoparticles for the Treatment of Complicated Ocular Disorders. Pharmaceutics 2021, 13, 1157. [Google Scholar] [CrossRef]
- Arora, W. Superparamagnetic Iron Oxide Nanoparticles: Magnetic Nanoplatforms as Drug Carriers. Int. J. Nanomed. 2012, 7, 3445–3471. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Sant, S.; Wang, B.; Laurent, S.; Sen, T. Superparamagnetic Iron Oxide Nanoparticles (SPIONs): Development, Surface Modification and Applications in Chemotherapy. Adv. Drug Deliv. Rev. 2011, 63, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, G.; Su, F.; Chu, Z.; Xu, L.; Zhang, Y.; Zhou, J.; Ding, Y. Regorafenib-Loaded Poly (Lactide-Co-Glycolide) Microspheres Designed to Improve Transarterial Chemoembolization Therapy for Hepatocellular Carcinoma. Asian J. Pharm. Sci. 2020, 15, 739–751. [Google Scholar] [CrossRef]
- Yue, M.; Yang, R.; Jiang, Y.; Yang, X. Precise Construction of Regorafenib-Loaded Gold Nanoparticles: Investigation of Antiproliferative Activity and Apoptosis Induction in Liver Cancer Cells. J. Exp. Nanosci. 2023, 18, 2254006. [Google Scholar] [CrossRef]
- Kyriakides, T.R.; Raj, A.; Tseng, T.H.; Xiao, H.; Nguyen, R.; Mohammed, F.S.; Halder, S.; Xu, M.; Wu, M.J.; Bao, S.; et al. Biocompatibility of Nanomaterials and Their Immunological Properties. Biomed. Mater. 2021, 16, 042005. [Google Scholar] [CrossRef]
- Fröhlich, E. The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Shastri, V.P. Non-Degradable Biocompatible Polymers in Medicine: Past, Present and Future. Curr. Pharm. Biotechnol. 2003, 4, 331–337. [Google Scholar] [CrossRef]
- He, C.; Jaffar Ali, D.; Li, Y.; Zhu, Y.; Sun, B.; Xiao, Z. Engineering of HN3 Increases the Tumor Targeting Specificity of Exosomes and Upgrade the Anti-Tumor Effect of Sorafenib on HuH-7 Cells. PeerJ 2020, 8, e9524. [Google Scholar] [CrossRef]
- Zhang, G.; Huang, X.; Xiu, H.; Sun, Y.; Chen, J.; Cheng, G.; Song, Z.; Peng, Y.; Shen, Y.; Wang, J.; et al. Extracellular Vesicles: Natural Liver-accumulating Drug Delivery Vehicles for the Treatment of Liver Diseases. J. Extracell. Vesicles 2020, 10, e12030. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Javius-Jones, K.; Hong, S.; Park, H. Cell-Based Drug Delivery Systems with Innate Homing Capability as a Novel Nanocarrier Platform. Int. J. Nanomed. 2023, 18, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Su, Y.-Y.; Jiang, X.-C.; Gao, J.-Q. Cell Membrane-Coated Nanoparticles: A Novel Multifunctional Biomimetic Drug Delivery System. Drug Deliv. Transl. Res. 2023, 13, 716–737. [Google Scholar] [CrossRef]
- Desai, N.; Tambe, V.; Pofali, P.; Vora, L.K. Cell Membrane-Coated Nanoparticles: A New Frontier in Immunomodulation. Adv. Nanobiomed. Res. 2024, 4, 2400012. [Google Scholar] [CrossRef]
- Xuan, M.; Shao, J.; Li, J. Cell Membrane-Covered Nanoparticles as Biomaterials. Natl. Sci. Rev. 2019, 6, 551–561. [Google Scholar] [CrossRef]
- Wang, D.; Liu, C.; You, S.; Zhang, K.; Li, M.; Cao, Y.; Wang, C.; Dong, H.; Zhang, X. Bacterial Vesicle-Cancer Cell Hybrid Membrane-Coated Nanoparticles for Tumor Specific Immune Activation and Photothermal Therapy. ACS Appl. Mater. Interfaces 2020, 12, 41138–41147. [Google Scholar] [CrossRef]
- Wu, Y.; Zhu, R.; Zhou, M.; Liu, J.; Dong, K.; Zhao, S.; Cao, J.; Wang, W.; Sun, C.; Wu, S.; et al. Homologous Cancer Cell Membrane-Camouflaged Nanoparticles Target Drug Delivery and Enhance the Chemotherapy Efficacy of Hepatocellular Carcinoma. Cancer Lett. 2023, 558, 216106. [Google Scholar] [CrossRef]
- Shao, T.; Chen, Y.-L. Stop Using the Misidentified Cell Line LO2 as a Human Hepatocyte. J. Hepatol. 2024, 80, e200–e201. [Google Scholar] [CrossRef]
- Yue, H.; Gou, L.; Tang, Z.; Liu, Y.; Liu, S.; Tang, H. Construction of pH-Responsive Nanocarriers in Combination with Ferroptosis and Chemotherapy for Treatment of Hepatocellular Carcinoma. Cancer Nano 2022, 13, 4. [Google Scholar] [CrossRef]
- Ferreira, J.V.; da Rosa Soares, A.; Ramalho, J.; Máximo Carvalho, C.; Cardoso, M.H.; Pintado, P.; Carvalho, A.S.; Beck, H.C.; Matthiesen, R.; Zuzarte, M.; et al. LAMP2A Regulates the Loading of Proteins into Exosomes. Sci. Adv. 2022, 8, eabm1140. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J.; Wen, R.; Li, Q.; Zhou, J.; Liu, X.; Wu, Z.; Lv, Y.; Wu, R. Improved Anti-Hepatocellular Carcinoma Effect by Enhanced Co-Delivery of Tim-3 siRNA and Sorafenib via Multiple pH Triggered Drug-Eluting Nanoparticles. Mater. Today Bio 2022, 16, 100350. [Google Scholar] [CrossRef]
- Zhang, C.; Zhong, W.; Cao, Y.; Liu, B.; Tao, X.; Li, Z. Sorafenib/2800Z Co-Loaded into Cholesterol and PEG Grafted Polylysine NPs for Liver Cancer Treatment. Pharmaceuticals 2023, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, L. Sorafenib-Loaded Hydroxyethyl Starch-TG100-115 Micelles for the Treatment of Liver Cancer Based on Synergistic Treatment. Drug Deliv. 2019, 26, 756–764. [Google Scholar] [CrossRef]
- Younis, M.A.; Khalil, I.A.; Elewa, Y.H.A.; Kon, Y.; Harashima, H. Ultra-Small Lipid Nanoparticles Encapsulating Sorafenib and Midkine-siRNA Selectively-Eradicate Sorafenib-Resistant Hepatocellular Carcinoma in Vivo. J. Control. Release 2021, 331, 335–349. [Google Scholar] [CrossRef]
- Zeng, Z.-M.; Mo, N.; Zeng, J.; Ma, F.-C.; Jiang, Y.-F.; Huang, H.-S.; Liao, X.-W.; Zhu, G.-Z.; Ma, J.; Peng, T. Advances in Postoperative Adjuvant Therapy for Primary Liver Cancer. World J. Gastrointest. Oncol. 2022, 14, 1604–1621. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Impact on NP | Effect on Drug Delivery | Ref. |
|---|---|---|---|
| Hydrophobic surface | Promotes binding of plasma protein and liver uptake | Higher accumulation in liver | [83,84,85,86] |
| Surface charge | Positively charged NPs → hepatocyte uptake; negatively charged NPs → Kupffer/endothelial uptake | Affects targeting efficiency and biodistribution | [87] |
| Size 150–200 nm | Enables crossing capillary fenestrations | Improves hepatocyte internalization | [88] |
| Size 12 nm | Optimal tumor accumulation via EPR effect | Minimizes nonspecific organ uptake | [90,91] |
| Size 6 nm | Rapid renal clearance | Reduces therapeutic utility | [90,91] |
| Kupfer cell uptake | Excretion into intestines | Limits drug delivery to tumor | [86] |
| Tumor-homing Kupffer cells | Migration to tumor tissues | Potential to enhance NP accumulation in tumors | [83,84] |
| Targeting moiety | Uses specific receptors on cancer cells | ~1.5× higher delivery efficiency than non-targeted systems | [89,94] |
| Target | Expression Pattern | Ligands | Challenges |
|---|---|---|---|
| Glypican-3 | Highly expressed in HCC, minimal in healthy liver and other tissues [102] (except placenta [103]) | L5 peptide, truncated L5 [104,105], recombinant human GPC3 core protein [106], antibodies, aptamers [107] | Limited ligand stability (peptides), potential off-target effects during pregnancy [103] |
| CXCR4 | Overexpressed in PLC, also found in normal tissues (bone marrow, lymphoid) [108] | Peptides (LFC131), small molecules (AMD3100 [109,110,111], cordycepin [112]) | Off-target effects, challenges in tumor specificity |
| Neuropilin-1 (NP-1) | Expressed in tumors and endothelial cells [113,114], debatable overexpression in PLC [115] | iRGD peptide [116,117,118] | Limited evidence in PLC, primarily studied in pancreatic cancer [119,120,121] |
| GRP78 | Overexpressed in drug-resistant PLC [122] | Peptides (SP94 [123], RGD) | Peptide ligands increase opsonization [124] |
| ASGPR | Predominantly in liver and PLC [125] | Saccharides (galactose, N-acetylgalactosamine [126]), glycoproteins, polymers (pullulan [127], arabinogalactan [128]) | Variable expression in PLC [129] |
| GLUT-1 | Elevated in PLC, increased in hypoxia-induced chemoresistant cells [130,131] | Saccharides (glucose [132], glucosamine [133], N-acetylglucosamine [134]) | Risk of BBB penetration, potential off-target effects [135] |
| LDLR | High in cancer and liver cells [136,137,138,139] | Apolipoproteins (Apo B, E) [80,136] | BBB permeability [140], incomplete understanding of liver targeting mechanisms [83,84], widely expressed among tissues [136,137,138,139] |
| CD44 | Low to moderate in PLC, high in stem-like PLC cells [141,142,143,144] (SK-HEP-1 [145]) | Hyaluronic acid [81,133] | Low baseline expression in PLC [145] |
| Folate receptor | Generally low in PLC [146,147], but overexpressed in specific cell lines (SMMC-7721, BEL-7402) [98,99,100] | Folic acid [148] | Limited PLC specificity [146,147], potential BBB penetration [149] |
| DDS | Outcome | Study Type | Ref. |
|---|---|---|---|
| Callispheres—PVA microspheres with SOR delivered during TACE |
| In vivo (humans), compared to standard TACE with sorafenib | [187] |
| Callispheres—PVA microspheres with regorafenib delivered during TACE |
| In vivo (humans) | [188] |
| NPs loaded with SOR targeting GPC3 via antibody, delivered during TACE |
| In vivo (humans), compared to standard therapy | [154] |
| NPs functionalized with hGC33 antibody, targeting GPC3, loaded with SOR |
| In vivo (mice) | [155] |
| PLGA-based NPs functionalized with GPC3-targeting peptide for SOR delivery |
| In vivo (mice) | [106] |
| Liposomal DDS with targeting antibody (anti-VEGFR) for SOR delivery |
| In vivo (mice), in vitro (humans) | [196] |
| Polymeric NPs functionalized with LFC131 peptide targeting CXCR4, co-delivering SOR and metapristone |
| In vivo (mice), in vitro (humans) | [111] |
| Pullulan-based NPs self-assembled with stearic acid, targeting ASGPR1 for SOR delivery |
| In vivo (mice), in vitro (humans) | [127] |
| Galactose-functionalized SLNPs for SOR delivery |
| In vivo (mice), in vitro (humans) | [197] |
| Zinc-based MOF loaded with SOR glucose oxidase and rhodamine B with N-acetylgalactosamine targeting ligand |
| In vivo (mice) and in vitro (humans) | [79] |
| SLNP coated with PEO and loaded with SOR and iron oxide NPs |
| In vivo (mice) and in vitro (humans) | [198] |
| Bismuth-based mesoporous nanomaterials loaded with SOR and coated with PEO-folic acid conjugate |
| In vivo (mice) and in vitro (humans), compared to sorafenib or radiotherapy alone | [98] |
| Lipid-based NPs co-delivering SOR and dihydroartemisinin with ApoB-100 coating targeting LDLRs |
| In vivo (mice) and in vitro (humans), compared to free drugs, single-drug NPs, or non-coated NPs | [136] |
| BSA NPs loaded with SOR and folic acid |
| In vivo (mice) and in vitro (humans), compared to non-targeted NPs | [99] |
| Glucosamine-functionalized PEO-disulfide–PCL micelles for SOR delivery |
| In vivo (mice), in vitro (humans) | [133] |
| HA-based dendronized micelles co-delivering SOR and PCX |
| In vivo (mice), in vitro (humans) | [81] |
| TPGS-modified dendrimers for SOR delivery |
| In vivo (mice), in vitro (humans) | [195] |
| Polymeric micelles with ApoE or mefenamate, containing SOR |
| In vivo (mice) | [80] |
| Regorafenib-loaded PLGA microspheres delivered during TACE |
| In vivo (mice), in vitro | [202] |
| Regorafenib–gold NP conjugates |
| In vitro | [203] |
| DDS | Outcome | Study Type | Ref. |
|---|---|---|---|
| Extracellular vesicles derived from red blood cells for SOR delivery |
| In vivo (mice) and in vitro (cells), compared to standard sorafenib therapy | [208] |
| Platelets as carriers for SOR/lenvatinib |
| In vivo (rats), compared to free sorafenib | [194] |
| Platelet-coated mesoporous silica NPs with PD-L1 antibody for SOR delivery |
| In vivo (mice), compared to non-coated NPs | [5] |
| Exosomes from normal epithelial cells with HN3 antibody for SOR delivery |
| In vitro (cells), compared to exosomes from cancer cells | [207] |
| Kiwi-derived exosomes for oral delivery of SOR |
| In vivo (mice) and in vitro (cells), compared to free sorafenib | [78] |
| PAE–PEG–NH2 NPs coated with biological membranes from PLC cells (SMMC-7721) for lenvatinib delivery |
| In vitro (human cells), In vivo (mice) | [214] |
| DDS | Outcome | Study Type | Ref. |
|---|---|---|---|
| Sorafenib-loaded PLGA microspheres with Fe3O4 NPs (MRI contrast agents) |
| In vitro, in vivo (rat model) | [190] |
| PLGA microspheres loaded with sorafenib and catalase |
| In vivo (rabbit VX2 liver tumor model) | [192] |
| Iron-based NPs loaded with sorafenib and iRGD peptide |
| In vitro (cells), in vivo (mouse model) | [116] |
| SP94-modified exosomes for delivery of siRNA targeting GPX4 and DHODH with SOR co-treatment |
| In vivo (mouse model) | [77] |
| SP94-functionalized NPs (copper-based MOF) with SOR and DOX |
| In vivo (xenograft HCC tumor model) | [165] |
| Amphiphilic polymer NPs with SOR (PEO, polylysine, cholesterol) with 2800Z |
| In vitro (cells), in vivo (mouse model) | [219] |
| Polymeric micelles based on PEO and hydroxyethyl starch for delivery of SOR and TG100-115 |
| In vitro (cells), in vivo (PLC-bearing mice) | [220] |
| Lipid-based NPs modified with PEO and SP94 for delivery of SOR + midkine-siRNA |
| In vitro (cells), in vivo (mouse model) | [221] |
| Prussian blue NPs (Fe2+, Fe3+, CN−) coated with chitosan for delivery of SOR |
| In vitro (cells), in vivo (mice) | [216] |
| Nanoconstruct (sorafenib and indocyanine via π–π interaction, coated with pluronic) |
| In vitro (Huh7 cells), in vivo (mice) | [193] |
| PEO polymer-based NPs for co-delivery of SOR and siRNA targeting Tim-3 |
| In vivo (PLC mouse model) | [218] |
| Janus particles for delivery of regorafenib and DOX (polycaprolactone, PLGA, embedded Fe3O4 nanocubes) for TACE |
| In vitro (HepG2 cells), in vivo (HCC rat model) | [189] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szyk, P.; Czarczynska-Goslinska, B.; Ziegler-Borowska, M.; Larrosa, I.; Goslinski, T. Sorafenib—Drug Delivery Strategies in Primary Liver Cancer. J. Funct. Biomater. 2025, 16, 148. https://doi.org/10.3390/jfb16040148
Szyk P, Czarczynska-Goslinska B, Ziegler-Borowska M, Larrosa I, Goslinski T. Sorafenib—Drug Delivery Strategies in Primary Liver Cancer. Journal of Functional Biomaterials. 2025; 16(4):148. https://doi.org/10.3390/jfb16040148
Chicago/Turabian StyleSzyk, Piotr, Beata Czarczynska-Goslinska, Marta Ziegler-Borowska, Igor Larrosa, and Tomasz Goslinski. 2025. "Sorafenib—Drug Delivery Strategies in Primary Liver Cancer" Journal of Functional Biomaterials 16, no. 4: 148. https://doi.org/10.3390/jfb16040148
APA StyleSzyk, P., Czarczynska-Goslinska, B., Ziegler-Borowska, M., Larrosa, I., & Goslinski, T. (2025). Sorafenib—Drug Delivery Strategies in Primary Liver Cancer. Journal of Functional Biomaterials, 16(4), 148. https://doi.org/10.3390/jfb16040148