Biomineralization of Fucoidan-Peptide Blends and Their Potential Applications in Bone Tissue Regeneration

Abstract
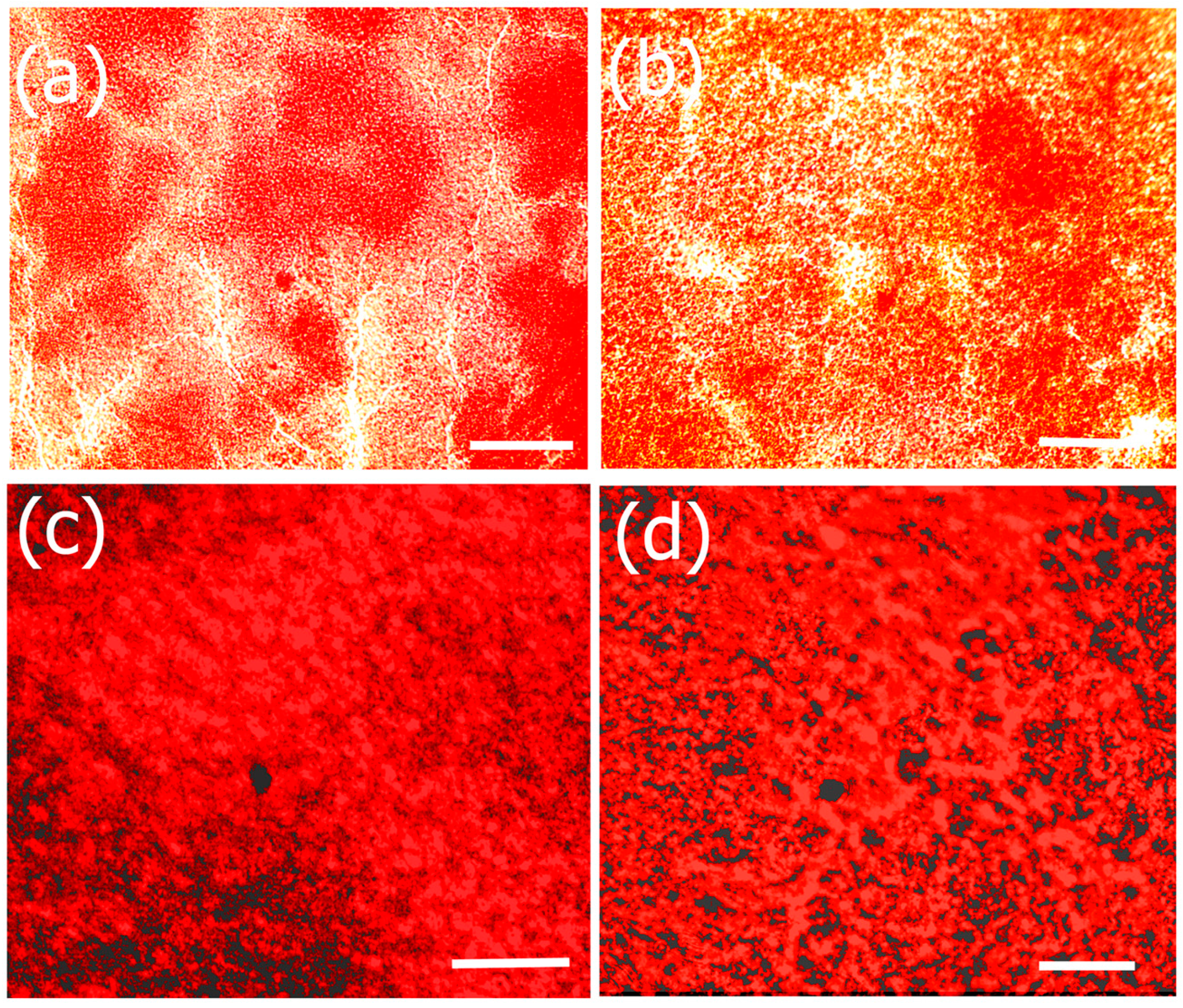
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Formation of Scaffold
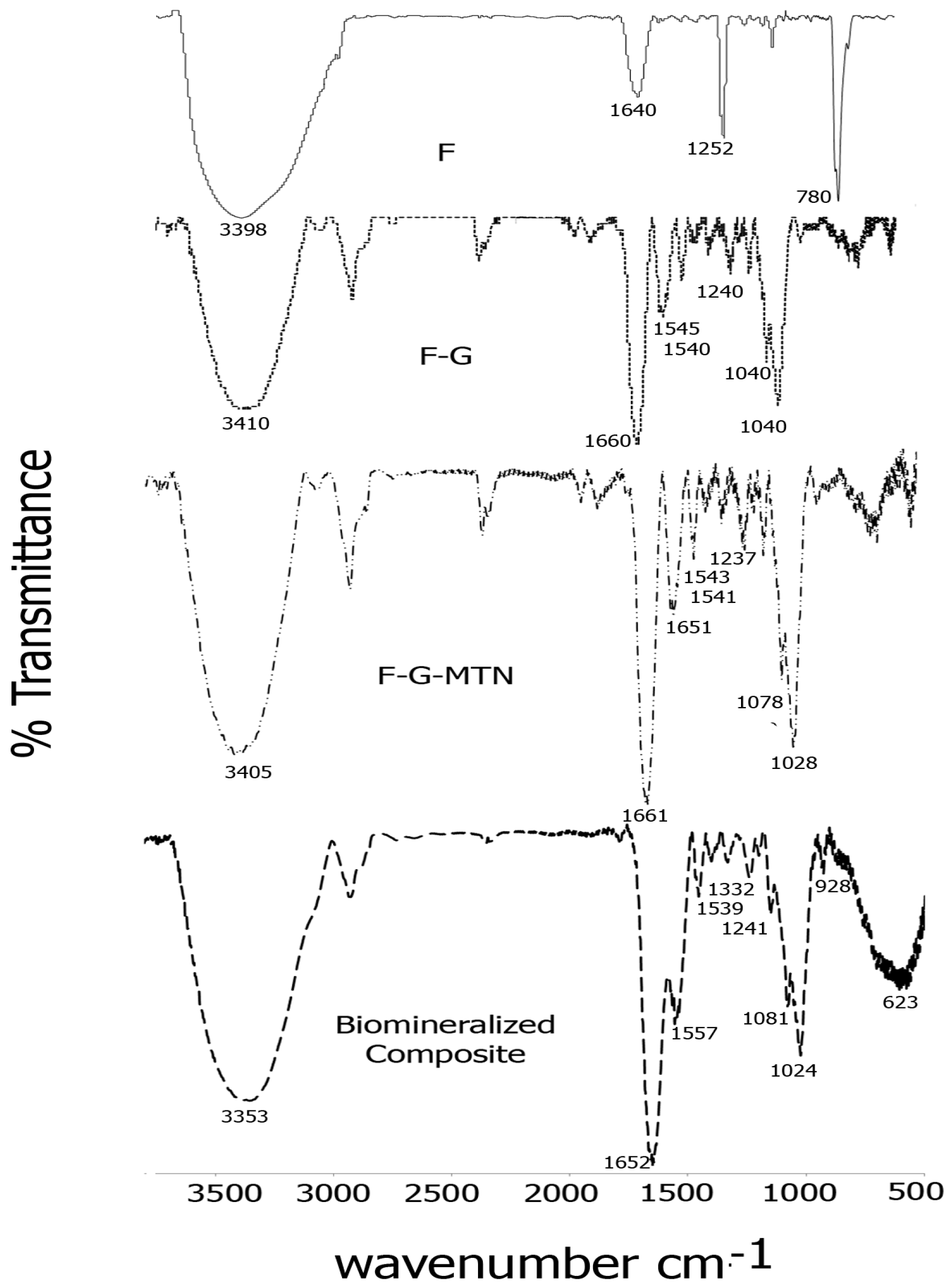
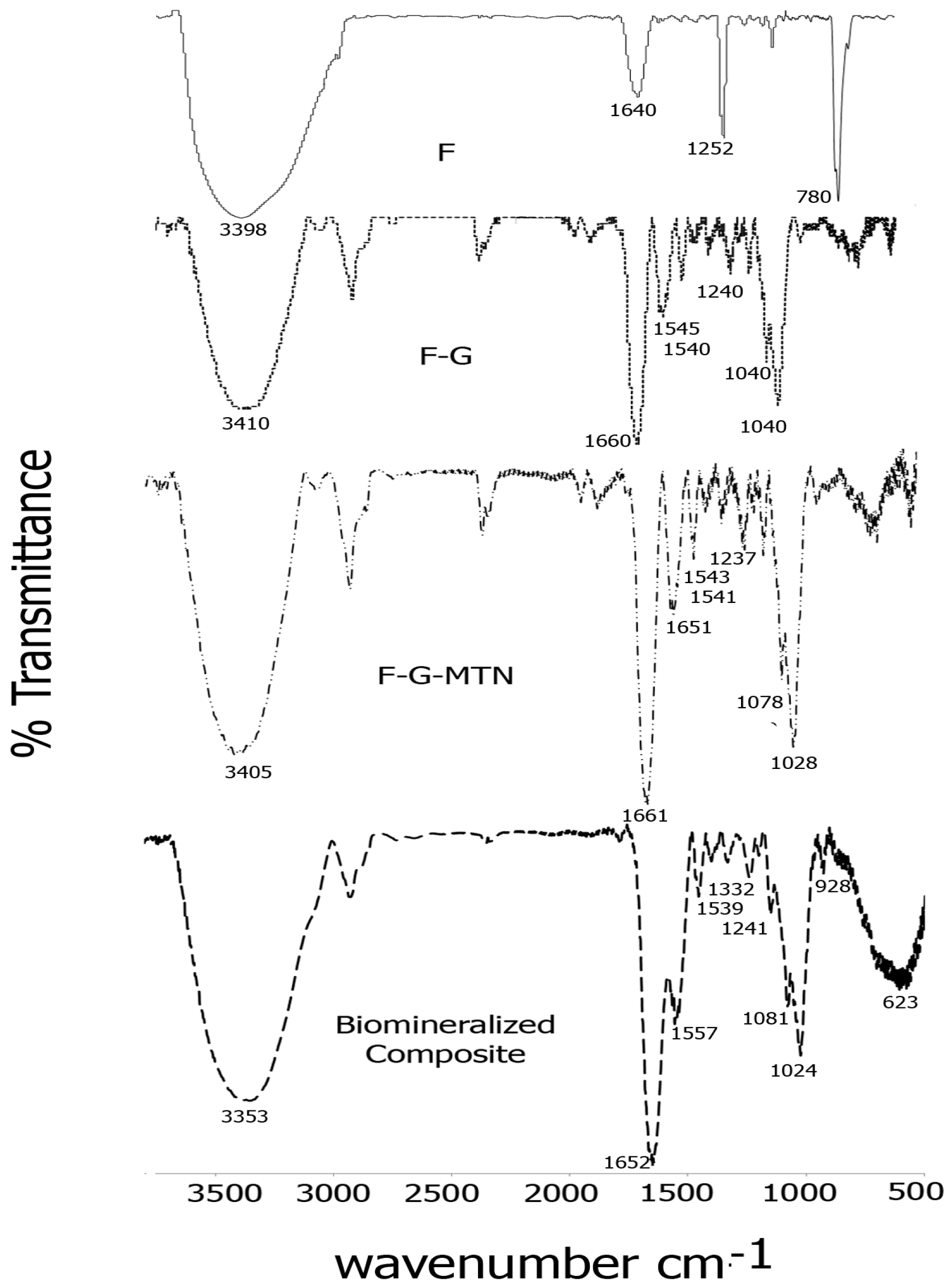
2.2. FTIR Spectroscopy
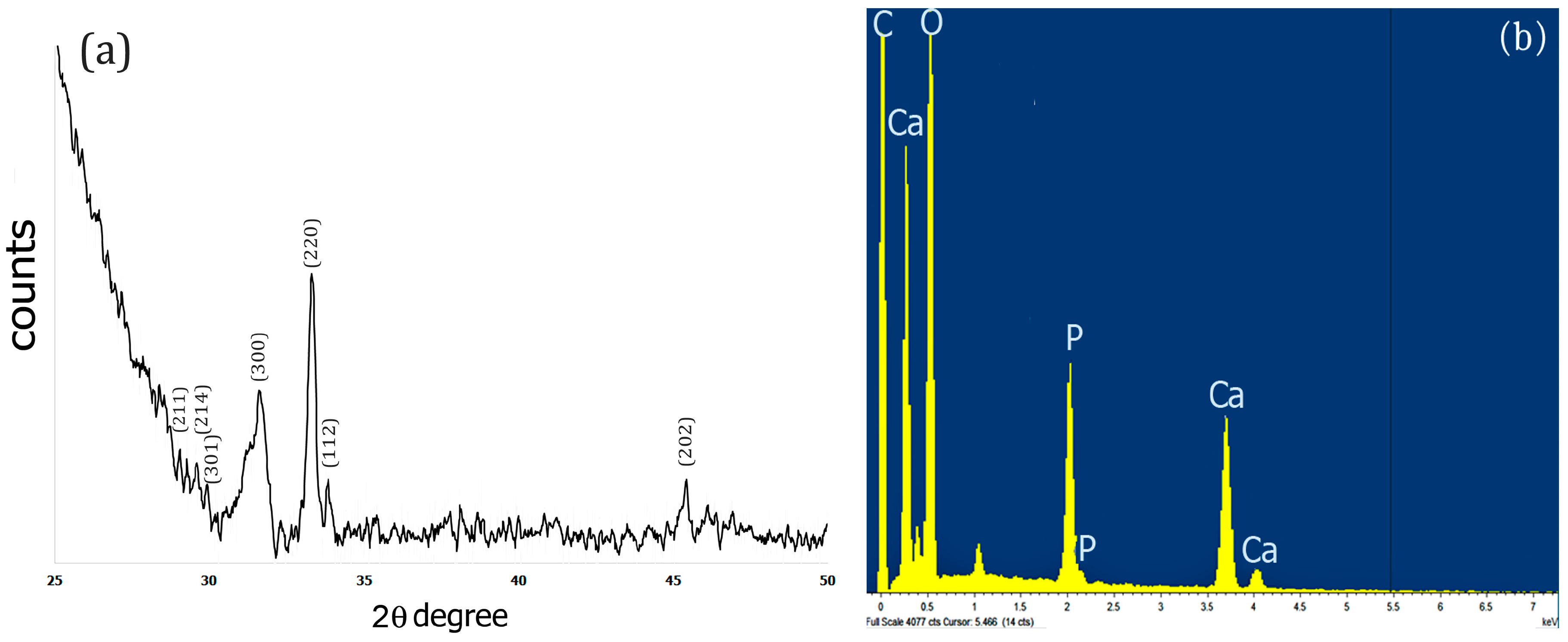
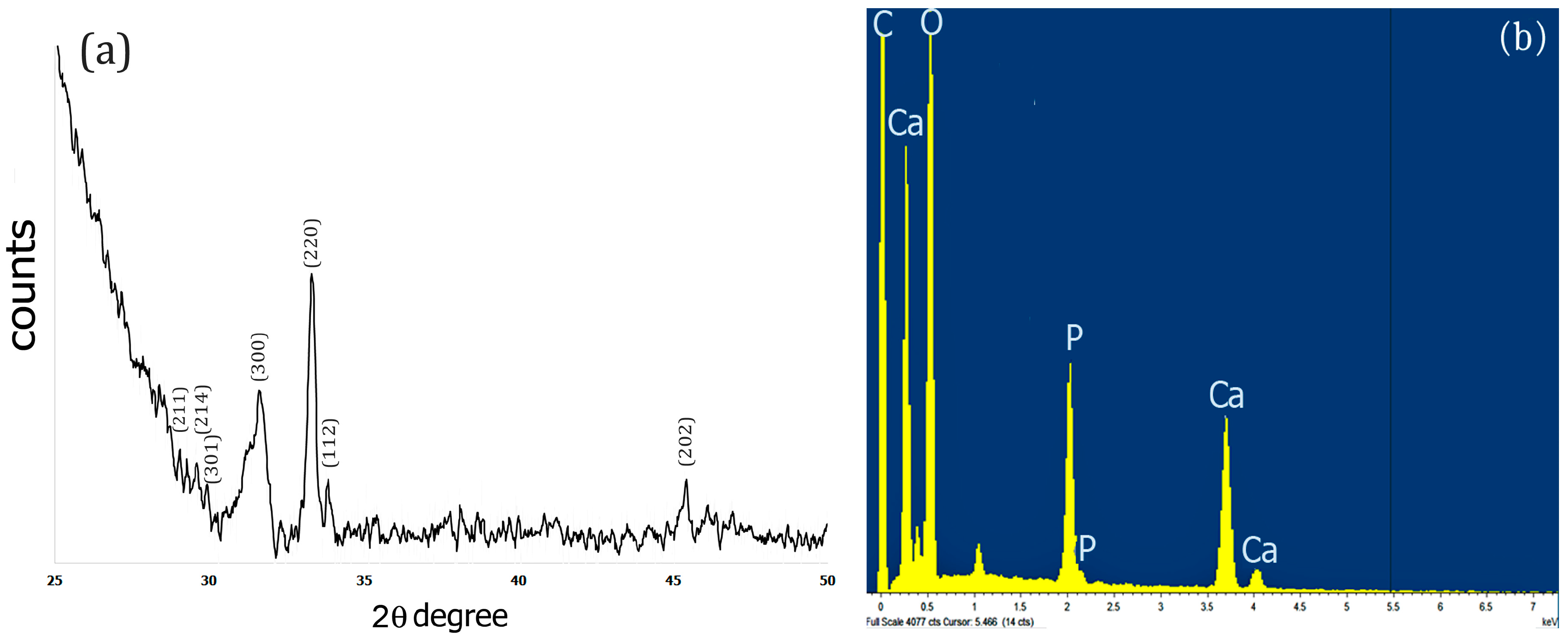
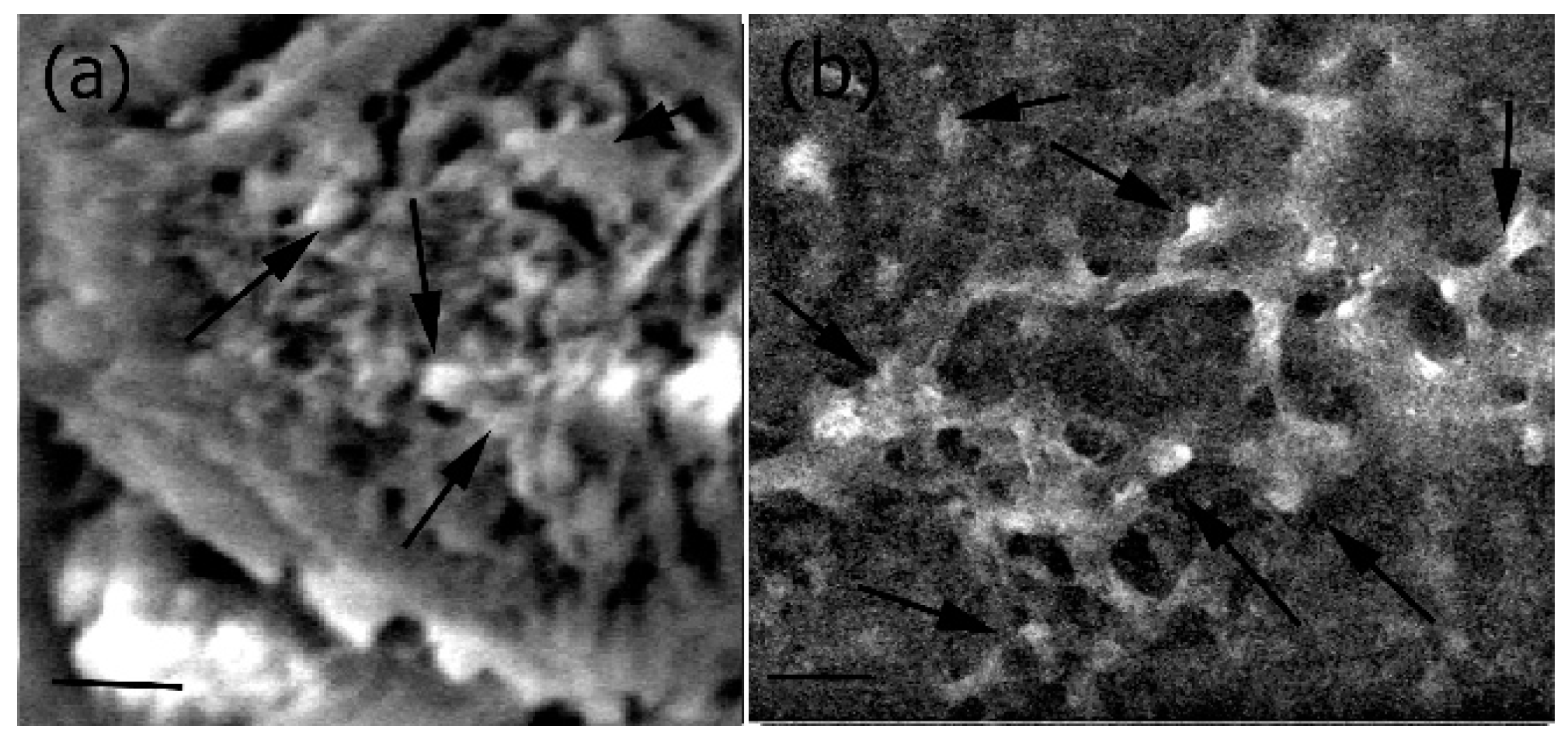
2.3. XRD and EDS Analysis
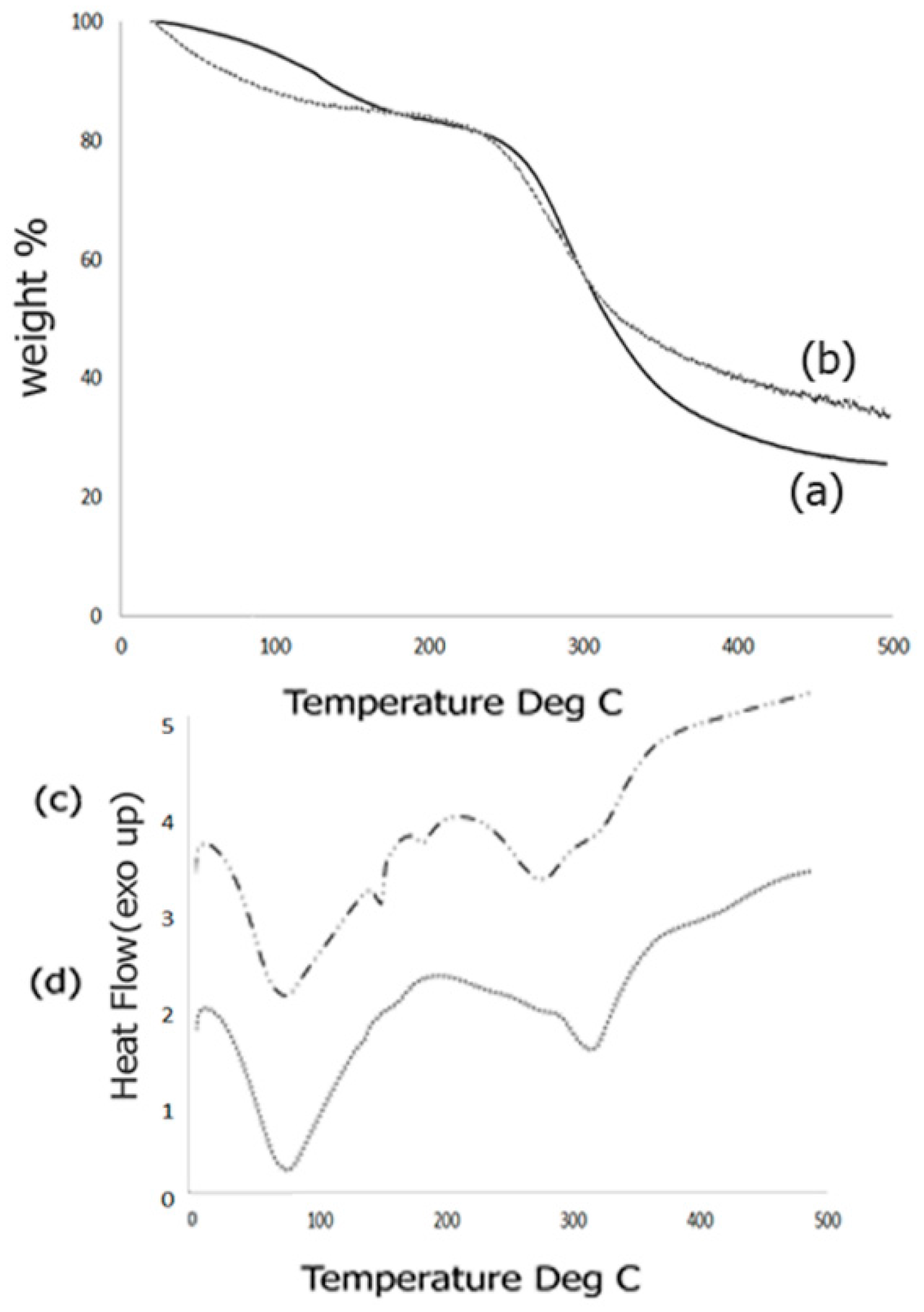
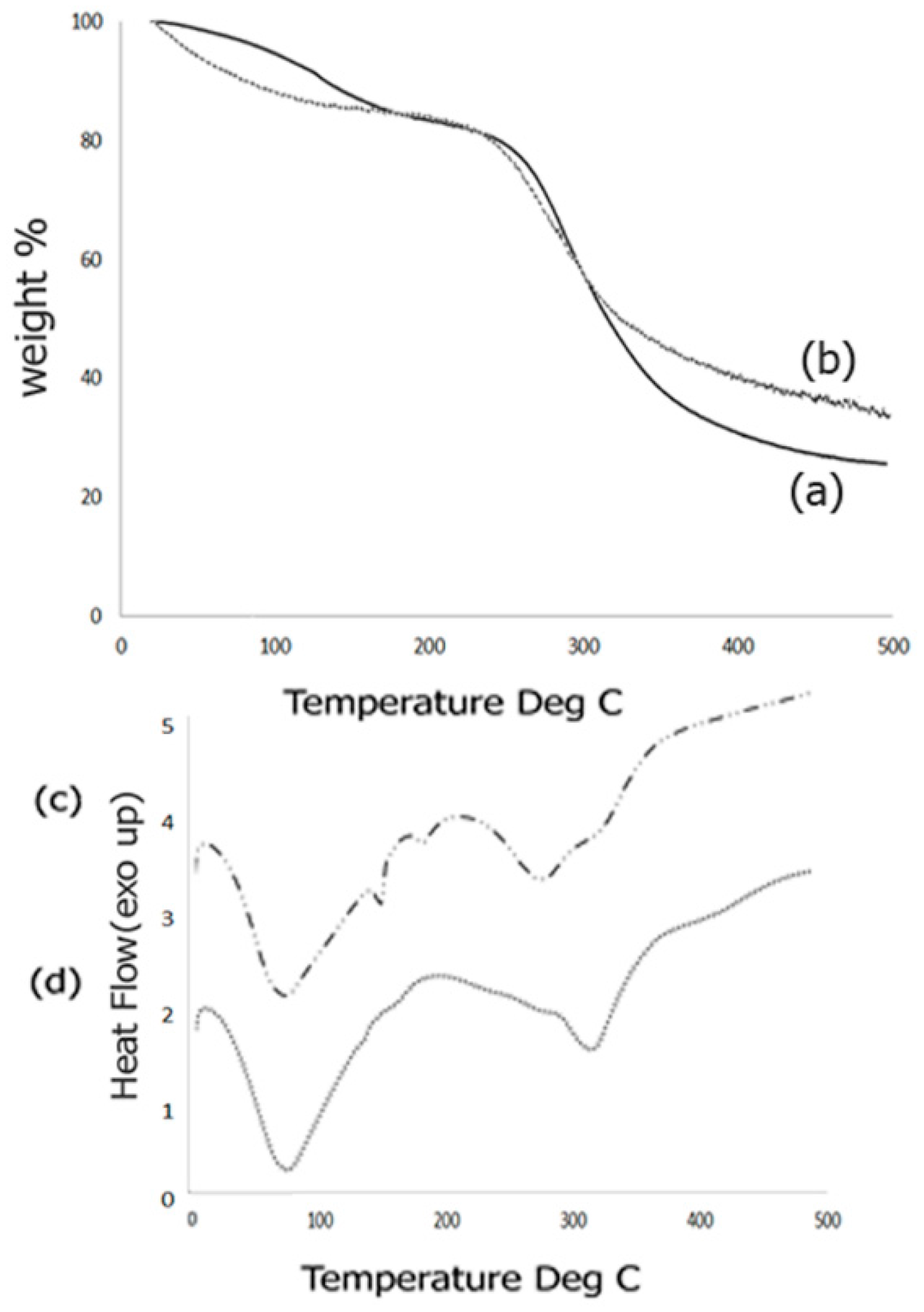
2.4. Thermal Analysis
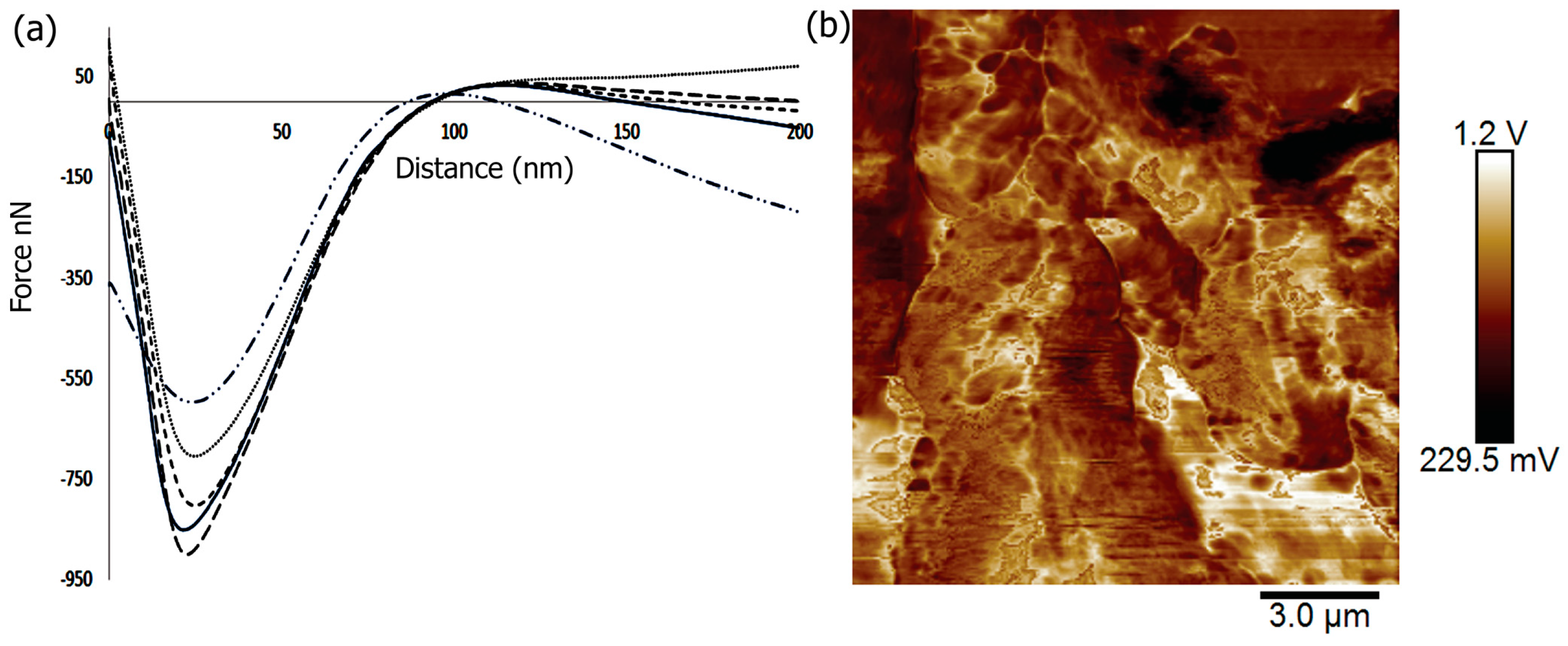
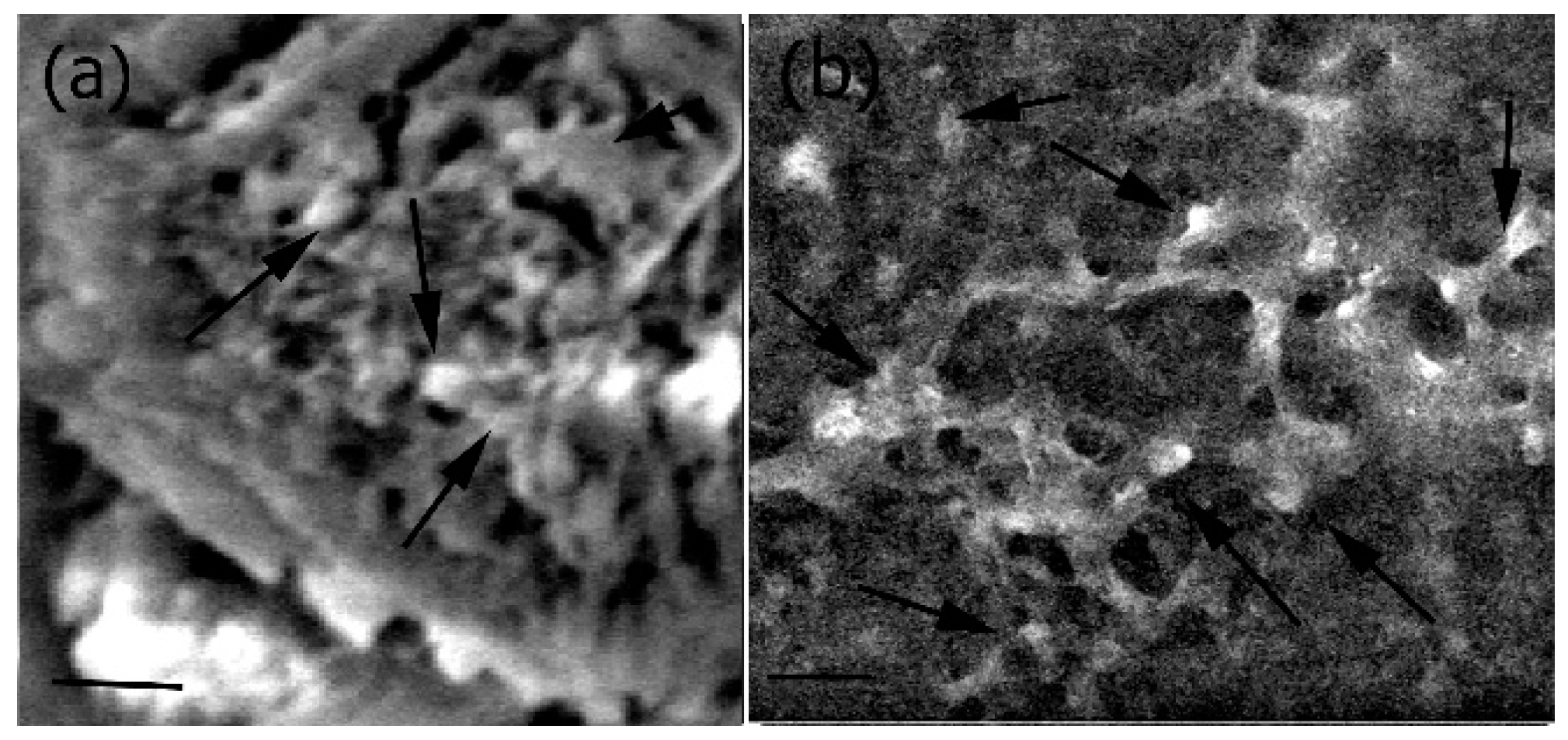
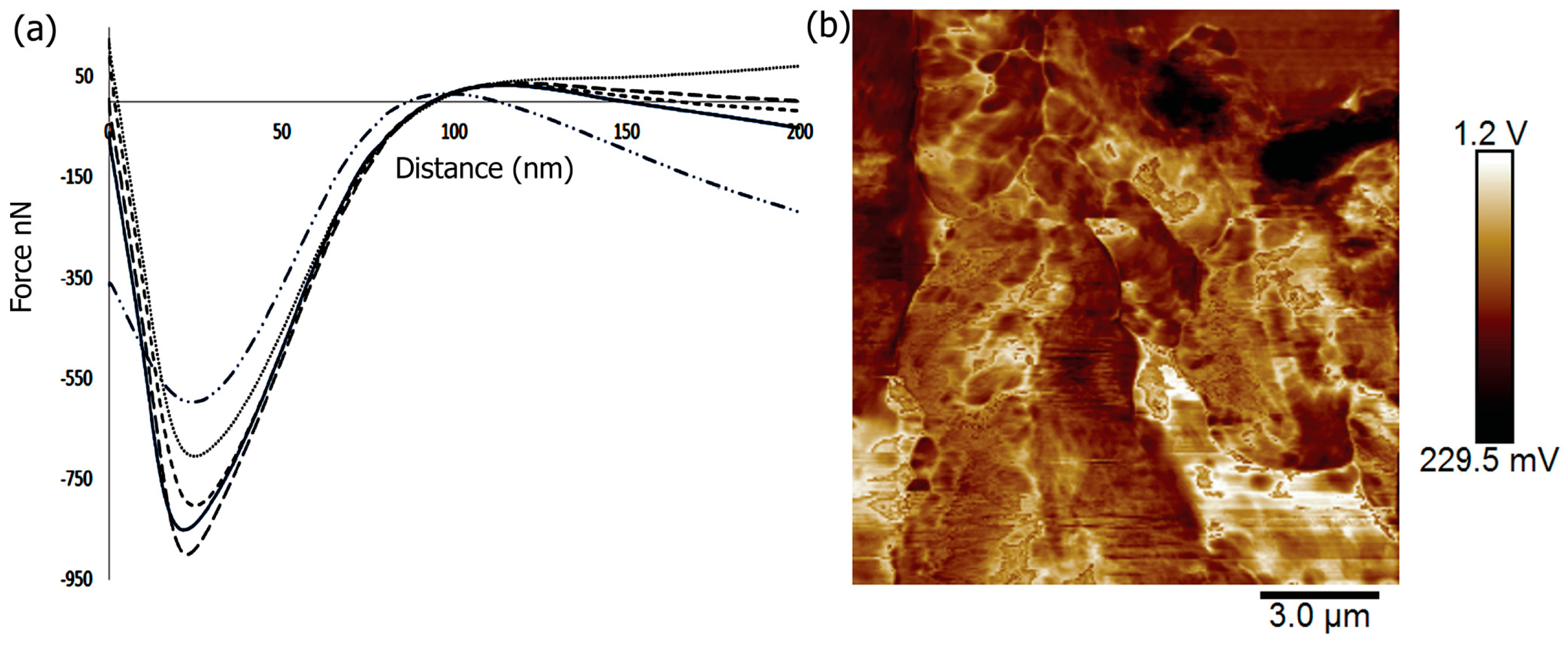
2.5. Mechanical and Surface Properties
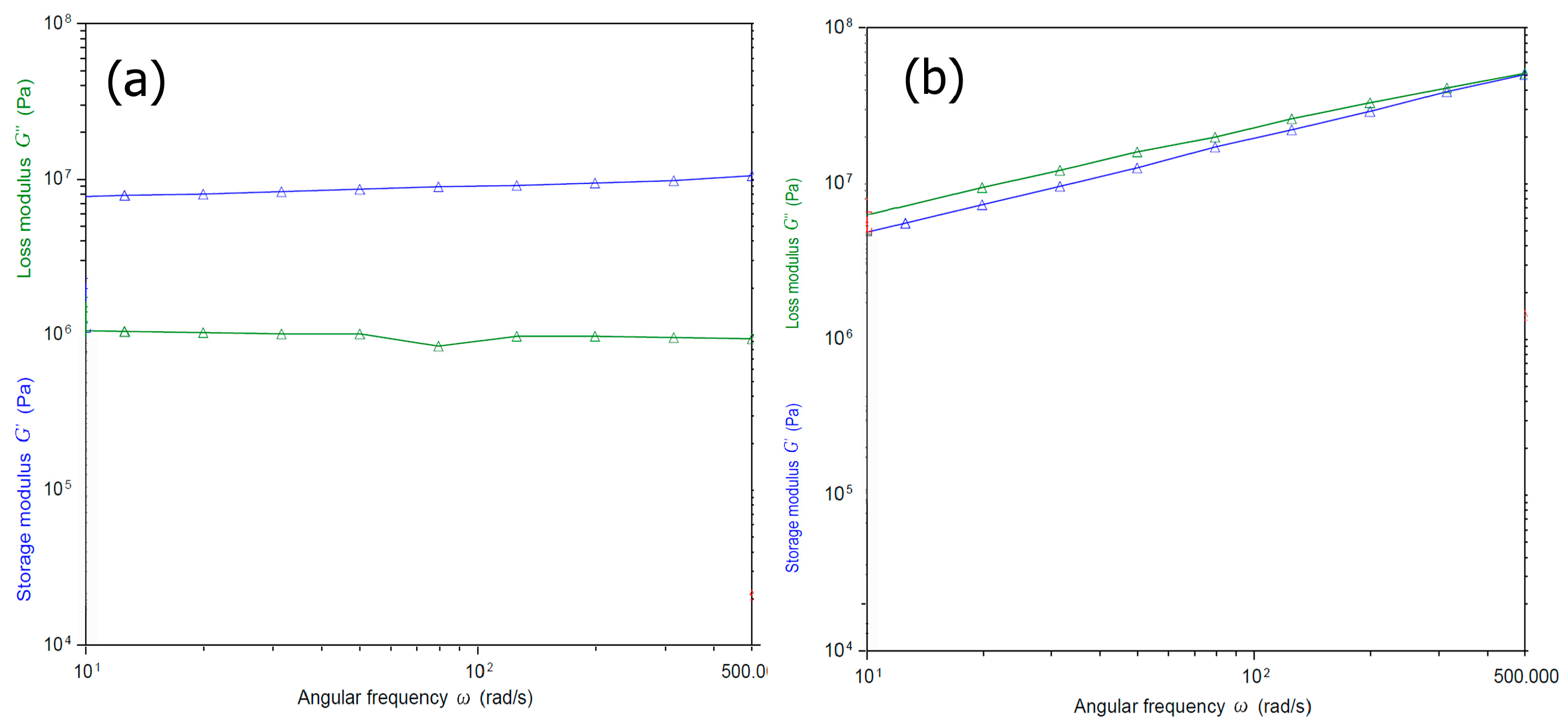
2.6. Rheology
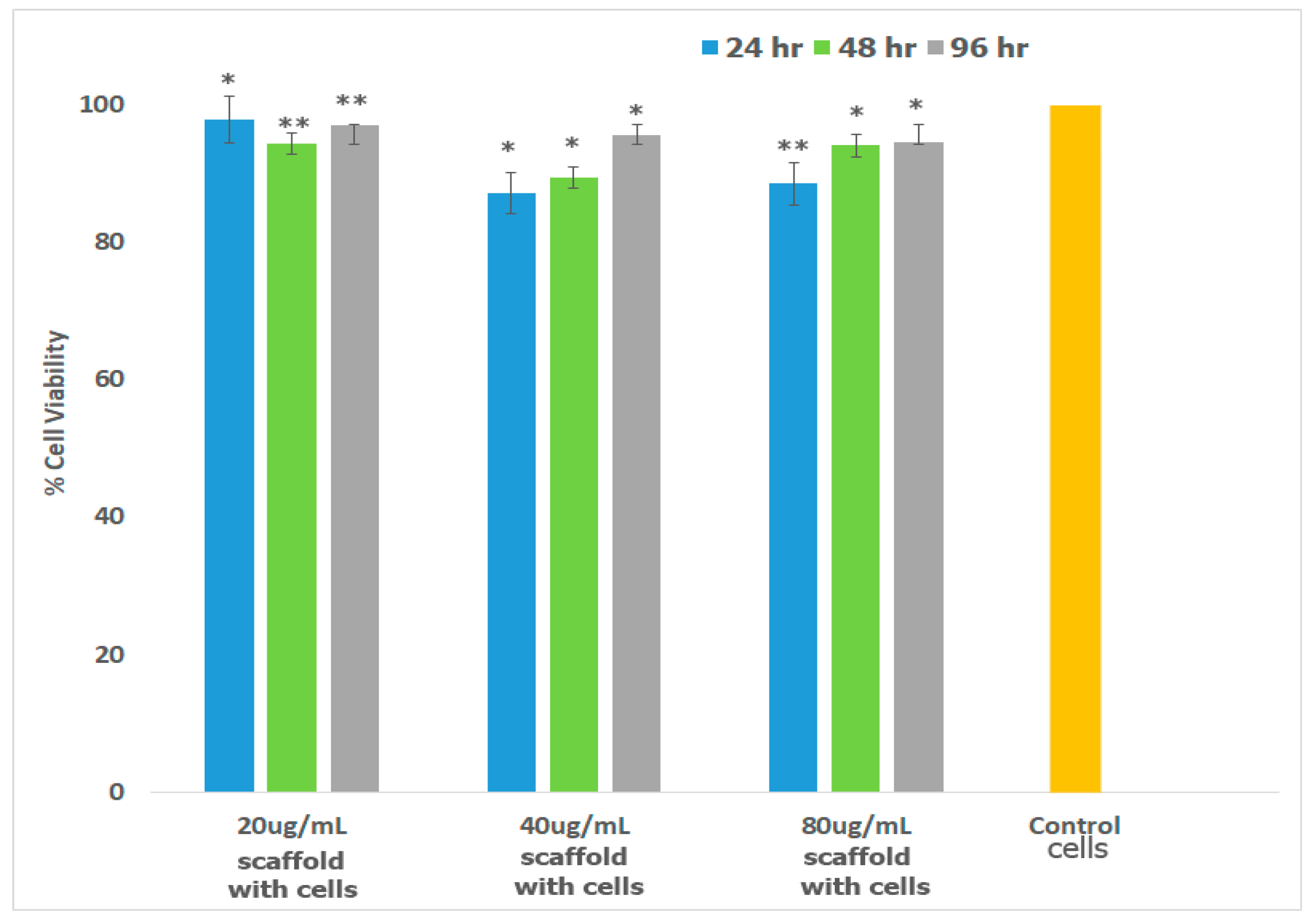
2.7. Cell Studies
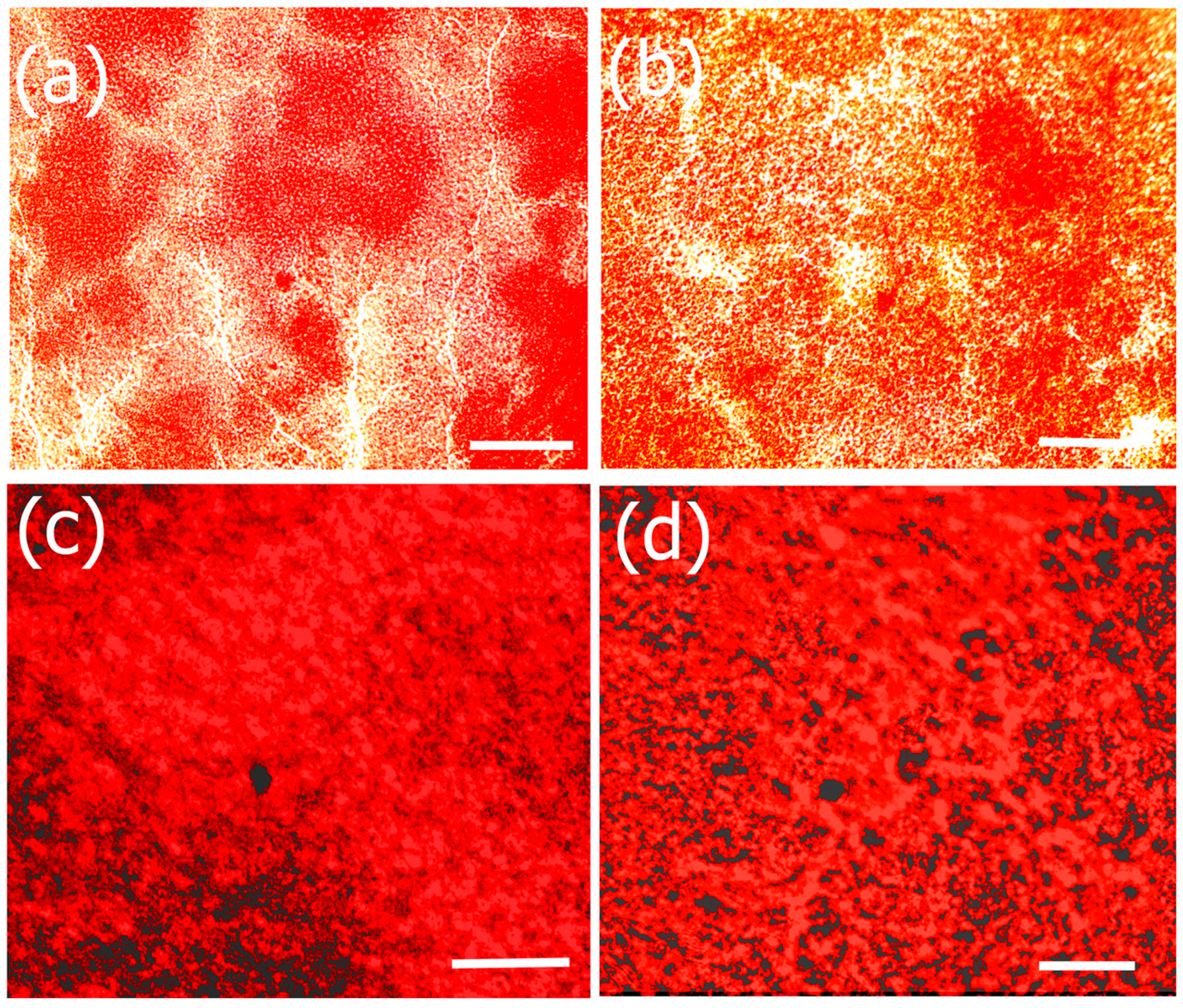
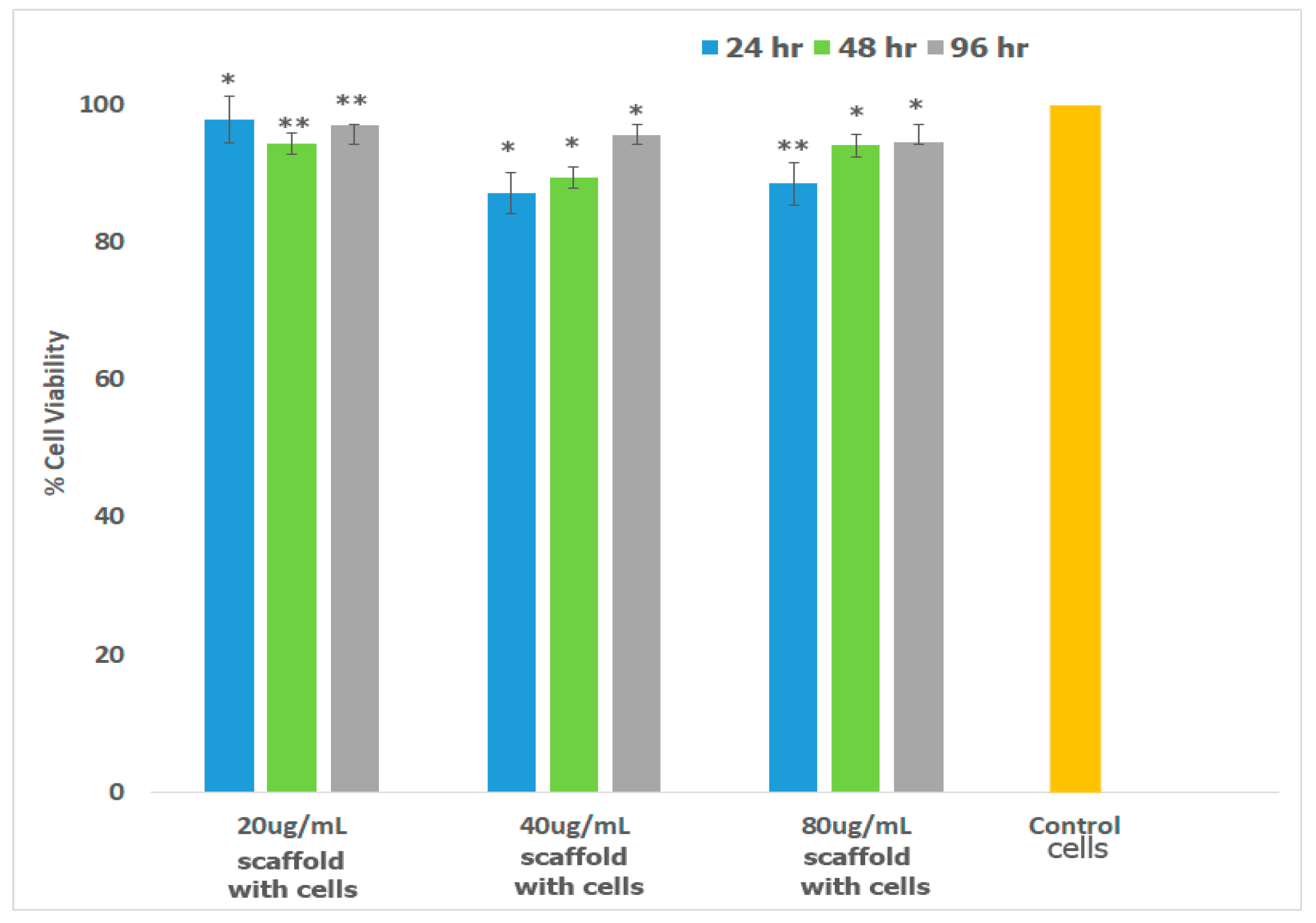
2.7.1. Cell Viability and Morphology Studies
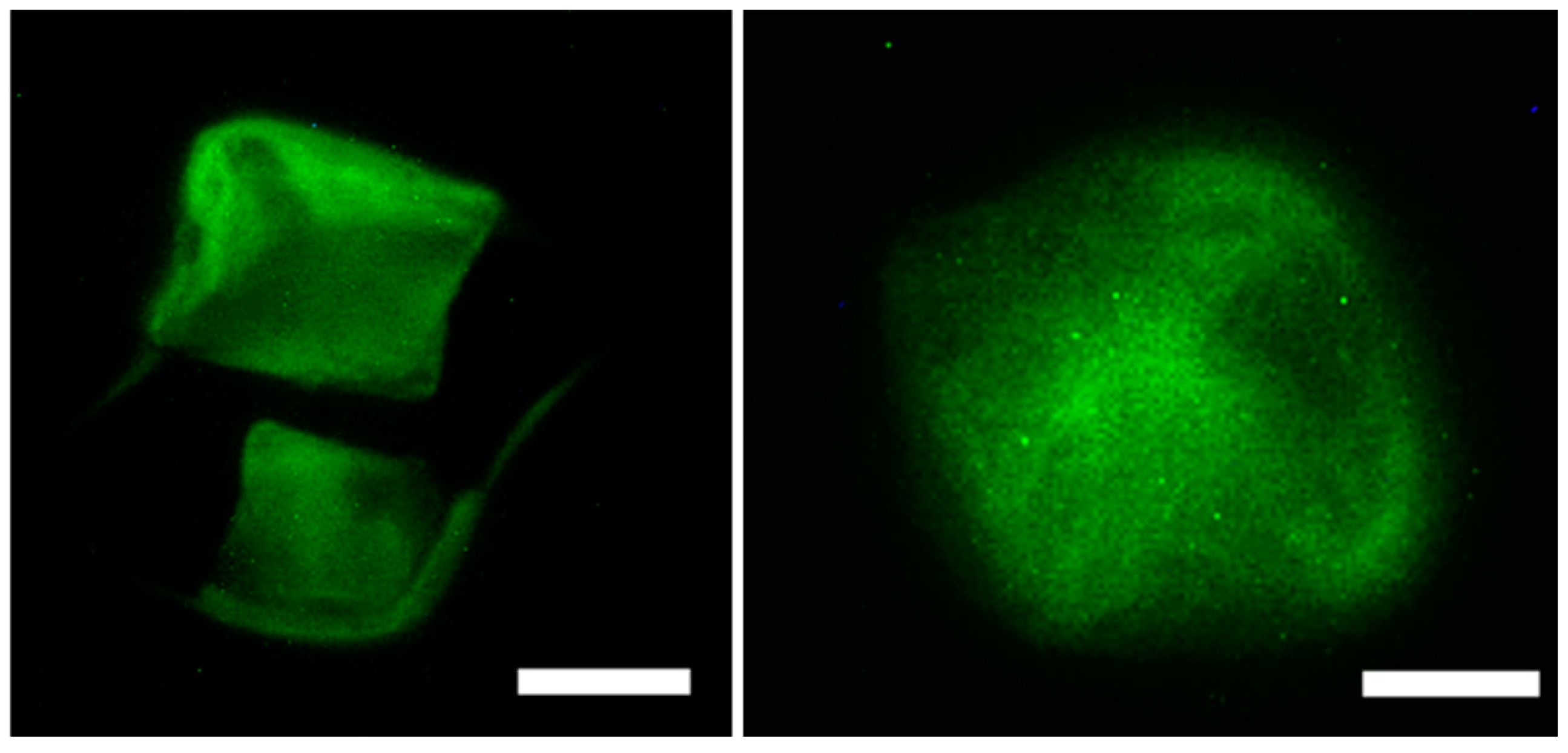
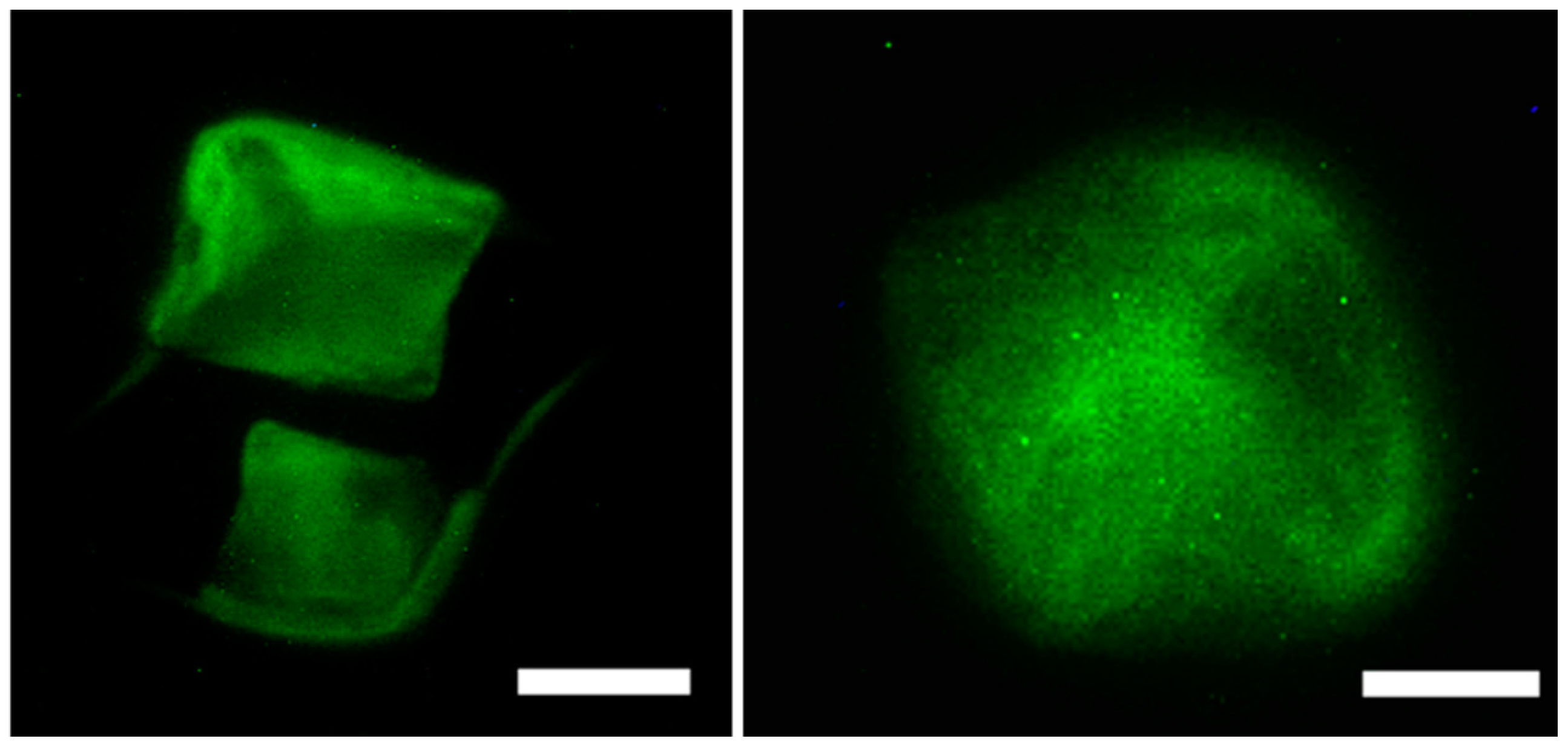
2.7.2. Cytoskeletal Staining (Phalloidin Assay)
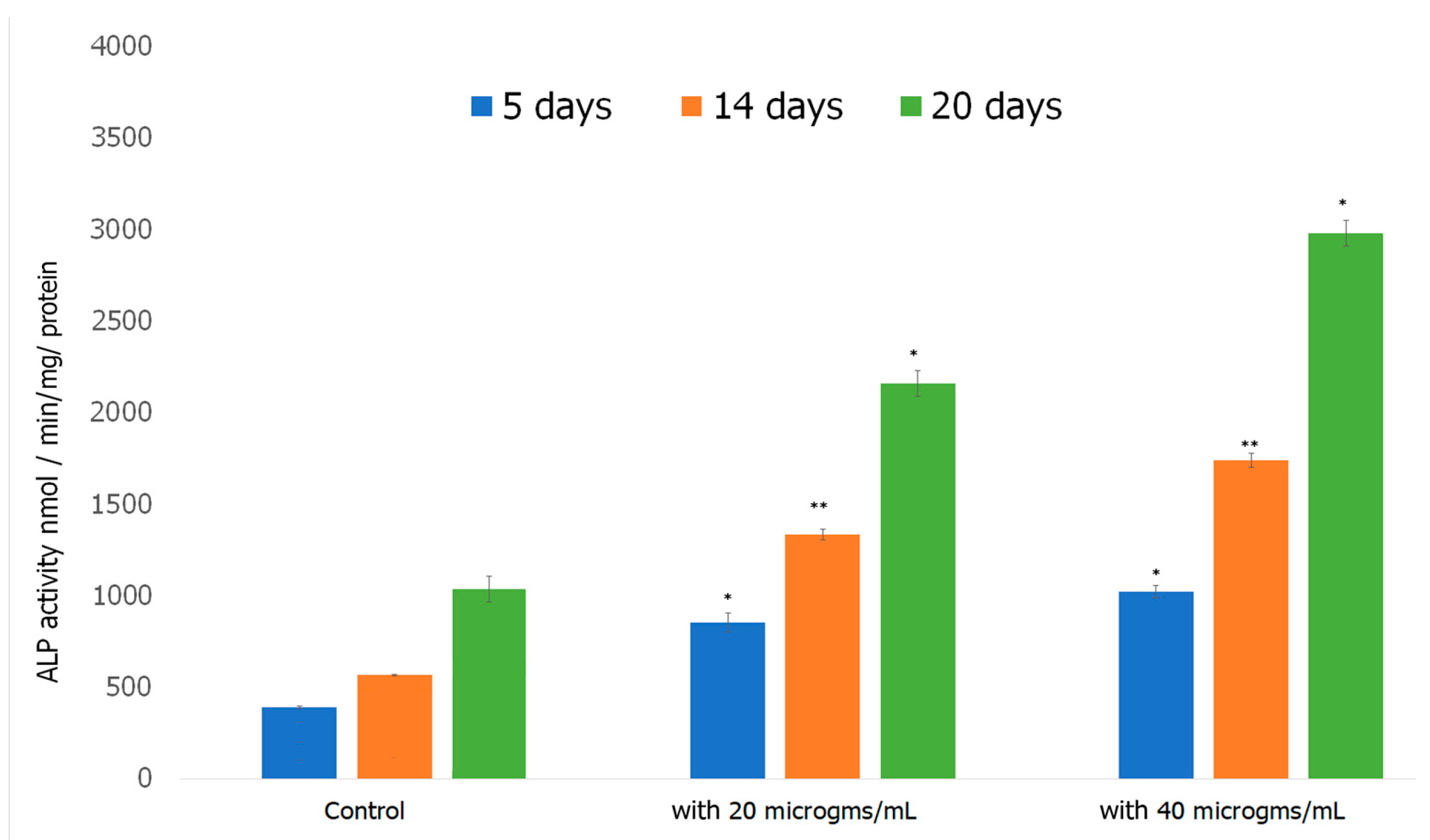
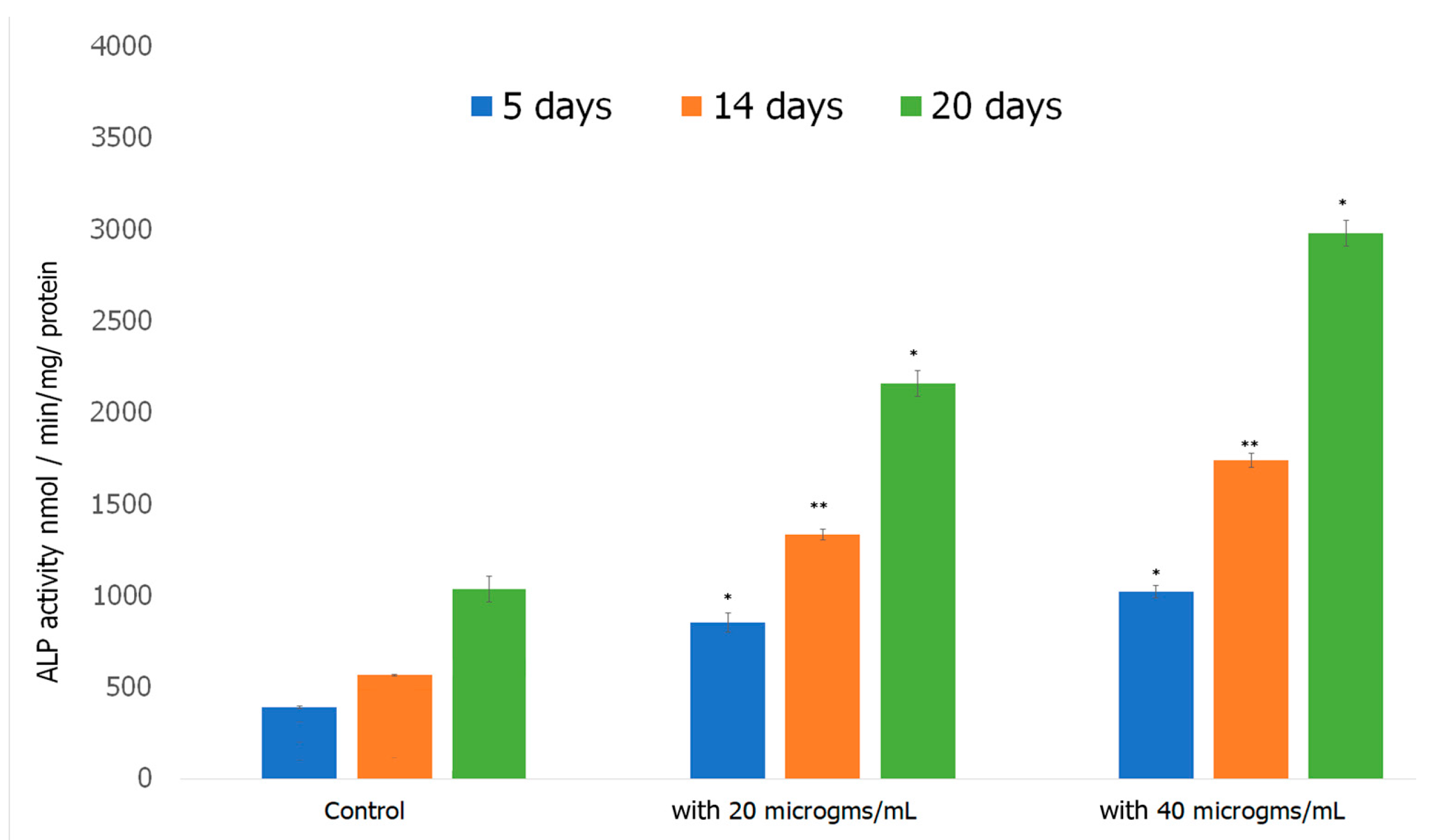
2.7.3. Alkaline Phosphatase Assay
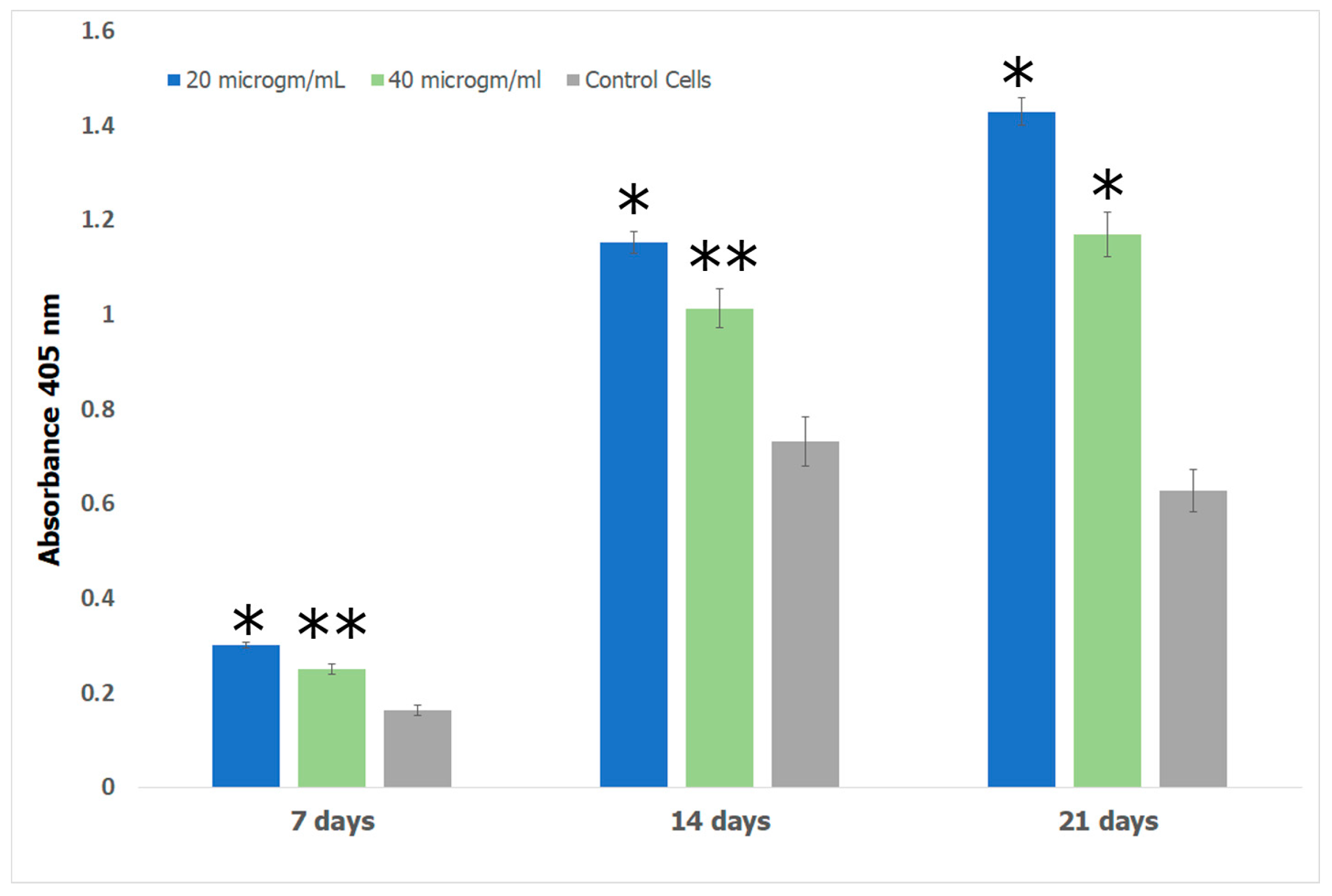
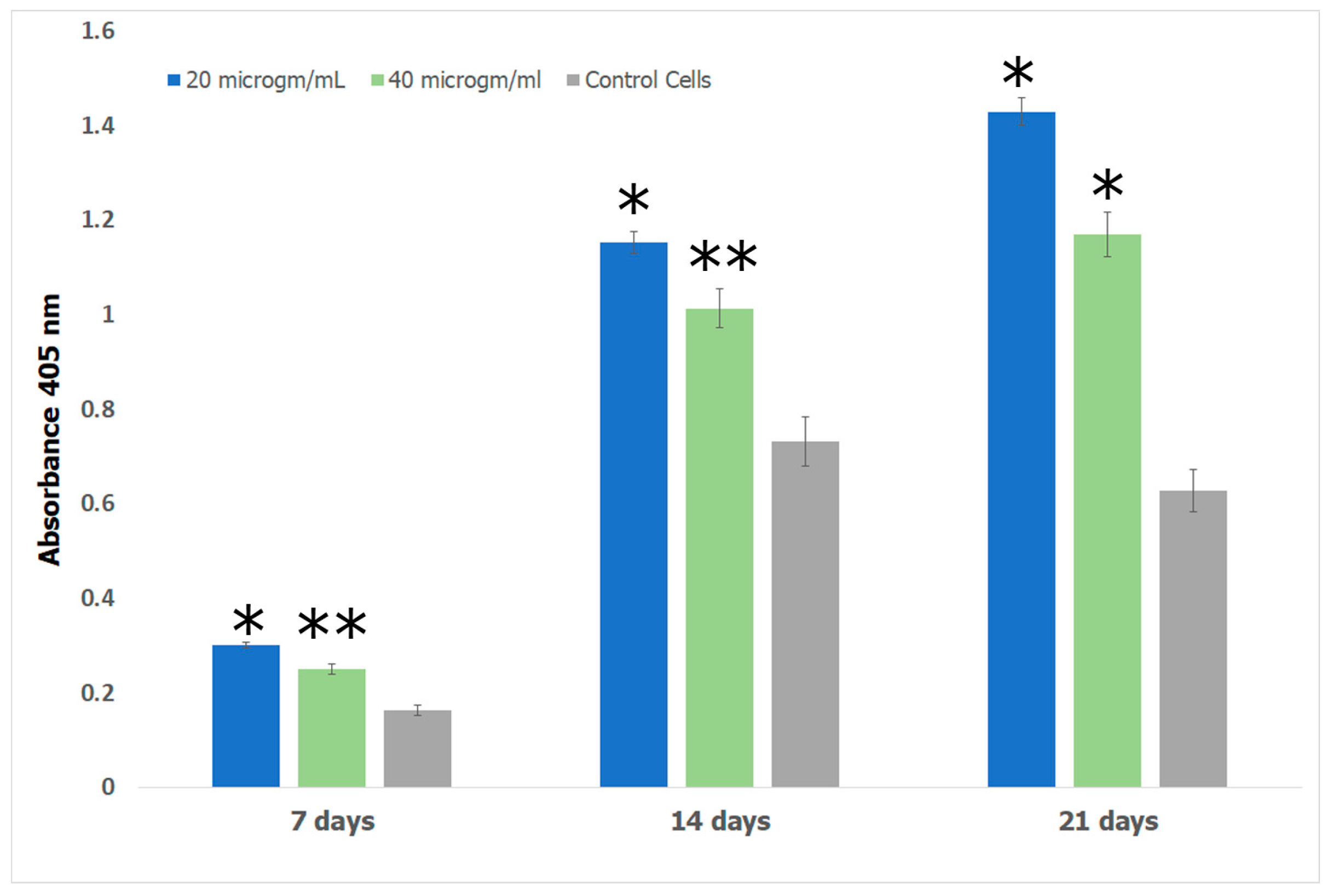
2.7.4. Alizarin Assay
3. Materials and Methods
3.1. Materials
3.2. Preparation of Scaffold
3.3. Characterization
3.3.1. Scanning Electron Microscopy (SEM)
3.3.2. Energy Dispersive X-ray Spectroscopy (EDS)
3.3.3. Fourier Transform Infrared (FTIR) Spectroscopy
3.3.4. Thermogravimetric Analysis
3.3.5. Differential Scanning Calorimetry (DSC)
3.3.6. X-ray Diffraction (XRD)
3.3.7. Atomic Force Microscopy
3.3.8. Rheology
3.4. Cell Studies
3.4.1. Cell Viability and Morphology Studies
SEM Imaging of Cells
3.4.2. Cytoskeletal Staining (Phalloidan Assay)
3.4.3. Alkaline Phosphatase Assay
3.4.4. Alizarin Assay
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Loi, F.; Córdova, L.A.; Pajarinen, J.; Lin, T.; Yao, Z.; Goodman, S.B. Inflammation, fracture and bone repair. Bone 2016, 86, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Uppuganti, S.; Granke, M.; Makowski, A.J.; Does, M.D.; Nyman, J.S. Age-related changes in the fracture resistance of male fischer F344 rat bone. Bone 2016, 83, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.A.; Manolagas, S.C. Effects of sex steroids on bones and muscles: Similarities, parallels, and putative interactions in health and disease. Bone 2015, 80, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.W.; Ulery, B.D.; Ashe, K.M.; Laurencin, C.T. Studies of bone morphogenetic protein based surgical repair. Adv. Drug Deliv. Rev. 2013, 64, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Finkemeier, C.G. Bone-grafting and bone-graft substitutes. J. Bone Jt. Surg. 2002, 84, 454–464. [Google Scholar] [CrossRef]
- Einhorn, T.A. Enhancement of fracture-healing. J. Bone Jt. Surg. 1995, 77, 940–956. [Google Scholar] [CrossRef]
- Gholizadeh, S.; Moztarzadeh, F.; Haghighipour, N.; Ghazizadeh, L.; Baghbani, F.; Shokrgozar, M.A.; Allahyari, Z. Preparation and characterization of novel functionalized multiwalled carbon nanotubes/chitosan/β-Glycerophosphate scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2017, 97, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Wahafu, T.; Jiang, G.; Liu, W.; Qiao, Y.; Peng, X.; Cheng, T.; Zhang, X.; He, G.; Liu, X. A novel open-porous magnesium scaffold with controllable microstructures and properties for bone regeneration. Sci. Rep. 2006, 6. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4829853/ (accessed on 15 September 2017). [CrossRef] [PubMed]
- Noori, A.; Ashrafi, S.J.; Vaez-Ghaemi, R.; Hatamian-Zaremi, A.; Webster, T.J. A review of fibrin and fibrin composites for bone tissue engineering. Int. J. Nanomed. 2017, 12, 4937–4961. [Google Scholar] [CrossRef] [PubMed]
- Sethu, S.N.; Namashivayam, S.; Devendran, S.; Nagarajan, S.; Tsai, W.B.; Narashiman, S.; Ramachandran, M.; Ambigapathi, M. Nanoceramics on Osteoblast Proliferation and Differentiation in Bone Tissue Engineering. Int. J. Biol. Macromol. 2017, 98, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.P.; Leong, K.W. Scaffolding in tissue engineering: General approaches and tissue-specific considerations. Eur. Spine J. 2008, 17, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Ao, C.; Niu, Y.; Zhang, X.; He, W.; Zhang, W.; Lu, C. Fabrication and characterization of electrospun cellulose/nano-hydroxyapatite nanofibers for bone tissue engineering. Int. J. Biol. Macromol. 2017, 97, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Unnithan, A.R.; Sasikala, A.R.; Park, C.H.; Kim, C.S. A unique scaffold for bone tissue engineering: An osteogenic combination of graphene oxide-hyaluronic acid-chitosan with simvastatin. J. Ind. Eng. Chem. 2017, 46, 182–191. [Google Scholar] [CrossRef]
- Chen, G.; Ushida, T.; Tateishi, T. Scaffold design for tissue engineering. Macromol. Biosci. 2008, 2, 67–77. [Google Scholar] [CrossRef]
- Amoabedin, G.; Salehi-Nik, N.; Heli, B. The role of biodegradable engineered scaffold in tissue engineering. In Biomaterials Sciences and Engineering; Pignatello, R., Ed.; InTech: Rijeka, Croatia, 2011; pp. 153–172. [Google Scholar]
- Thurner, P.J.; Chen, C.G.; Ionova-Martin, S.; Sun, L.; Harman, A.; Porter, A.; Ager, J.W.; Ritchie, R.O.; Alliston, T. Osteopontin deficiency increases bone fragility but preserves bone mass. Bone 2010, 46, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Khafaji, M.; Naji, M.; Vossoughi, M.; Alemzadeh, I.; Haghighpour, N. A biomimetic heparinized composite silk-based vascular scaffold with sustained antithrombogenicity. Sci. Rep. 2017, 7. Available online: http://www.nature.com/articles/s41598-017-04510-1 (accessed on 15 September 2017). [CrossRef] [PubMed]
- O’Brien, F.J. Biomaterials & scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Chang, Y.; Yang, S.T.; Liu, J.H.; Dong, E.; Wang, Y.; Cao, A.; Liu, Y.; Wang, H. In vitro toxicity evaluation of graphene oxide on A549 cells. Toxicol. Lett. 2011, 200, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Amini, A.R.; Laurencin, C.T.; Nukavarapu, S.P. Bone tissue engineering: Recent advances and challenges. Crit. Rev. Biomed. Eng. 2012, 40, 363–408. [Google Scholar] [CrossRef] [PubMed]
- Pistone, A.; Iannazzo, D.; Panseri, S.; Montesi, M.; Tampieri, A.; Galvagno, S. Hydroxyapatite-magnetite-MWCNT nanocompositre as a biocompatible drug delivery system for bone tissue engineering. Nanotechnology 2014, 25, 425701. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.X.; He, Y.; Bi, L.; Qu, Z.H.; Zou, J.W.; Pan, Z.; Fan, J.J.; Chen, L.; Dong, X.; Liu, X.N.; et al. Enhancing the bioactivity of poly(lactic-co-glycolic acid) scaffold with a nano-hydroxyapatite coating for the treatment of segmental bone defect in a rabbit model. Int. J. Nanomed. 2013, 8, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Pistone, A.; Iannazzo, D.; Espro, C.; Galvagno, S.; Tampieri, A.; Montesi, M.; Panseri, S.; Sandri, M. Tethering of Gly-Arg-Gly-Asp-Ser-Pro-Lys peptides on Mg-doped hydroxyapatite. Engineering 2017, 3, 55–59. [Google Scholar] [CrossRef]
- Sepulveda, P.; Jones, J.R.; Hench, L.L. In vitro dissolution of melt-derived 45S5 and sol-gel derived 58S bioactive glasses. J. Biomed. Res. 2002, 61, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Lakhkar, N.J.; Park, J.H.; Mordan, N.J.; Salih, V.; Wall, I.B.; Kim, H.W.; King, S.P.; Hanna, J.V.; Martin, R.A.; Addison, O.; et al. Titanium phosphate glass microspheres for bone tissue engineering. Acta Biomater. 2012, 11, 4181–4190. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Rahaman, M.N.; Bal, B.S.; Bonewald, L.F.; Kuroki, K.; Brown, R.F. Silicate, borosilicate, and borate bioactive glass scaffolds with controllable degradation rate for bone tissue engineering applications. II. In vitro and in vivo biological evaluation. J. Biomed. Mater. Res. A 2010, 95, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Mellonig, T.J. Bone allografts in periodontal therapy. Clin. Orthop. Relat. Res. 2006, 324, 116–125. [Google Scholar] [CrossRef]
- Canter, H.I.; Vargel, I.; Mavili, M.E. Reconstruction of mandibular defects using autografts combined with demineralized bone matrix and cancellous allograft. J. Craniofac. Surg. 2007, 18, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Rui, X.; Pan, H.; Shao, S.; Xu, X. Anti-tumor and anti-angiogenic effects of Fucoidan on prostate cancer: Possible JAK-STAT3 pathway. MBC Complement. Altern. Med. 2017, 17, 378. [Google Scholar] [CrossRef] [PubMed]
- Guangling, J.; Guangli, Y.; Junzeng, Z.; Ewart, H.S. Chemical Structures and bioactivites of sulfated polysachharides from marine algae. Mar. Drugs 2011, 9, 196–233. [Google Scholar] [CrossRef]
- Ale, M.T.; Mikkelsen, J.D.; Meyer, A.S. Important determinants for fucoidan bioactivity: A critical review of structure-function relations and extraction methods for fucose-containing sulfated polysachharides from brown seaweeds. Mar. Drugs 2011, 9, 2106–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, G.; Lee, H.; Shin, S.; Kim, H.; Jung, J. Anticancer effect of Fucoidan on DU-145 prostate cancer cells through inhibition of PI3K/Akt and MAPK pathway expression. Mar. Drugs 2016, 14, E126. [Google Scholar] [CrossRef] [PubMed]
- Venkatasan, J.; Bahtnagar, I.; Kim, S.K. Chitosan-alginate biocomposite containing fucoidan for bone tissue engineering. Mar. Drugs 2014, 12, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Subramanium, P.; Raghavendran, H.B.; Malliga, S.T.; Murali, R.; Mahmood, S.A.; Singh, S.; Kamarul, T. Incorporation of fucoidan in β-tricalcium phosphate-chitosan scaffold prompts the differentiation of human bone marrow stromal cells into osteogenic lineage. Nat. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Bogue, R. Conditions affecting the hydrolysis of collagen to gelatin. Ind. Eng. Chem. 1923, 15, 1154–1159. Available online: https://www.nature.com/articles/srep24202 (accessed on 15 September 2017). [CrossRef]
- Robinson, J.J. Comparative biochemical analysis of sea urchin perisotme and rat tail tendon collagen. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 117, 307–313. [Google Scholar] [CrossRef]
- Liu, X.H.; Ma, P.X. Phase separation, pore structure, and properties of nanofibrous gelatin scaffolds. Biomaterials 2009, 30, 4094–4103. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Suzuki, N. Isolation of collagen from fish waste material-skin, bone and fins. Food Chem. 2000, 68, 277–281. [Google Scholar] [CrossRef]
- Chuang, C.; Lin, R.; Tien, H.; Chu, Y.; Melero-Martin, J.; Chen, Y. Enzymatic regulation of functional vascular networks using gelatin hydrogels. Acta Biomater. 2015, 19, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Neffe, A.T.; Loebus, A.; Zaupa, A.; Stoetzel, C.; Müller, F.A.; Lendlein, A. Gelatin functionalization with tyrosine derived moieties to increase the interaction with hydroxyapatite fillers. Acta Biomater. 2011, 7, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.S.; Mano, J.F.; Reis, R.L. Potential applications of natural origin polymer-based systems in soft tissue regeneration. Crit. Rev. Biotechnol. 2010, 30, 200–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Fu, X.B. Naturally derived materials-based cell and drug delivery systems in skin regeneration. J. Control. Release 2010, 142, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.; Yates, K.; Vogt, C.; Qian, Z.; Frost, M.; Zhao, F. Increasing mechanical strength of gelatin hydrogels by divalent metal ion removal. Sci. Rep. 2014, 4. Available online: https://www.nature.com/articles/srep04706 (accessed on 15 September 2017). [CrossRef] [PubMed]
- Bode, F.; da Silva, M.A.; Drake, A.F.; Ross-Murphy, S.B.; Dreiss, C.A. Enzymatically cross-linked tilapia gelatin hydrogels: Physical, chemical, and hybrid networks. Biomacromolecules 2011, 12, 3741–3752. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.; de la Caba, K.; Eceiza, A.; Ruseckaite, R.; Mondragon, I. Enhancing water repellence and mechanical properties of gelatin films by tannin addition. Bioresour. Technol. 2010, 101, 6836–6842. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.; Bohidar, H.B. Systematic of alcohol-induced simple coacervation in aqueous gelatin solutions. Biomacromolecules 2003, 4, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.; Bohidar, H.B. Microscopic structure of gelatin coacervates. Int. J. Biol. Macromol. 2005, 36, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Lewit-Bentley, A.; Rety, S. EF-hand calcium binding proteins. Curr. Opin. Struct. Biol. 2000, 10, 637–643. [Google Scholar] [CrossRef]
- Jamalian, A.; Sneekes, E.J.; Dekker, L.J.M.; Ursem, M.; Luider, T.M.; Burgers, P.C. Dimerization of Peptides by Calcium Ions: Investigation of a Calcium-Binding Motif. Int. J. Proteom. 2014. Available online: https://www.hindawi.com/journals/ijpro/2014/153712/ (accessed on 15 September 2017). [CrossRef] [PubMed]
- Sanchez, C.; Arribart, H.; Guille, M. Biomimetism and bioinspiration as tools for the design of innovative materials and systems. Nat. Mater. 2005, 4, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Gungormus, M.; Fong, H.; Kim, W.; Evans, J.S.; Tamerler, C.; Sarikaya, M. Regulation of in vitro calcium phosphate mineralization by combainatorially selected hydroxyapatite-binding peptides. Biomacromolecules 2007, 9, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H. Chitin and collagen as universal and alternative templates in biomineralization. Int. Geol. Rev. 2010, 52, 661–669. [Google Scholar] [CrossRef]
- Hattan, S.J.; Laue, T.M.; Chasteen, N.D. Purification and characterization of a novel calcium-binding protein from the extrapallial fluid of the mollusk, Mytilus edulis. J. Biol. Chem. 2000, 276, 4461–4468. [Google Scholar] [CrossRef] [PubMed]
- Raj, P.A.; Johnsson, M.; Levine, M.J.; Nancollas, G.H. Salivary statherin: Dependence on sequence, charge, hydrogen bonding potency, and helical conformation for adsorption to hydroxyapatite and inhibition of mineralization. J. Biol. Chem. 1992, 267, 5968–5976. [Google Scholar] [PubMed]
- Sarikaya, M.; Tamerler, C.; Jen, A.K.; Schulten, K.; Baneyx, F. Molecular biomimetics: Nanotechnology through biology. Nat. Mater. 2003, 9, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, S.M.; Fath, K.R.; Phekoo, A.P.; Knoll, G.A.; Banerjee, I.A. Layer-by-layer assembly of peptide based bioorganic-inorganic hybrid scaffolds and their interactions with osteoblastic MC3T3-E1 cells. Mater. Sci. Eng. C 2015, 51, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Ghione, C.; Tonda-Turo, C.; Kalaskar, D.M. Peptide functionalisation of nanocomposite polymer for bone tissue engineering using plasma surface polymerization. RSC Adv. 2015, 5, 80039–80047. [Google Scholar] [CrossRef]
- Gentile, P.; Ferreiera, A.M.; Callaghan, J.T.; Miller, C.A.; Atkinson, J.; Freeman, C.; Hatton, P.V. Multilayer Nanoscale Encapsulation of Biofunctional Peptides to Enhance Bone Tissue Regeneration In Vivo. Adv. Healthc Mater. 2017, 6, 1601182. [Google Scholar] [CrossRef] [PubMed]
- Nonoyama, T.; Ogasawara, H.; Tanaka, M.; Higuchi, M.; Kinoshita, T. Calcium phosphate biomineralization in peptide hydrogels for injectable bone-filling materials. Soft Matter 2012, 8, 11531–11536. [Google Scholar] [CrossRef]
- Pawelec, K.M.; Kluijtmans, S.G.J.M. Biomineralization of Recombinatn Peptide Scaffolds; Interplay among Chemistry, Architecture, and Mechanics. ACS Biomater. Sci. Eng. 2017, 3, 1100–1108. [Google Scholar] [CrossRef]
- Lee, M.J.; Park, J.B.; Kim, H.H.; Ki, C.S.; Park, S.Y.; Kim, H.J.; Park, Y.H. Surface coating of hydroxyapatite on silk nanofiber through biomineralization using ten times concentrated simulated body fluid and the evaluation of bone regeneration. Macromol. Res. 2014, 22, 710–716. [Google Scholar] [CrossRef]
- Hamai, R.; Shirosaki, Y.; Miyazaki, T. Biomineralization behavior of a vinylphosphonic acid-based copolymer added with polymerization accelerator in simulated body fluid. J. Asian Ceram. Soc. 2015, 3, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Kokubo, T.; Kushitani, H.; Sakka, S.; Kitusugi, T.; Yamamuro, T. Solutions able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W. J. Biomed. Mater. Res. 1990, 24, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.A.; Oh, Y.S.; Park, H.J. Preparation and characterization of aminated Gelatin-Fucoidan microparticles. Korean J. Food Sci. Technol. 2012, 44, 191–195. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Panzavolta, S.; Roveri, N.; Rubini, K. Bonelike apatite growth on hydroxyapatite-gelatin sponges from simulated body fluid. J. Biomed. Res. 2002, 59, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Acri, T.; Geary, S.; Salem, A.K. Biomimetic Mineralization of Biomaterials Using Simulated Body Fluids for Bone Tissue Engineering and Regenerative Medicine. Tissue Eng. Part A 2017. accepted. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.; Saxena, G.; Sell, S.; Bowlin, G. Mineralization and characterization of composite lyophilized gelatin sponges intended for early bone regeneration. Bioengineering 2014, 1, 62–84. [Google Scholar] [CrossRef]
- Wei, G.; Ma, P.X. Macroporous and nanofibrous polymer scaffolds and polymer/bone-like apatite composite scaffolds generated by sugar spheres. J. Biomed. Mater. Res. 2006, 78A, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Tomoalia, G.; Pasca, R.D. On the Collagen Mineralization. A Review. Clujul Med. 2015, 88, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Zadpoor, A.A. Relationship between in vitro apatite-forming ability measured using simulated body fluid and in vivo bioactivity of biomaterials. Mater. Sci. Eng. C 2014, 35, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ma, P.X. Biomimetic polymer/apatite composite scaffolds for mineralized tissue engineering. Macromol. Biosci. 2004, 4, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Goonoo, N.; Bhaw-Luximon, A.; Passanha, P.; Esteves, S. Biomineralization potential and cellular response of PHB and PHBV blends with natural anionic polysachharides. Mater. Sci. Eng. C 2017, 76, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Max, J.J.; Chapados, C. Glucose and fructose hydrates in aqueous solutions by IR spectroscopy. J. Phys. Chem. A 2007, 111, 2679–2689. [Google Scholar] [CrossRef] [PubMed]
- Kanou, M.; Kameoka, T.; Suehara, K.I.; Hashimoto, A. Mid-infrared spectorscopic analysis of saccharides in aqueous solutions with sodium chloride. Biosci. Biotechnol. Biochem. 2017, 81, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Macquarrie, D. Microwave assisted extraction of sulfated polysachhraides (fucoidan) from Ascophyllum nodosum and its antioxidant activity. Carbohydr. Polym. 2015, 129, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Figueriro, S.D.; Macedo, A.A.M.; Melo, M.R.S.; Freitas, A.L.P.; Moreira, R.A.; de Oliveria, R.S.; Goes, J.C.; Sombra, A.S.B. On the dielectric behavior of collagen-algal sulfated polysaccharide blends: Effect of glutaraldehdye crosslinking. Biophys. Chem. 2006, 120, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Sinurat, E.; Rosmawaty, P.; Saepudin, E. Characterization of fucoidan extracted from Binuangeun’s Brown Seaweeds. Int. J. Chem. Environ. Biol. Sci. 2015, 3, 329–332. [Google Scholar]
- Meejoo, S.; Maeeprakorn, W.; Winotai, P. Phase and thermal stability of nanocrystalline hydroxyapatite prepared via microwave heating. Thermochim. Acta 2006, 447, 115–120. [Google Scholar] [CrossRef]
- Destainville, A.; Champion, E.; Bernache-Assollante, D.; Laborde, E. Synthesis, characterization and thermal behaviour of apatite tricalcium phosphate. Mater. Chem. Phys. 2003, 80, 269–277. [Google Scholar] [CrossRef]
- Han, J.K.; Song, H.Y.; Saito, F.; Lee, B.T. Synthesis of high purity nano-sized hydroxyapaptite powder by microwave hydrothermal method. Mater. Chem. Phys. 2006, 99, 235–239. [Google Scholar] [CrossRef]
- Ishikawa, K.; Ducheyene, P.; Radin, S. Determinaion of Ca/P ratio in calcium-deficient hydroxyapatite using X-ray diffraction analysis. J. Mater. Sci. Mater. Med. 1993, 4, 165–168. [Google Scholar] [CrossRef]
- Serra, I.R.; Fradique, R.; Vallejo, M.C.S.; Correia, T.R.; Miguel, S.P.; Correia, I.J. Production and characterization of chitosan/gelatin/β-TCP scaffolds for improved bone tissue regeneration. Mater. Sci. Eng. C 2015, 55, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Gallinetti, S.; Canal, C.; Ginebra, M.P. Development and Characterization of Biphasic Hydroxyapatite-β-TCP cements. J. Am. Ceram. Soc. 2014, 97, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Venkatesan, J.; Kim, S.K. Hydroxyapatite-fucoidan nanocomposites for bone tissue engineering. Int. J. Biol. Macromol. 2013, 57, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Barreto, P.L.M.; Pires, A.T.N.; Soldi, V. Thermal degradation of edible films based on milk proteins and gelatin in inert atmosphere. Polym. Degrad. Stab. 2003, 79, 147–152. [Google Scholar] [CrossRef]
- Perumal, R.K.; Perumal, S.; Ramar, T.; Gopinath, A.; Ramadass, S.K.; Balaraman, M.; Sivasubramanian, S. Collagen-fucoidan blend film with the potential to induce fibroblast proliferation for regenerative applications. Int. J. Biol. Macromol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.S.; Wang, J.; Pontoon, C.B.; Marquis, P.M. An improvement in processing of hydroxyapatite ceramics. J. Mater. Sci. 1995, 30, 3061–3074. [Google Scholar] [CrossRef]
- Lee, J.; Yun, H.S. Hydroxyapatite-containing gelatin/chitosan microspheres for controlled release of lysozyme and enhanced cytocompatibility. J. Mater. Chem. B 2014, 2, 1255–1263. [Google Scholar] [CrossRef]
- Liji Sobhana, S.S.; Sundaraseelan, J.; Sekar, S.; Shastry, T.P.; Mandal, A.B. Gelatin-Chitosan composite capped gold nanoparticles a matrix for the growth of hydroxyapatite. J. Nanopart. Res. 2009, 11, 333–340. [Google Scholar] [CrossRef]
- Wang, S.; Hu, Y.; Uzoejinwa, B.B.; Cao, B.; He, Z.; Wang, Q.; Xu, S. Pyrolysis mechanisms of typical seaweed polysaccharides. J. Anal. Appl. Pyrolysis 2017, 124, 373–383. [Google Scholar] [CrossRef]
- Xu, X.; Xue, C.; Chang, Y.; Wang, J.; Jiang, K. Chain conformational and physicochemical properties of fucoidans from sea cucumber. Carbohydr. Polym. 2016, 152, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Z.M.; Xu, X.; Lim, C.T.; Ramkrishna, S. Preparation of core-shell structure PCL-r-gelatin bi-component nanofibers by coaxial electrospinning. Chem. Mater. 2004, 15, 3406–3409. [Google Scholar] [CrossRef]
- Zhang, F.; Song, Q.; Huang, X.; Li, F.; Wang, K.; Tang, Y.; Hou, C.; Shen, H. A novel high mechanical property PLGA composite matrix loaded with nanodiamond-phospholipid compound for bone tissue engineering. ACS Appl. Mater. Interfaces 2016, 8, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, T.N.; Zhang, J.; Ovaert, T.C.; Roeder, R.K.; Biebur, G.L. Direct comparison of nanoindentation and macroscopic measurements of bone viscosity. J. Mech. Behav. Biomed. Mater. 2011, 4, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Hengsberger, S.; Kulik, A.; Zysset, P. A combined atomic force microscopy and nanoindentation technique to investigate the elastic properties of bone structural units. Eur. Cells Mater. 2001, 12–17. [Google Scholar] [CrossRef]
- Dinis, J.C.; Moraes, T.F.; Amorim, P.H.; Moreno, M.R.; Nunes, A.A.; Silva, J.V. POMES: An open-source software tool to generate porous/roughness on surface. Proced. CIRP 2016, 49, 178–182. [Google Scholar] [CrossRef]
- Brett, P.M.; Harle, J.; Salih, V.; Mihoc, R.; Olsen, I.; Jones, F.; Tonetti, M. Roughness response genes in osteoblasts. Bone 2004, 35, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Sammons, R.L.; Lumbikanonda, N.; Gross, M.; Cantzler, P. Comparison of osteoblast spreading on microstructured dental implant surfaces and cell behaviour in an explant model of osseointegration. Clin. Oral Implants Res. 2005, 16, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Tako, M. Rheological characteristics of fucoidan isolated from commercially cultured Cladosiphon Okamuranus. Bot. Mater. 2003, 46, 465. [Google Scholar] [CrossRef]
- Gisler, T.; Ball, R.C.; Weitz, D.A. Strain Hardening of fractal colloidal gels. Phys. Rev. Lett. 1999, 82, 1064–1067. [Google Scholar] [CrossRef]
- Shin, Y.C.; Lee, J.H.; Jin, O.S.; Kang, S.H.; Hong, S.W.; Kim, B.; Park, J.; Han, D. Synergistic effects of reduced graphene oxide and hydroxyapatite on osteogenic differentiation of MC3T3-E1 preosteoblasts. Carbon 2015, 95, 1051–1060. [Google Scholar] [CrossRef]
- Chen, Z.; Klein, T.; Murray, R.Z.; Crawford, R.; Chang, J.; Wu, C.; Xiao, Y. Osteoimmunomodulation for the development of advanced bone biomaterials. Mater. Today 2016, 19, 304–321. [Google Scholar] [CrossRef]
- Hotulainen, P.; Lappalainen, P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 2006, 173, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Fath, K.; Kelly, T.; Nuckolls, G.; Turner, C. Focal adhesions: Transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu. Rev. Cell Dev. Biol. 1988, 4, 487–525. [Google Scholar] [CrossRef] [PubMed]
- Galow, A.M.; Rebl, A.; Koczan, D.; Bonk, S.M.; Baumann, W.; Gimsa, J. Increased osteoblast viability at alkaline pH in vitro provides a new perspective on bone regeneration. Biochem. Biophys. Rep. 2017, 10, 17–25. [Google Scholar] [CrossRef]
- Nijhuis, A.W.; van den Beucken, J.J.; Jansen, J.A.; Leeuwenburgh, S.C. In vitro response to alkaline phosphatase coatings immobilized onto titanium implants using electrospray deposition or polydopamine-assisted deposition. J. Biomed. Mater. Res. Part A 2014, 102, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Zioupos, P.; Currey, J.D.; Hamer, A.J. The role of collagen in the declining mechanical properties of aging human cortical bone. J. Biomed. Mater. Res. 1999, 45, 108–116. [Google Scholar] [CrossRef]
- Thyberg, J. Electron microscopic studies on the initial phases of calcification in guinea pig epiphyseal cartilage. J. Ultrastruct. Res. 1974, 46, 206–216. [Google Scholar] [CrossRef]
- Wu, L.N.; Ishikawa, Y.; Sauer, G.R.; Genge, B.R.; Mwale, F.; Mishima, H.; Wuthier, R.E. Morphological and biochemical characterization of mineralizing primary cultures of avian growth plate chondrocytes: Evidence for cellular processing of Ca2+ and Pi prior to matrix mineralization. J. Cell. Biochem. 1995, 57, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, J.E.; Lee, J.H.; Jeong, J.H.; Kim, J. A biodegradation study of SBA-15 microparticles in simulated body fluid. Langmuir 2015, 31, 6457–6462. [Google Scholar] [CrossRef] [PubMed]
- Barak, L.S.; Yocum, R.R.; Nothnagel, E.A.; Webb, W.W. Fluorescence staining of the actin cytoskeleton in living cells with 7-nitrobenz-2-oxa-1,3-diazole-phallacidin. Proc. Natl. Acad. Sci. USA 1980, 77, 980–984. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajovich, H.T.; Banerjee, I.A. Biomineralization of Fucoidan-Peptide Blends and Their Potential Applications in Bone Tissue Regeneration. J. Funct. Biomater. 2017, 8, 41. https://doi.org/10.3390/jfb8030041
Pajovich HT, Banerjee IA. Biomineralization of Fucoidan-Peptide Blends and Their Potential Applications in Bone Tissue Regeneration. Journal of Functional Biomaterials. 2017; 8(3):41. https://doi.org/10.3390/jfb8030041
Chicago/Turabian StylePajovich, Harrison T., and Ipsita A. Banerjee. 2017. "Biomineralization of Fucoidan-Peptide Blends and Their Potential Applications in Bone Tissue Regeneration" Journal of Functional Biomaterials 8, no. 3: 41. https://doi.org/10.3390/jfb8030041
APA StylePajovich, H. T., & Banerjee, I. A. (2017). Biomineralization of Fucoidan-Peptide Blends and Their Potential Applications in Bone Tissue Regeneration. Journal of Functional Biomaterials, 8(3), 41. https://doi.org/10.3390/jfb8030041