

A Review on the Different Aspects and Challenges of the Dry Reforming of Methane (DRM) Reaction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
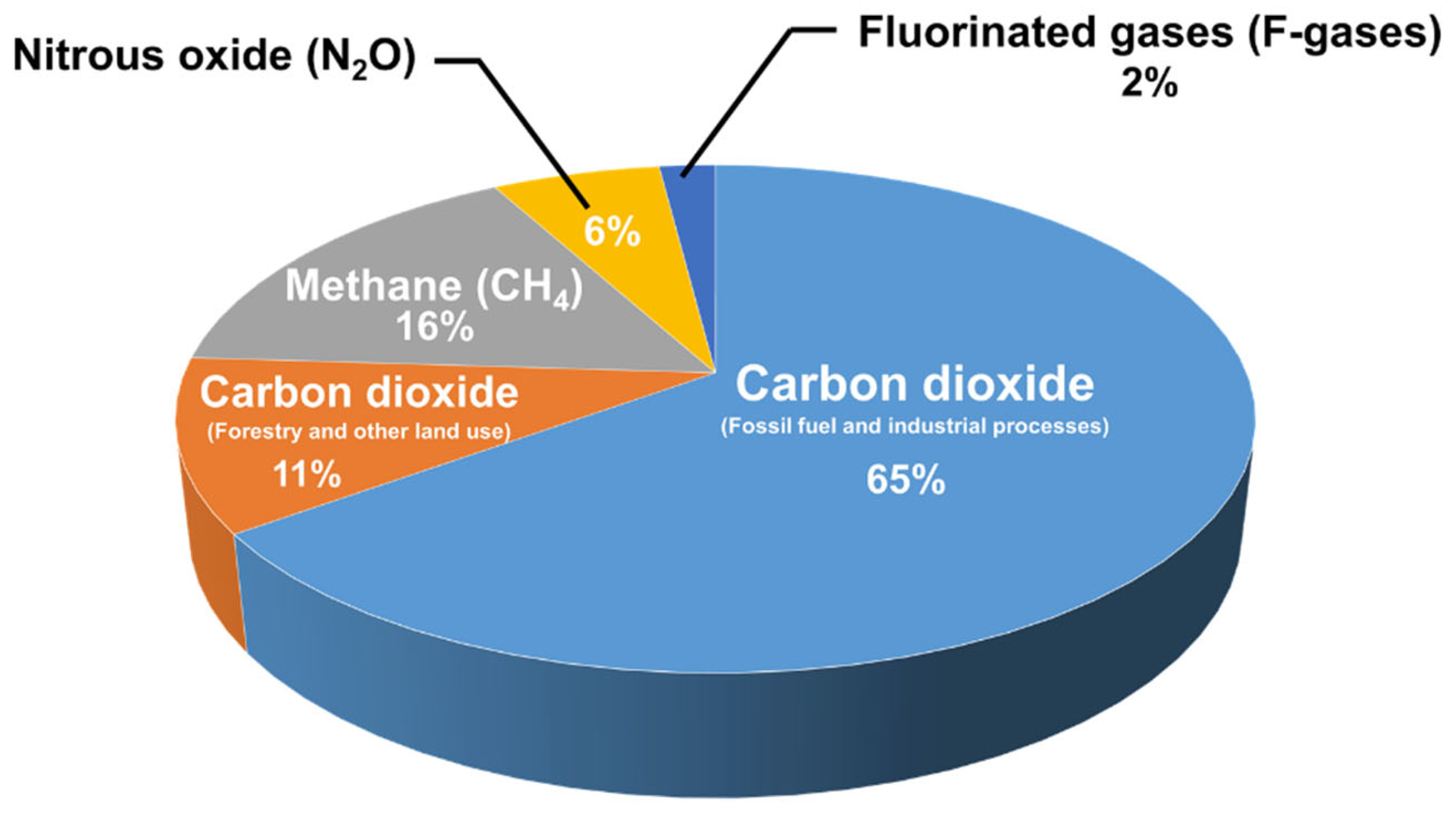
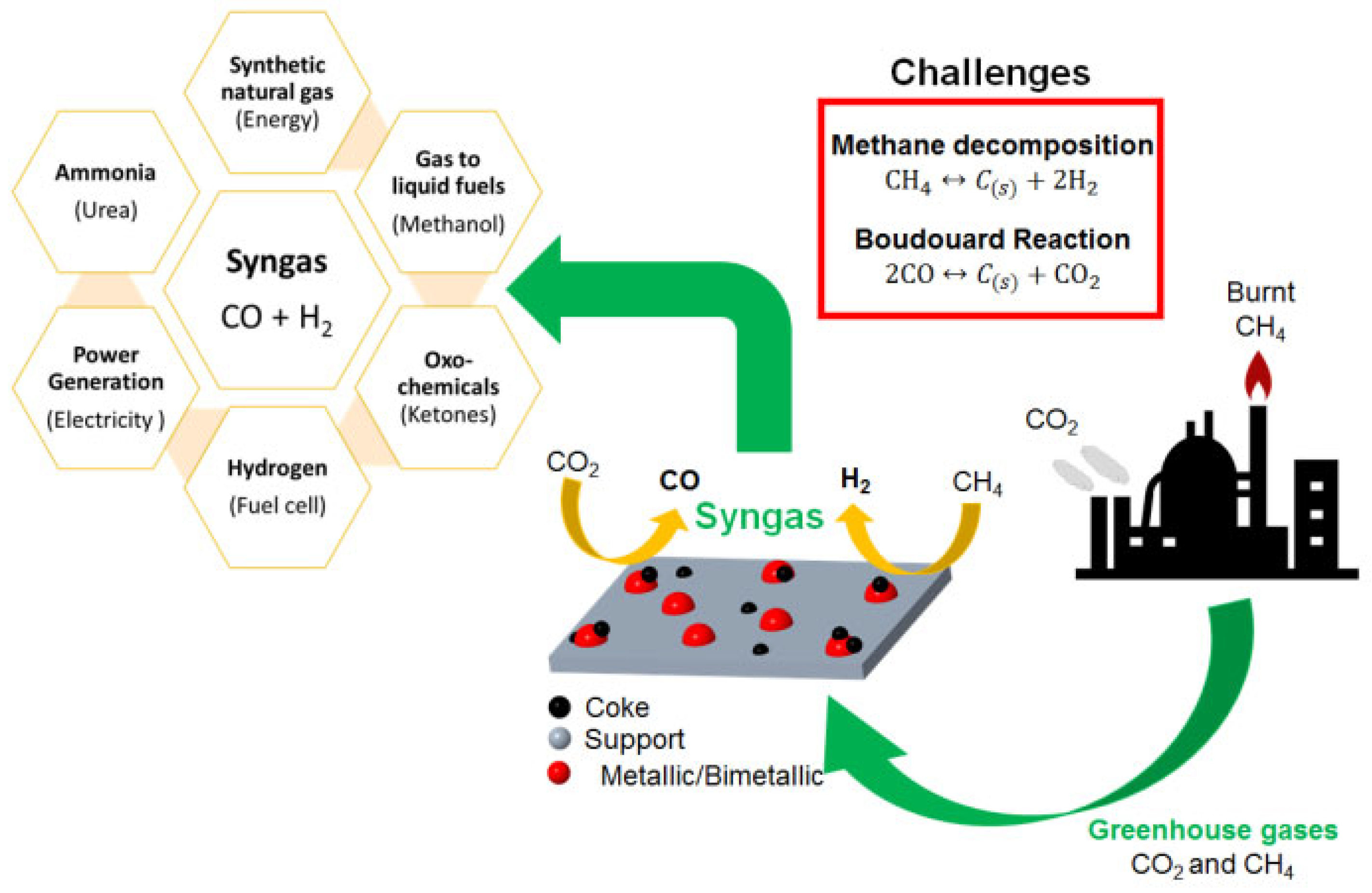
1. Introduction
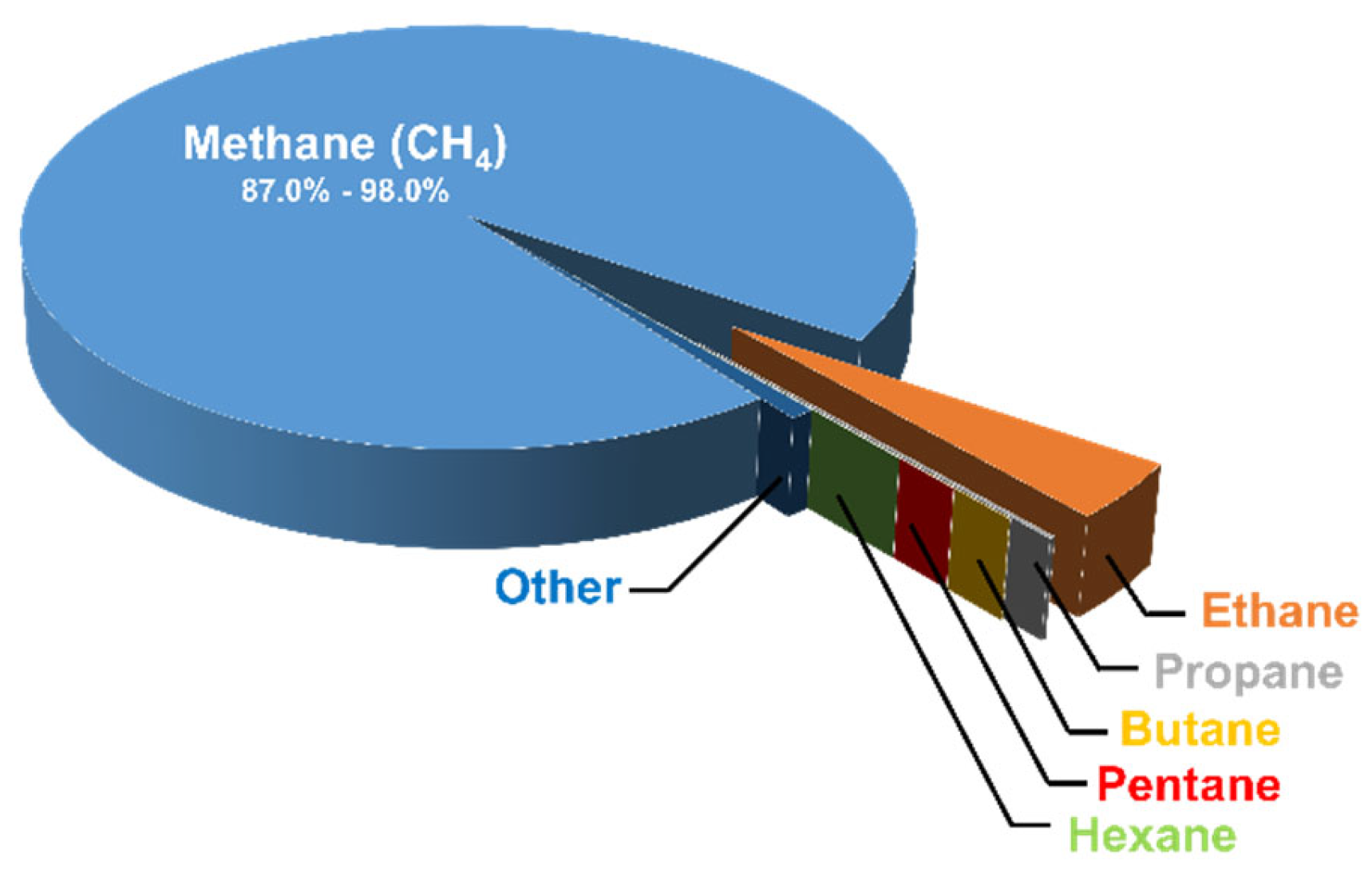
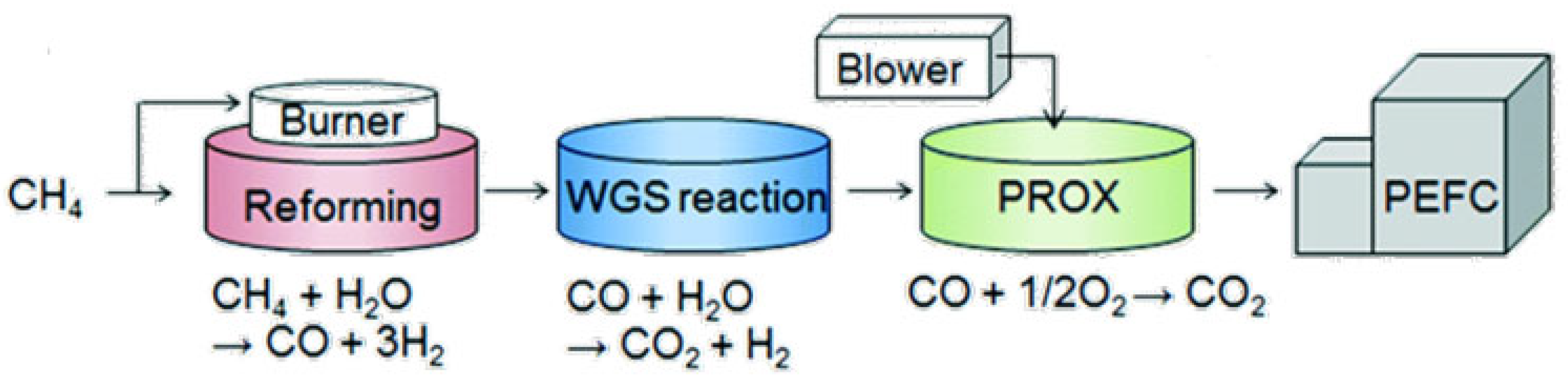
2. Natural Gas and Reforming Technologies for Syngas Production
2.1. Steam Reforming
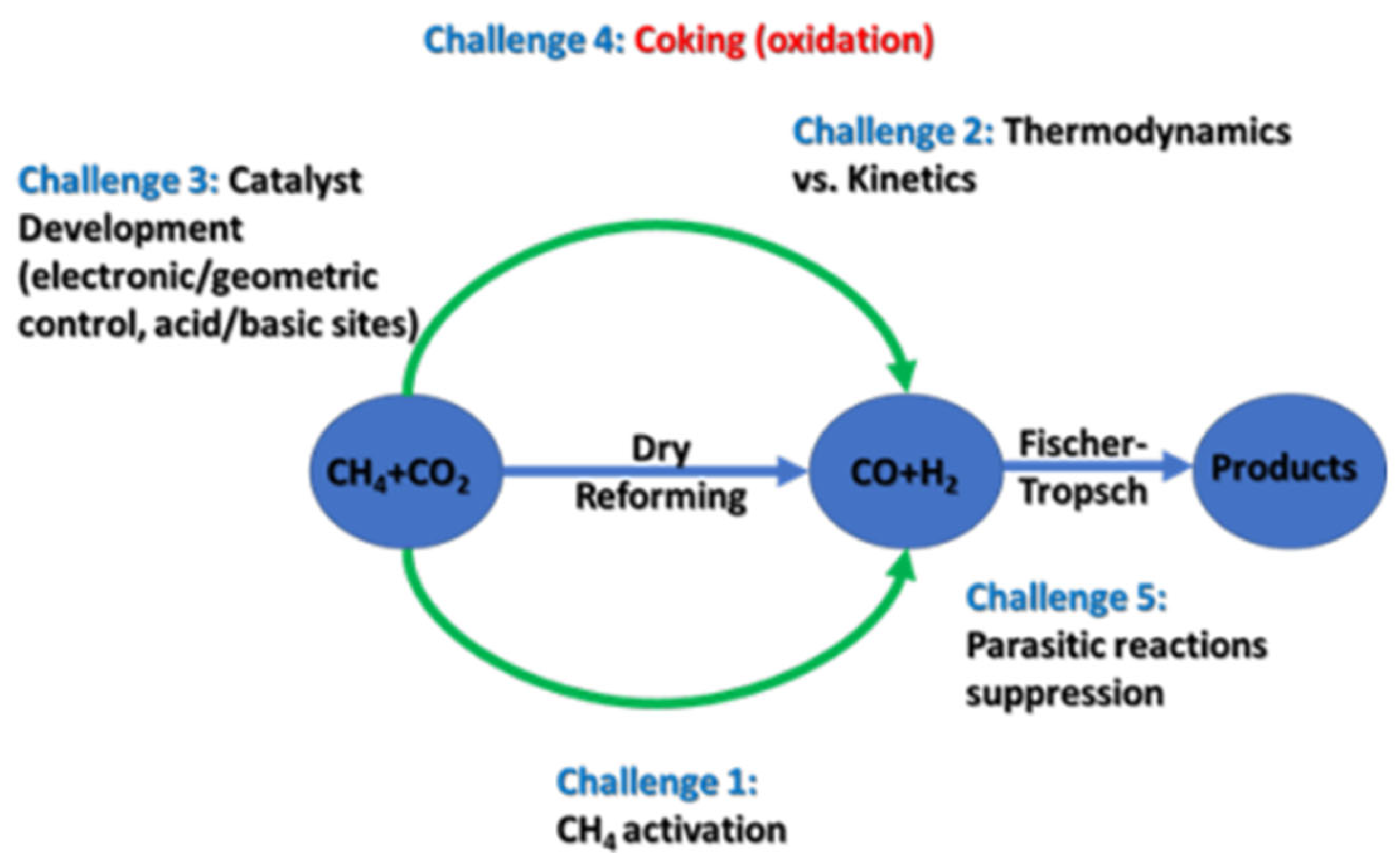
2.2. Dry Reforming of Methane (DRM)
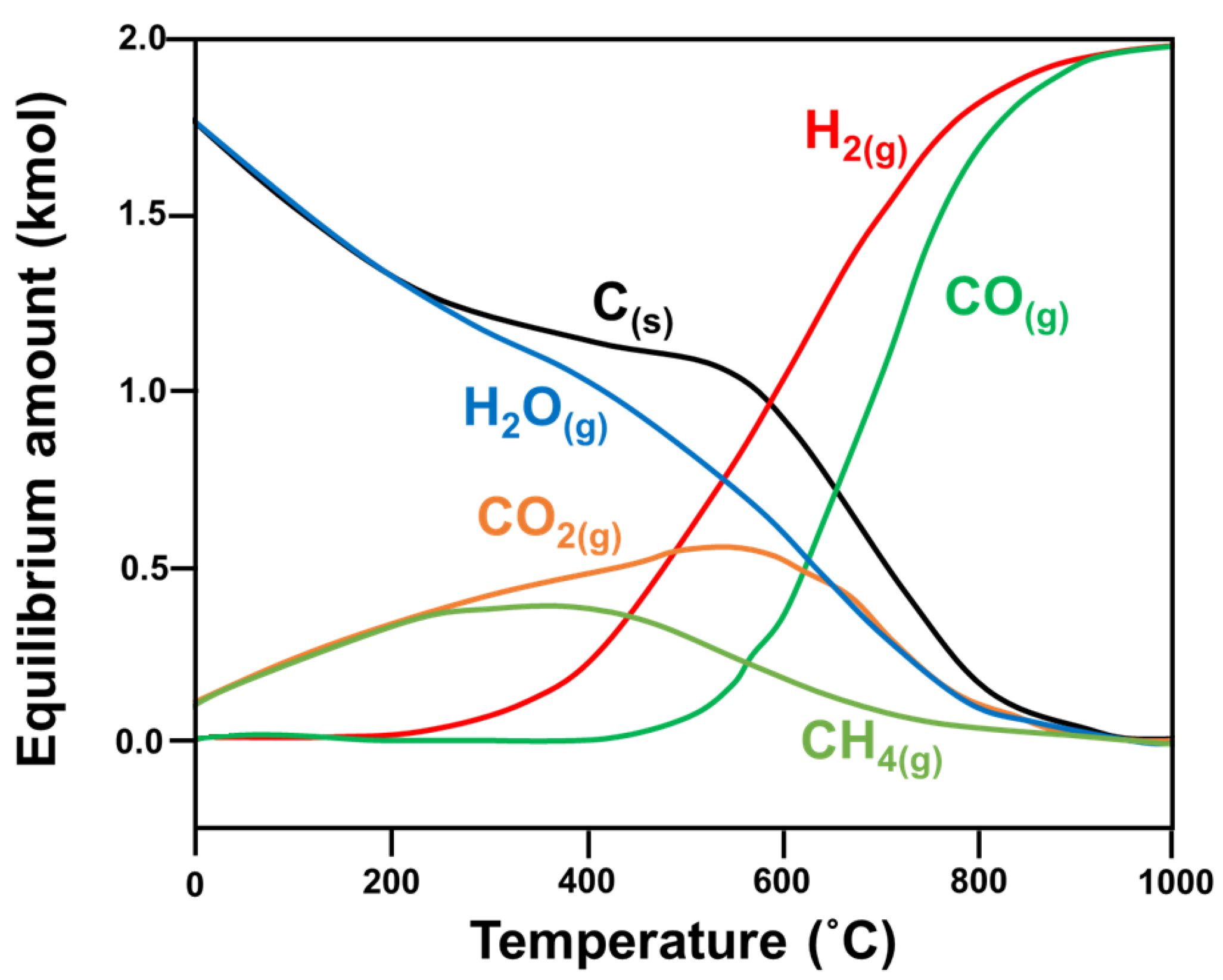
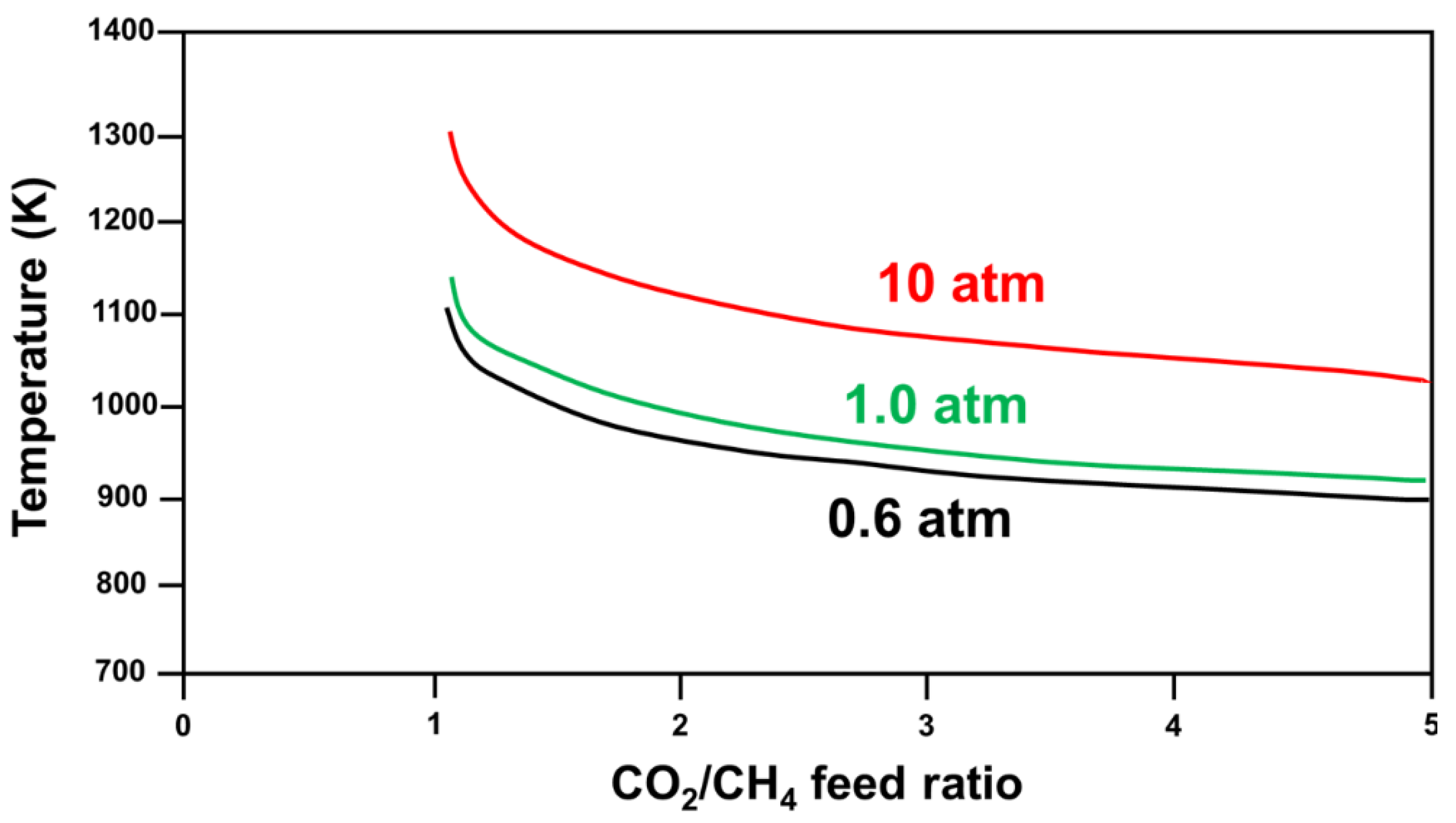
2.2.1. Brief on DRM Thermodynamics
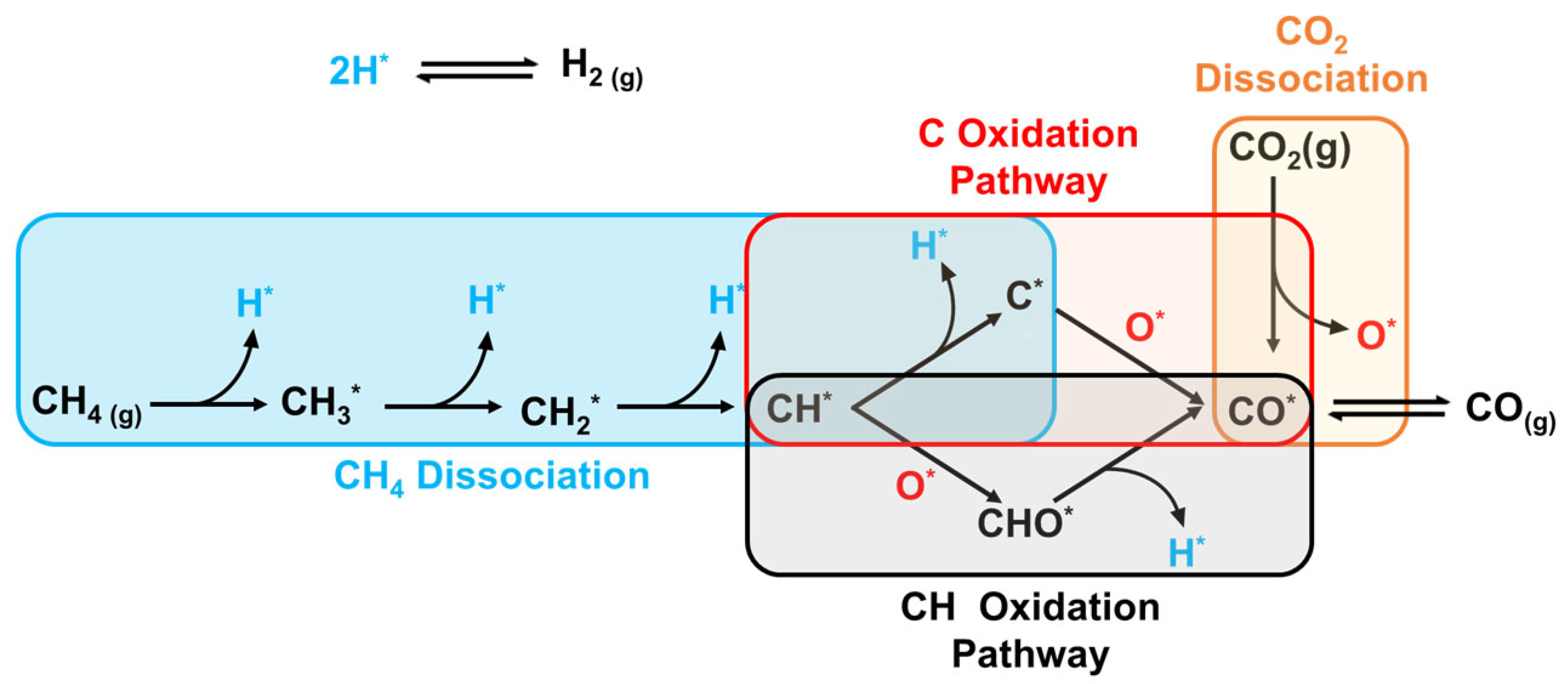
2.2.2. Brief on DRM Kinetics
2.3. Partial Oxidation
2.4. Autothermal Reforming
2.5. Critical Comparison of the Different Reforming Technologies
3. Challenges in DRM Technology
3.1. Metal Sintering
3.2. Coking
3.2.1. Metal Sites and Their Impact on Coking Formation
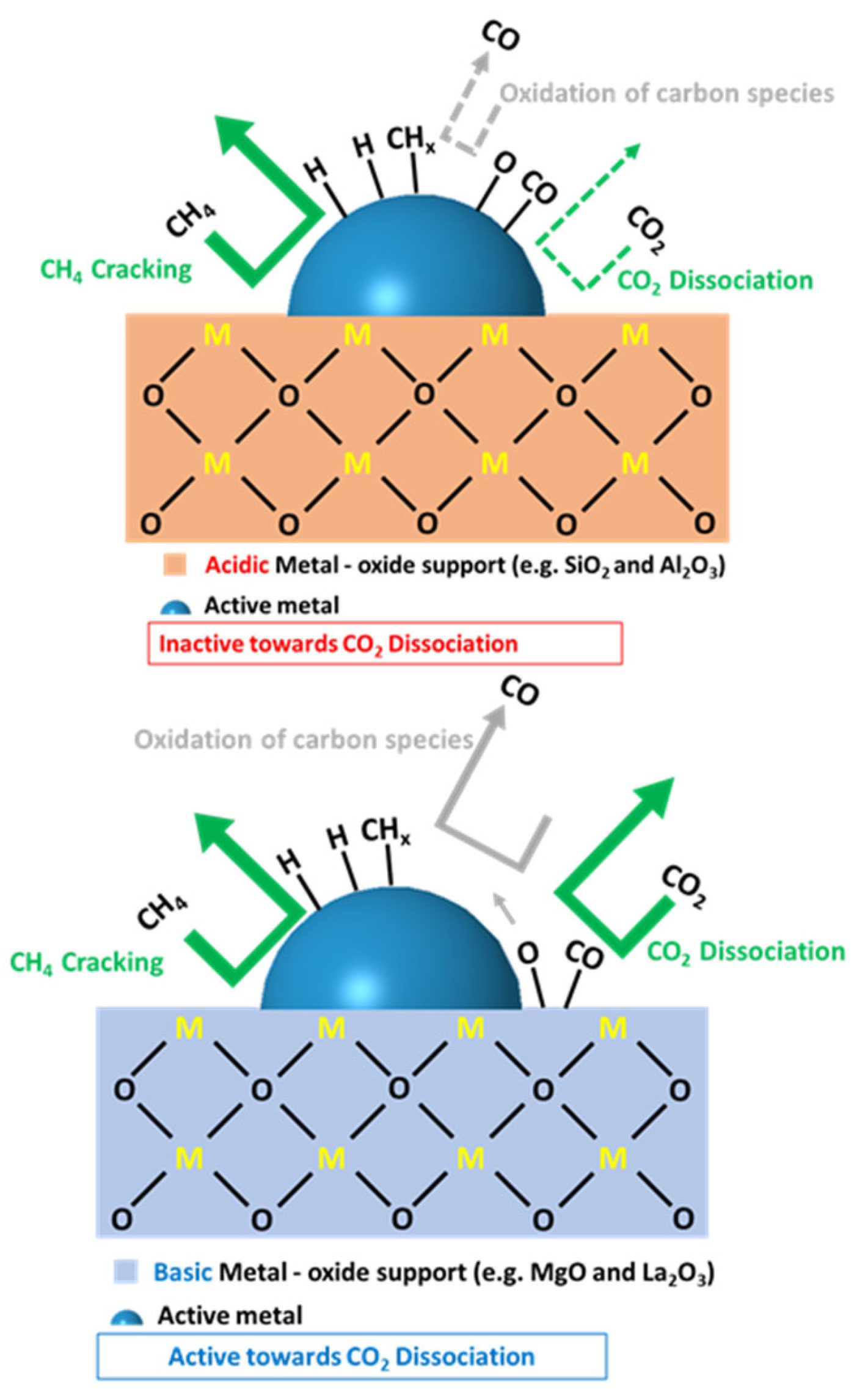
3.2.2. Support Effect
3.2.3. Types of Carbon Formed
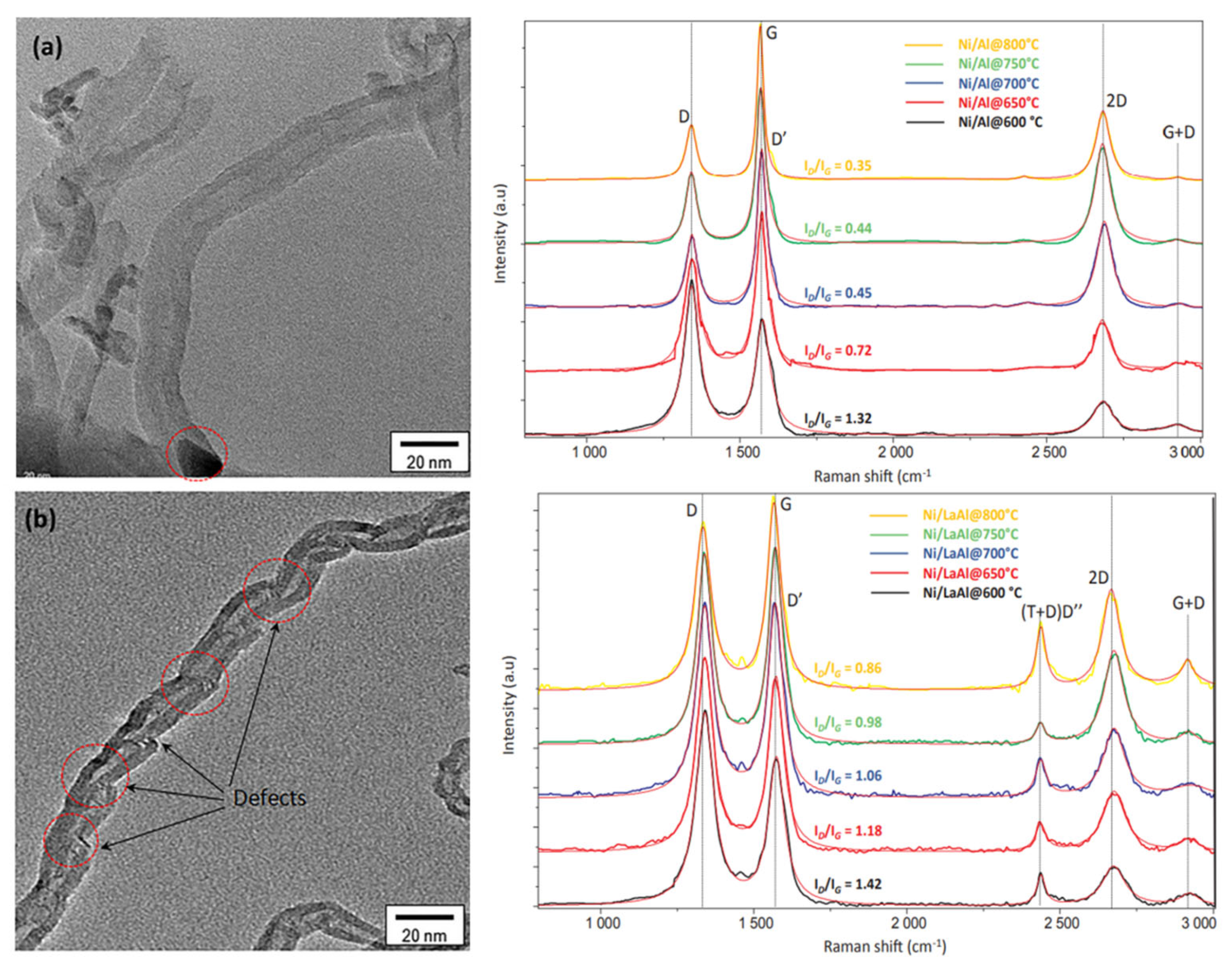
3.2.4. Characterization of Coking
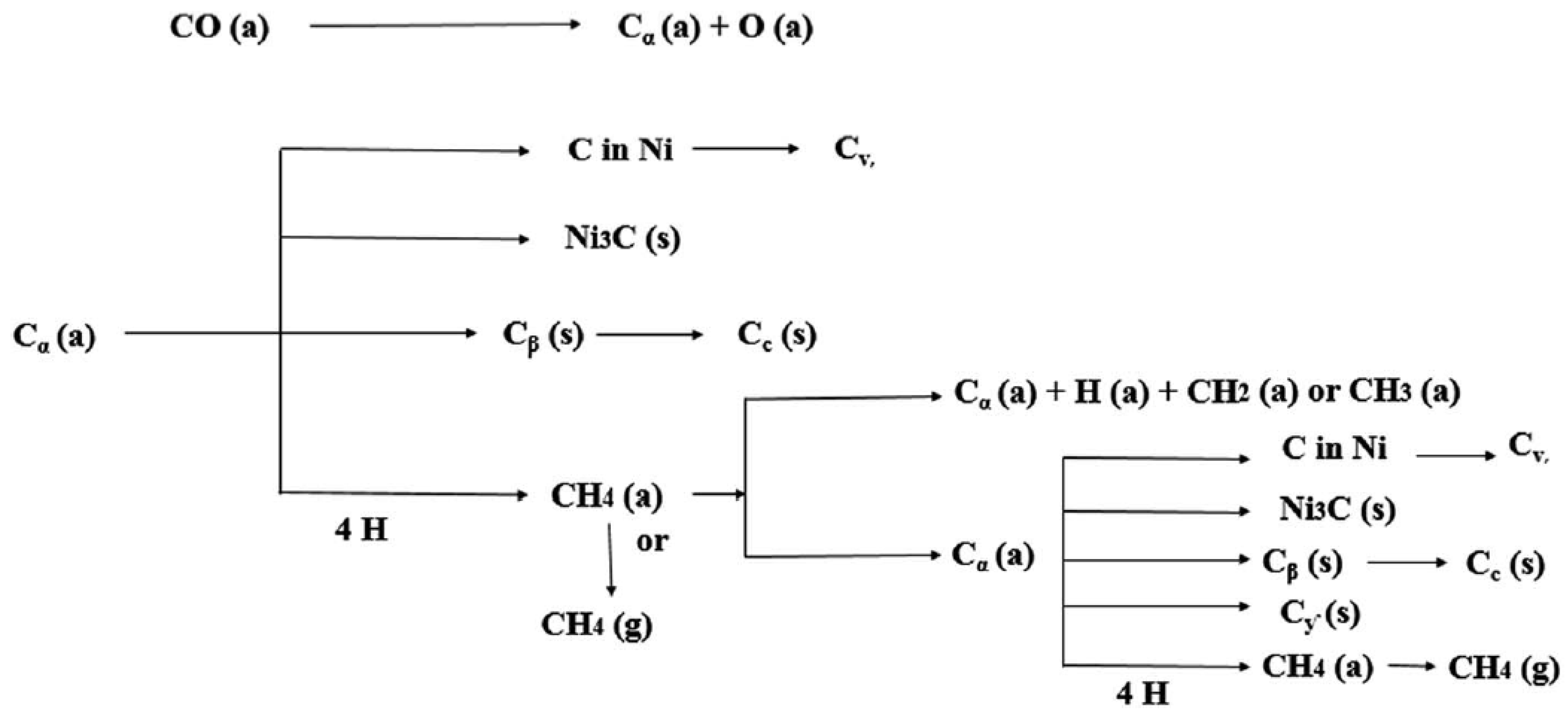
3.2.5. SSITKA-DRIFTS as a Tool to Track the Coking Pathways
3.2.6. Regeneration of Spent Catalyst
4. Learning-Driven Design of DRM Catalysts
4.1. Noble Metals: Their Role in Controlling the Coking
4.2. Non-Noble-Metal (Ni-Based) Catalysts
4.3. Other Transition Metal Catalysts
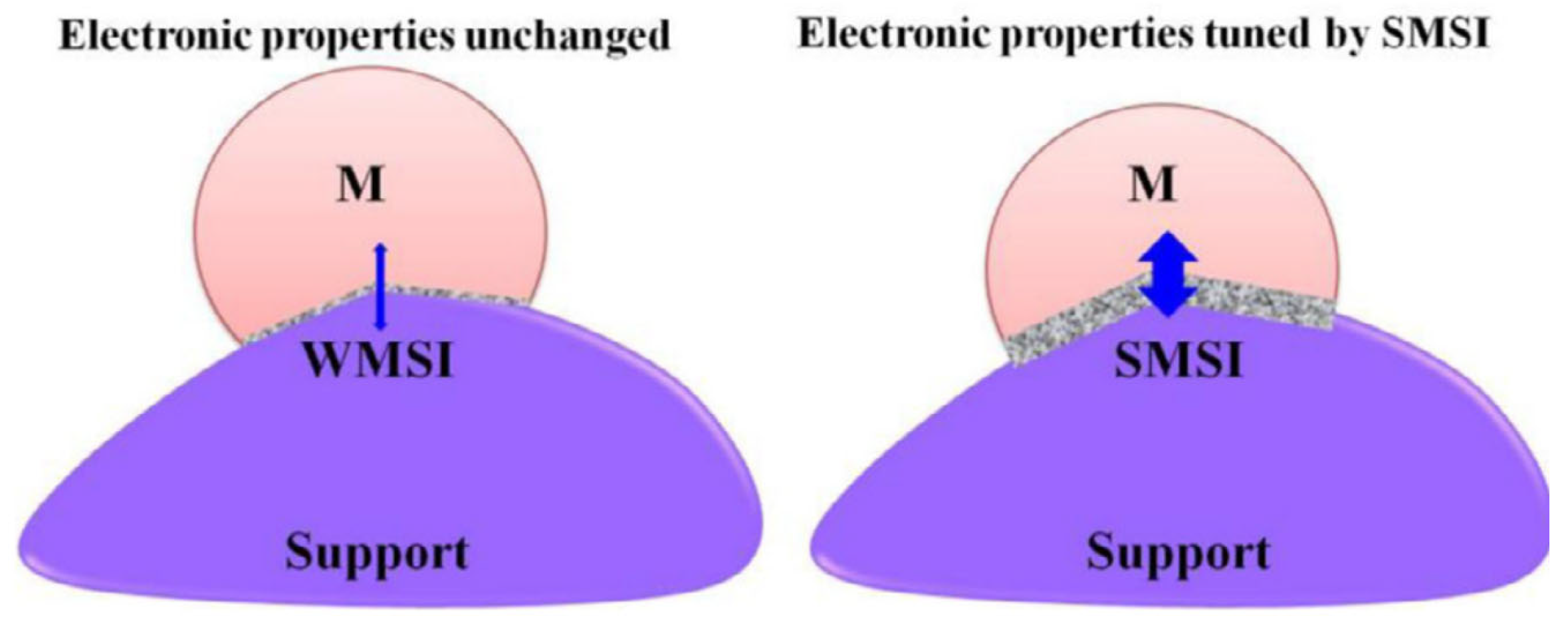
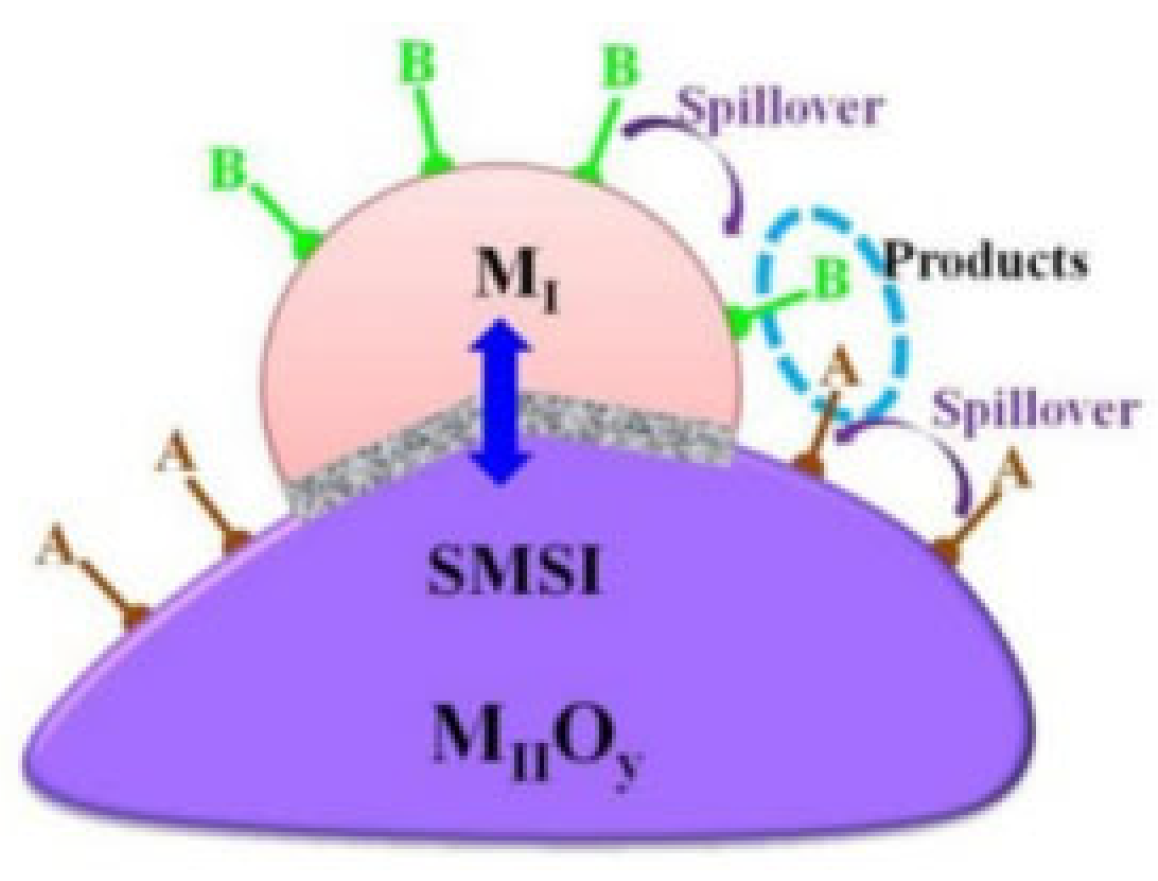
4.4. Strong Metal–Support Interaction (SMSI)
4.4.1. Electronic Effect
4.4.2. Geometric Effect
4.4.3. Bifunctional Effect
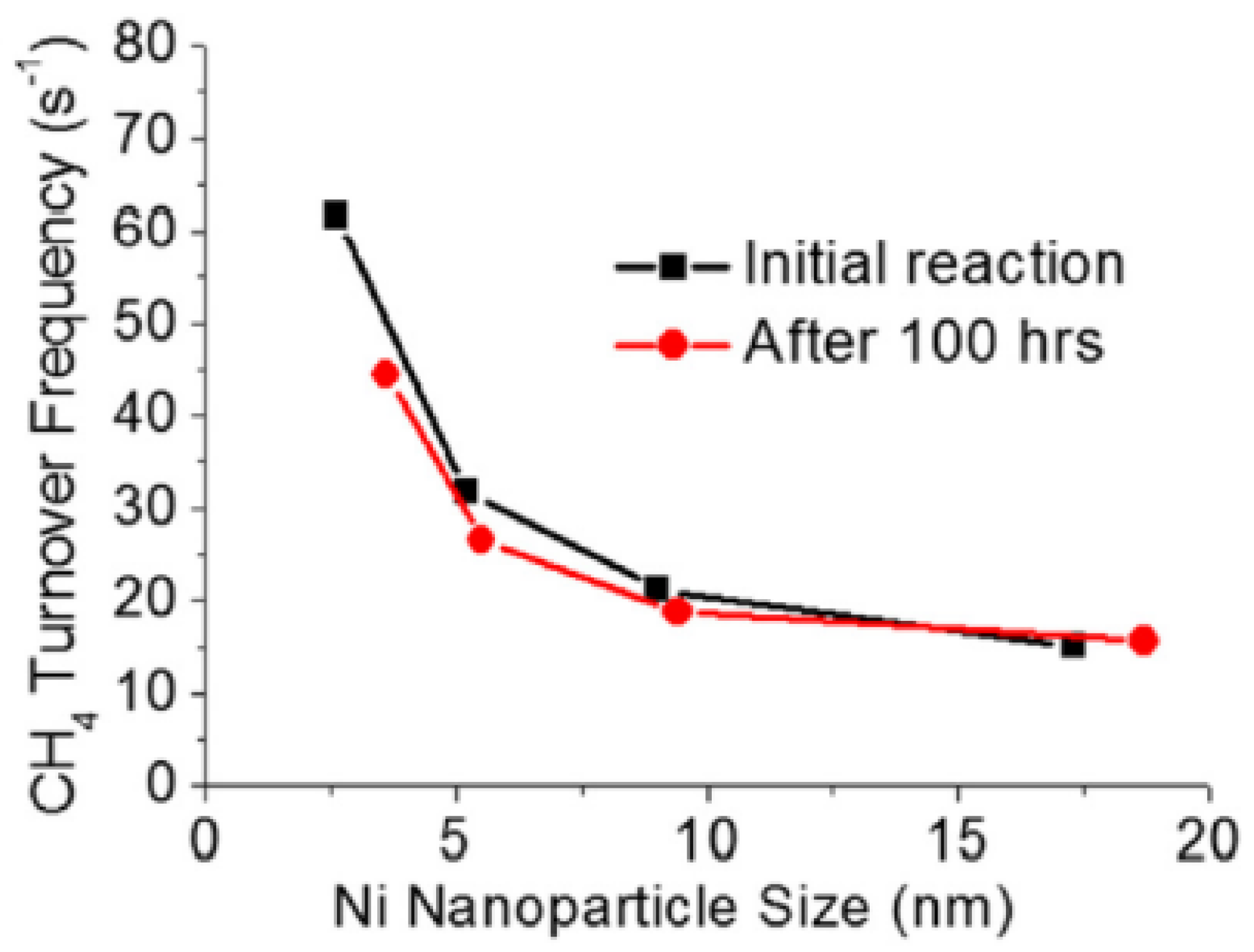
4.5. Metal Dispersion and Carbon Formation
5. Support Types for DRM Reaction
5.1. Metal Oxide Supports: Intrinsic Characteristics
Labile Oxygen Species
5.2. Zeolites
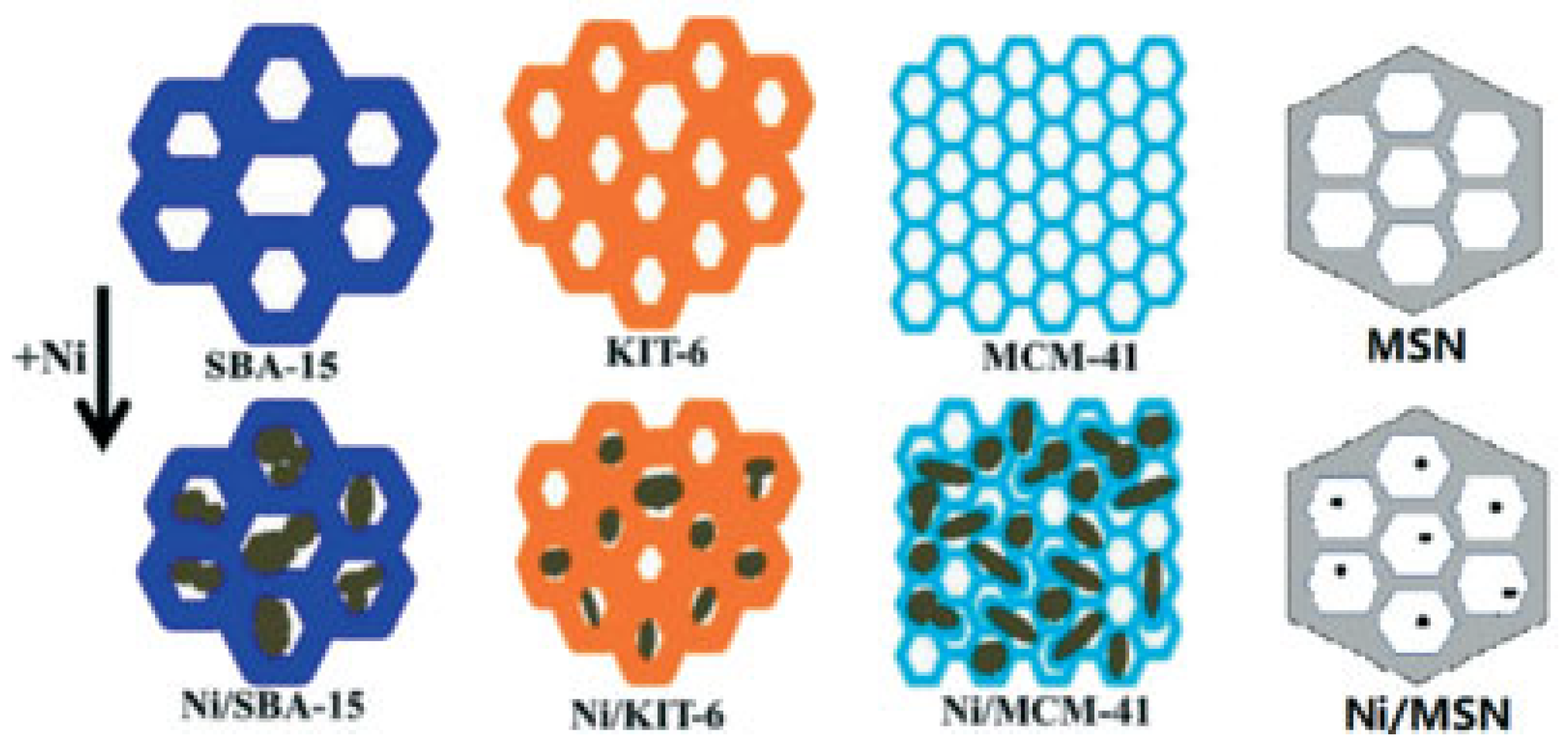
5.3. Mesoporous Silicas

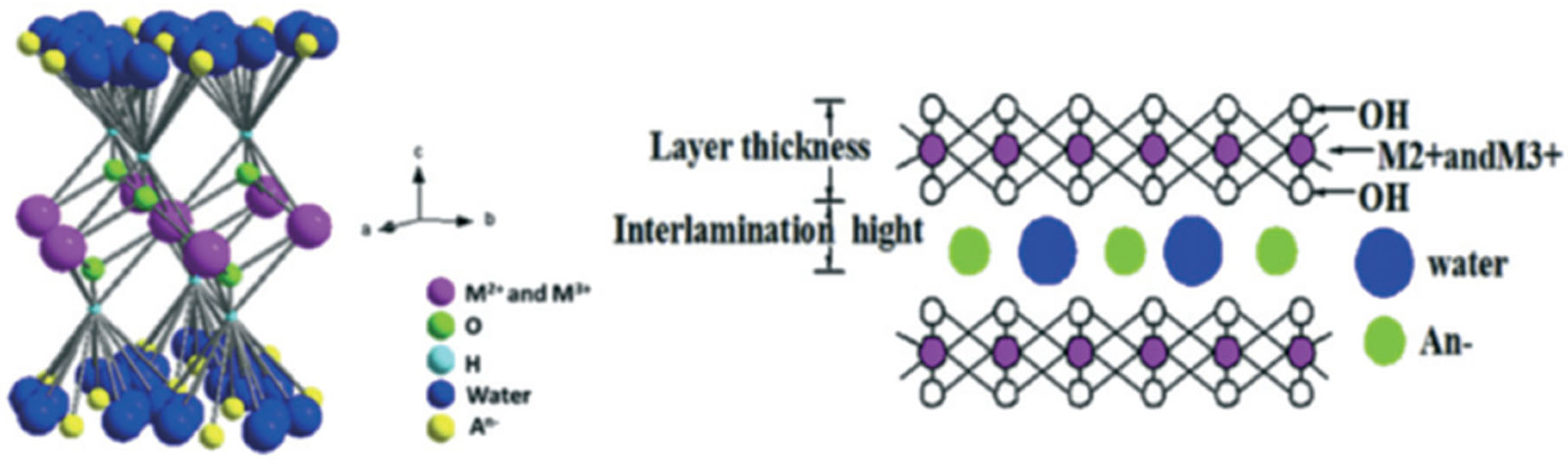
5.4. Layered Double Hydroxides (LDHs)

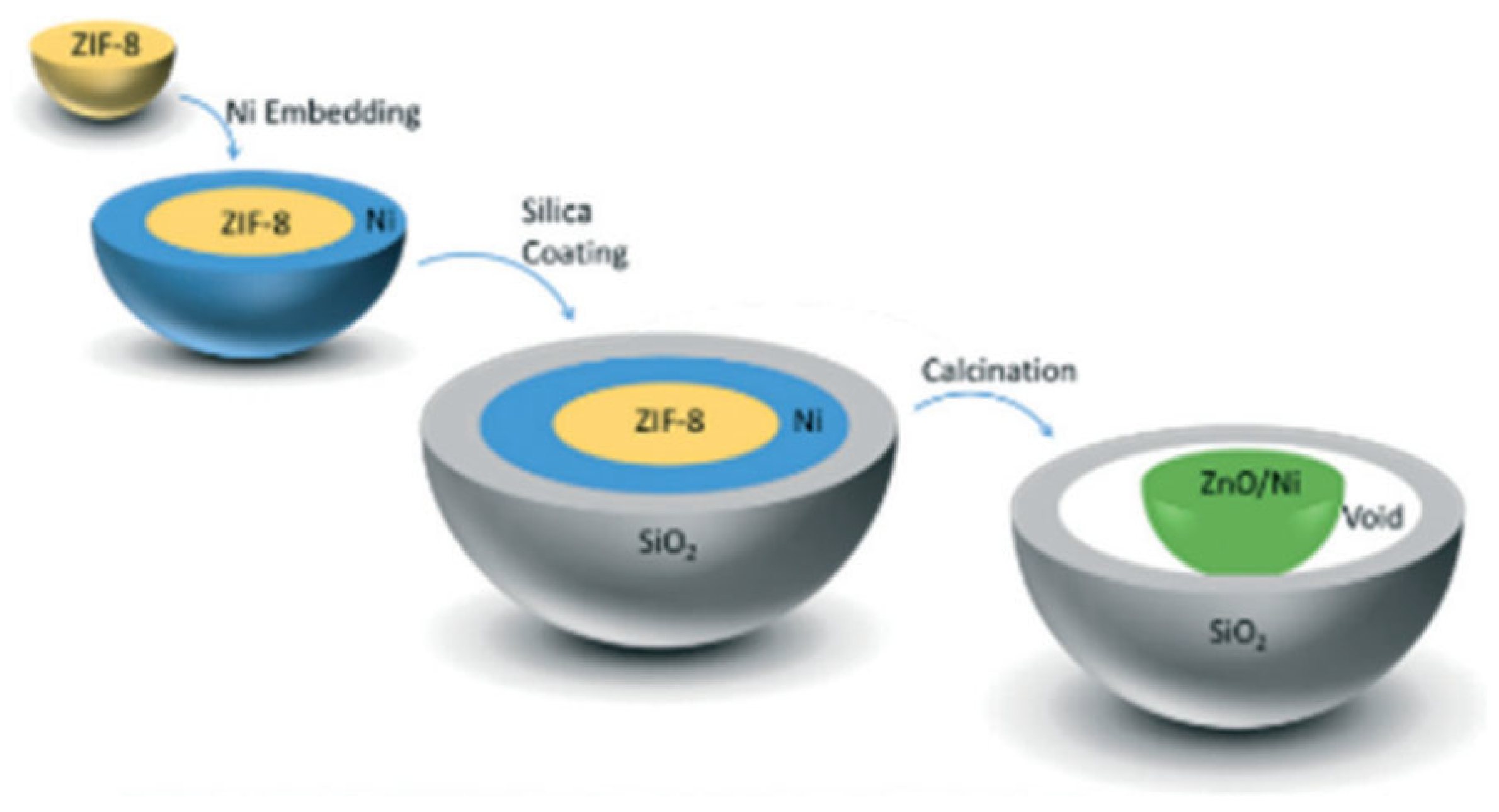
5.5. Metal–Organic Frameworks (MOFs)
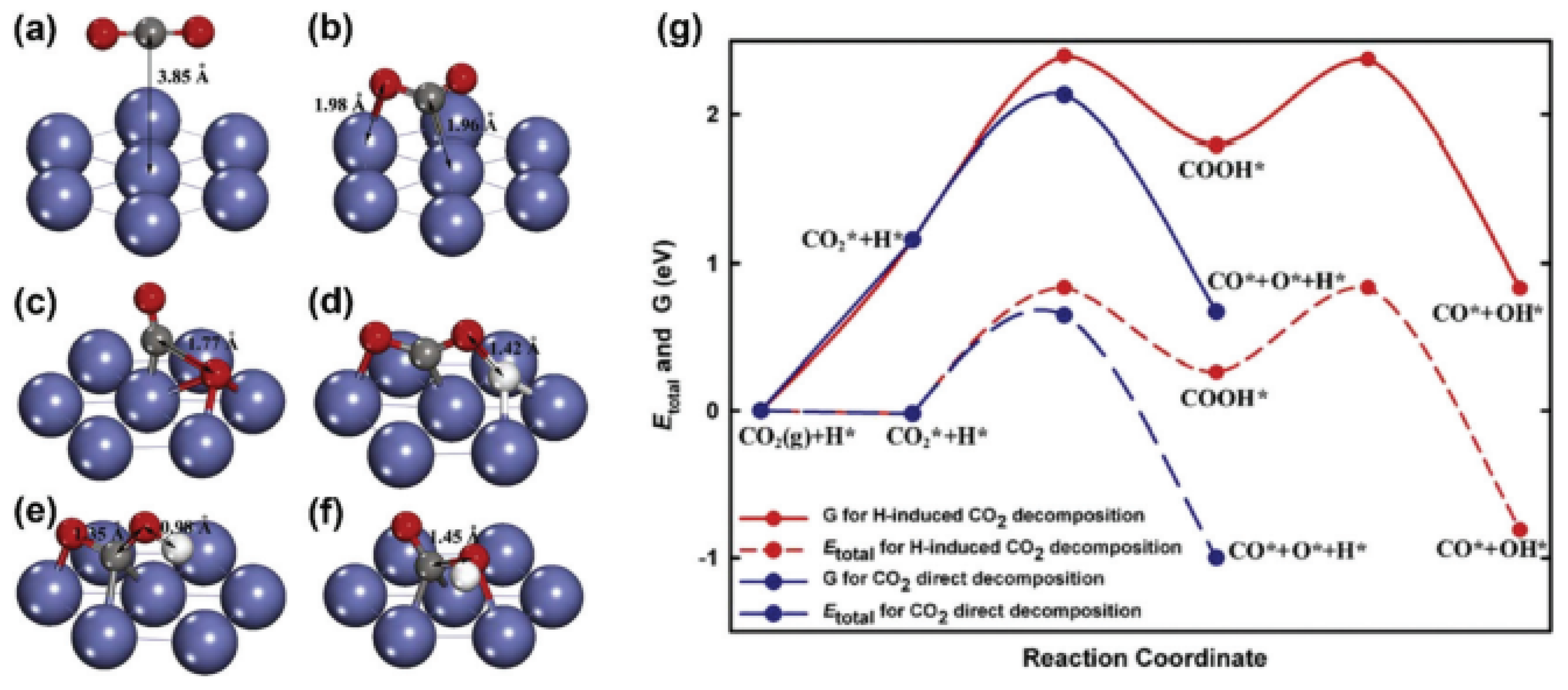
6. Density-Functional Theory (DFT)
7. From Funded Research to Technological Innovation
8. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hansen, J.; Ruedy, R.; Sato, M.; Lo, K. Global Surface Temperature Change. Rev. Geophys. 2010, 48, RG4004. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a Chemical Feedstock: Opportunities and Challenges. Dalt. Trans. 2007, 2975. [Google Scholar] [CrossRef] [PubMed]
- Muraza, O.; Galadima, A. A Review on Coke Management during Dry Reforming of Methane. Int. J. Energy Res. 2015, 39, 1196–1216. [Google Scholar] [CrossRef]
- Intergovernmental Panel on Climate Change. Climate Change 2014 Mitigation of Climate Change. 2014. Available online: https://keneamazon.net/Documents/Publications/Virtual-Library/Impacto/157.pdf (accessed on 6 March 2022).
- Sanz-Pérez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct Capture of CO2 from Ambient Air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef]
- Arora, S.; Prasad, R. An Overview on Dry Reforming of Methane: Strategies to Reduce Carbonaceous Deactivation of Catalysts. RSC Adv. 2016, 6, 108668. [Google Scholar] [CrossRef]
- Chemical Composition of Natural Gas. Available online: https://www.enbridgegas.com/about-enbridge-gas/learn-about-natural-gas (accessed on 6 March 2022).
- Casas-Ledón, Y.; Arteaga-Perez, L.E.; Morales-Perez, M.C.; Peralta-Suárez, L.M. Thermodynamic Analysis of the Hydrogen Production from Ethanol: First and Second Laws Approaches. ISRN Thermodyn. 2012, 2012, 1–8. [Google Scholar] [CrossRef][Green Version]
- Potocnik, P. (Ed.) Natural Gas; InTechOpen: London, UK, 2010; ISBN 9789533071121. [Google Scholar]
- Anderson, J.R.; Boudart, M. (Eds.) Catalysis, Science and Technology; Springer: Berlin/Heidelberg, Germany, 1996; ISBN 9783642646669. [Google Scholar]
- Armor, J.N. The Multiple Roles for Catalysis in the Production of H2. Appl. Catal. A Gen. 1999, 176, 159–176. [Google Scholar] [CrossRef]
- Tada, S.; Kikuchi, R. Mechanistic Study and Catalyst Development for Selective Carbon Monoxide Methanation. Catal. Sci. Technol. 2015, 5, 3061–3070. [Google Scholar] [CrossRef]
- Gallucci, F.; Comite, A.; Capannelli, G.; Basile, A. Steam Reforming of Methane in a Membrane Reactor: An Industrial Case Study. Ind. Eng. Chem. Res. 2006, 45, 2994–3000. [Google Scholar] [CrossRef]
- Istadi, I. Thermodynamic Analysis of Synthesis Gas and Higher Hydrocarbons Production from Methane. In Syngas: Production, Applications and Environmental Impact; Nova Science Publishers: New York, NY, USA, 2013; pp. 99–120. [Google Scholar]
- Pakhare, D.; Spivey, J. A Review of Dry (CO2) Reforming of Methane over Noble Metal Catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Ginsburg, J.M.; Piña, J.; El Solh, T.; de Lasa, H.I. Coke Formation over a Nickel Catalyst under Methane Dry Reforming Conditions: Thermodynamic and Kinetic Models. Ind. Eng. Chem. Res. 2005, 44, 4846–4854. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q.; Millar, G.J. Carbon Dioxide Reforming of Methane to Produce Synthesis Gas over Metal-Supported Catalysts: State of the Art. Energy Fuels 1996, 10, 896–904. [Google Scholar] [CrossRef]
- Bradford, M.C.J.; Vannice, M.A. CO2 Reforming of CH 4. Catal. Rev. 1999, 41, 1–42. [Google Scholar] [CrossRef]
- Jang, W.-J.; Jeong, D.-W.; Shim, J.-O.; Kim, H.-M.; Roh, H.-S.; Son, I.H.; Lee, S.J. Combined Steam and Carbon Dioxide Reforming of Methane and Side Reactions: Thermodynamic Equilibrium Analysis and Experimental Application. Appl. Energy 2016, 173, 80–91. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Dalai, A. Development of Stable Bimetallic Catalysts for Carbon Dioxide Reforming of Methane. J. Catal. 2007, 249, 300–310. [Google Scholar] [CrossRef]
- Nikoo, M.K.; Amin, N.A.S. Thermodynamic Analysis of Carbon Dioxide Reforming of Methane in View of Solid Carbon Formation. Fuel Process. Technol. 2011, 92, 678–691. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Dalai, A.K. Kinetic Studies of Carbon Dioxide Reforming of Methane over Ni–Co/Al–Mg–O Bimetallic Catalyst. Ind. Eng. Chem. Res. 2009, 48, 677–684. [Google Scholar] [CrossRef]
- Zhang, Z.; Verykios, X.E. Mechanistic Aspects of Carbon Dioxide Reforming of Methane to Synthesis Gas over Ni Catalysts. Catal. Lett. 1996, 38, 175–179. [Google Scholar] [CrossRef]
- Slagtern, S.; Olsbye, U.; Blom, R.; Dahl, I.M. The Influence of Rare Earth Oxides on Ni/Al2O3 Catalysts during CO2 Reforming of CH4. Stud. Surf. Sci. Catal. 1997, 107, 497–502. [Google Scholar]
- Hu, Y.H.; Ruckenstein, E. Transient Response Analysis via a Broadened Pulse Combined with a Step Change or an Isotopic Pulse. Application to CO2 Reforming of Methane over NiO/SiO2. J. Phys. Chem. B 1997, 101, 7563–7565. [Google Scholar] [CrossRef]
- Xu, J.; Froment, G.F. Methane Steam Reforming, Methanation and Water-Gas Shift: I. Intrinsic Kinetics. AIChE J. 1989, 35, 88–96. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Isotopic and Kinetic Assessment of the Mechanism of Reactions of CH4 with CO2 or H2O to Form Synthesis Gas and Carbon on Nickel Catalysts. J. Catal. 2004, 224, 370–383. [Google Scholar] [CrossRef]
- Osaki, T.; Horiuchi, T.; Suzuki, K.; Mori, T. Kinetics, Intermediates and Mechanism for the CO2-Reforming of Methane on Supported Nickel Catalysts. J. Chem. Soc. Faraday Trans. 1996, 92, 1627. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Abdulrasheed, A.; Jalil, A.A.; Gambo, Y.; Ibrahim, M.; Hambali, H.U.; Shahul Hamid, M.Y. A Review on Catalyst Development for Dry Reforming of Methane to Syngas: Recent Advances. Renew. Sustain. Energy Rev. 2019, 108, 175–193. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Batchu, R.; Galvita, V.V.; Poelman, H.; Marin, G.B. Carbon Gasification from Fe-Ni Catalysts after Methane Dry Reforming. Appl. Catal. B Environ. 2016, 185, 42–55. [Google Scholar] [CrossRef]
- Luo, J.Z.; Yu, Z.L.; Ng, C.F.; Au, C.T. CO2/CH4 Reforming over Ni–La2O3/5A: An Investigation on Carbon Deposition and Reaction Steps. J. Catal. 2000, 194, 198–210. [Google Scholar] [CrossRef]
- Osaki, T.; Mori, T. Role of Potassium in Carbon-Free CO2 Reforming of Methane on K-Promoted Ni/Al2O3 Catalysts. J. Catal. 2001, 204, 89–97. [Google Scholar] [CrossRef]
- Kroll, V.C.H.; Swaan, H.M.; Lacombe, S.; Mirodatos, C. Methane Reforming Reaction with Carbon Dioxide over Ni/SiO2 Catalyst. J. Catal 1997, 398, 387–398. [Google Scholar]
- Schuurman, Y.; Kroll, V.C.H.; Ferreira-Aparicio, P.; Mirodatos, C. Use of Transient Kinetics Techniques for Studying the Methane Reforming by Carbon Dioxide. Catal. Today 1997, 38, 129–135. [Google Scholar] [CrossRef]
- Chang, J.; Park, S.; Yoo, J.W.; Park, J. Catalytic Behavior of Supported KNiCa Catalyst and Mechanistic Consideration for Carbon Dioxide Reforming of Methane. J. Catal. 2000, 195, 1–11. [Google Scholar] [CrossRef]
- Bradford, M.C.J.; Vannice, M.A. Catalytic Reforming of Methane with Carbon Dioxide over Nickel Catalysts II. Reaction Kinetics. Appl. Catal. A Gen. 1996, 142, 97–122. [Google Scholar] [CrossRef]
- Nandini, A.; Pant, K.K.; Dhingra, S.C. Kinetic Study of the Catalytic Carbon Dioxide Reforming of Methane to Synthesis Gas over Ni-K/CeO2-Al2O3 Catalyst. Appl. Catal. A Gen. 2006, 308, 119–127. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, H.; Xu, H.; Li, W. Kinetic Study of the Catalytic Reforming of CH4 with CO2 to Syngas over Ni/α-Al2O3 Catalyst: The Effect of Temperature on the Reforming Mechanism. Appl. Catal. A Gen. 2007, 318, 79–88. [Google Scholar] [CrossRef]
- De Smet, C.R.H.; Berger, R.J.; Slaa, J.C.; Marin, G.B. Kinetic Modelling of the Partial Oxidation of Methane to Syn-Gas at High Temperatures. Stud. Surf. Sci. Catal. 1998, 119, 825–829. [Google Scholar]
- Fathi, M.; Bjorgum, E.; Viig, T.; Rokstad, O.A. Partial Oxidation of Methane to Synthesis Gas—Elimination of Gas Phase Oxygen. Catal. Today 2000, 63, 489–497. [Google Scholar] [CrossRef]
- Peña, M.A.; Gómez, J.P.; Fierro, J.L.G. New Catalytic Routes for Syngas and Hydrogen Production. Appl. Catal. A Gen. 1996, 144, 7–57. [Google Scholar] [CrossRef]
- Ayabe, S.; Omoto, H.; Utaka, T.; Kikuchi, R.; Sasaki, K.; Teraoka, Y.; Eguchi, K. Catalytic Autothermal Reforming of Methane and Propane over Supported Metal Catalysts. Appl. Catal. A Gen. 2003, 241, 261–269. [Google Scholar] [CrossRef]
- Wilhelm, D.; Simbeck, D.; Karp, A.; Dickenson, R. Syngas Production for Gas-to-Liquids Applications: Technologies, Issues and Outlook. Fuel Process. Technol. 2001, 71, 139–148. [Google Scholar] [CrossRef]
- Palm, C.; Cremer, P.; Peters, R.; Stolten, D. Small-Scale Testing of a Precious Metal Catalyst in the Autothermal Reforming of Various Hydrocarbon Feeds. J. Power Sources 2002, 106, 231–237. [Google Scholar] [CrossRef]
- Chiesa, P. Advanced Technologies for Syngas and Hydrogen (H2) Production from Fossil-Fuel Feedstocks in Power Plants. In Advanced Power Plant Materials, Design and Technology; Woodhead Publishing: Sawston, UK, 2010; pp. 383–411. [Google Scholar]
- Chen, W.-H.; Lin, M.-R.; Lu, J.-J.; Chao, Y.; Leu, T.-S. Thermodynamic Analysis of Hydrogen Production from Methane via Autothermal Reforming and Partial Oxidation Followed by Water Gas Shift Reaction. Int. J. Hydrogen Energy 2010, 35, 11787–11797. [Google Scholar] [CrossRef]
- Ross, J.R.H. Natural Gas Reforming and CO2 Mitigation. Catal. Today 2005, 100, 151–158. [Google Scholar] [CrossRef]
- Ayodele, F.O.; Mohammad, N.; Mustapa, S.I.; Ayodele, B.V. An Overview of Economic Analysis and Environmental Impacts of Natural Gas Conversion Technologies. Sustainability 2020, 12, 10148. [Google Scholar] [CrossRef]
- Gaddalla, A.M.; Sommer, M.E. Carbon Dioxide Reforming of Methane on Nickel Catalysts. Chem. Eng. Sci. 1989, 44, 2825–2829. [Google Scholar] [CrossRef]
- Gallego, J.; Batiot-Dupeyrat, C.; Barrault, J.; Mondragón, F. Severe Deactivation of a LaNiO3 Perovskite-Type Catalyst Precursor with H2S during Methane Dry Reforming. Energy Fuels 2009, 23, 4883–4886. [Google Scholar] [CrossRef]
- Gallego, G.S.; Batiot-Dupeyrat, C.; Barrault, J.; Florez, E.; Mondragón, F. Dry Reforming of Methane over LaNi1−yByO3±δ (B = Mg, Co) Perovskites Used as Catalyst Precursor. Appl. Catal. A Gen. 2008, 334, 251–258. [Google Scholar] [CrossRef]
- Inderwildi, O.R.; Jenkins, S.J.; King, D.A. Mechanistic Studies of Hydrocarbon Combustion and Synthesis on Noble Metals. Angew. Chem. Int. Ed. 2008, 47, 5253–5255. [Google Scholar] [CrossRef]
- Kapokova, L.; Pavlova, S.; Bunina, R.; Alikina, G.; Krieger, T.; Ishchenko, A.; Rogov, V.; Sadykov, V. Dry Reforming of Methane over LnFe0.7Ni0.3O3−δ Perovskites: Influence of Ln Nature. Catal. Today 2011, 164, 227–233. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Tsipouriari, V.A.; Efstathiou, A.M.; Verykios, X.E. Reforming of Methane with Carbon Dioxide to Synthesis Gas over Supported Rhodium Catalysts. J. Catal. 1996, 158, 51–63. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Au, C.-T. CH4/CD4 Isotope Effects in the Carbon Dioxide Reforming of Methane to Syngas over SiO2-Supported Nickel Catalysts. Catal. Lett. 1996, 38, 77–79. [Google Scholar] [CrossRef]
- Kawi, S.; Kathiraser, Y.; Ni, J.; Oemar, U.; Li, Z.; Saw, E.T. Progress in Synthesis of Highly Active and Stable Nickel-Based Catalysts for Carbon Dioxide Reforming of Methane. ChemSusChem 2015, 8, 3556–3575. [Google Scholar] [CrossRef] [PubMed]
- Saedodin, S.; Zamzamian, S.A.H.; Nimvari, M.E.; Wongwises, S.; Jouybari, H.J. Performance Evaluation of a Flat-Plate Solar Collector Filled with Porous Metal Foam: Experimental and Numerical Analysis. Energy Convers. Manag. 2017, 153, 278–287. [Google Scholar] [CrossRef]
- Son, I.H.; Lee, S.J.; Roh, H.-S. Hydrogen Production from Carbon Dioxide Reforming of Methane over Highly Active and Stable MgO Promoted Co–Ni/γ-Al2O3 Catalyst. Int. J. Hydrogen Energy 2014, 39, 3762–3770. [Google Scholar] [CrossRef]
- Anchieta, C.G.; Assaf, E.M.; Assaf, J.M. Effect of Ionic Liquid in Ni/ZrO2 Catalysts Applied to Syngas Production by Methane Tri-Reforming. Int. J. Hydrogen Energy 2019, 44, 9316–9327. [Google Scholar] [CrossRef]
- Abbas, H.F.; Wan Daud, W.M.A. Hydrogen Production by Methane Decomposition: A Review. Int. J. Hydrogen Energy 2010, 35, 1160–1190. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Argyle, M.D. Advances in Catalyst Deactivation and Regeneration. Catalysts 2015, 5, 949–954. [Google Scholar] [CrossRef]
- Oemar, U.; Hidajat, K.; Kawi, S. Pd-Ni Catalyst over Spherical Nanostructured Y2O3 Support for Oxy-CO2 Reforming of Methane: Role of Surface Oxygen Mobility. Int. J. Hydrogen Energy 2015, 40, 12227–12238. [Google Scholar] [CrossRef]
- Pakhare, D.; Wu, H.; Narendra, S.; Abdelsayed, V.; Haynes, D.; Shekhawat, D.; Berry, D.; Spivey, J. Characterization and Activity Study of the Rh-Substituted Pyrochlores for CO2 (Dry) Reforming of CH4. Appl. Petrochem. Res. 2013, 3, 117–129. [Google Scholar] [CrossRef]
- Seo, H.O. Recent Scientific Progress on Developing Supported Ni Catalysts for Dry (CO2) Reforming of Methane. Catalysts 2018, 8, 110. [Google Scholar] [CrossRef]
- Alipour, Z.; Rezaei, M.; Meshkani, F. Effect of Alkaline Earth Promoters (MgO, CaO, and BaO) on the Activity and Coke Formation of Ni Catalysts Supported on Nanocrystalline Al2O3 in Dry Reforming of Methane. J. Ind. Eng. Chem. 2014, 20, 2858–2863. [Google Scholar] [CrossRef]
- Tsipouriari, V.A.; Verykios, X.E. Kinetic Study of the Catalytic Reforming of Methane with Carbon Dioxide to Synthesis Gas over Ni/La2O3 Catalyst. Catal. Today 2001, 64, 83–90. [Google Scholar] [CrossRef]
- Tsipouriari, V.A.; Verykios, X.E. Carbon and Oxygen Reaction Pathways of CO2 Reforming of Methane over Ni/La2O3 and Ni/Al2O3 Catalysts Studied by Isotopic Tracing Techniques. J. Catal. 1999, 187, 85–94. [Google Scholar] [CrossRef]
- Das, S.; Sengupta, M.; Patel, J.; Bordoloi, A. A Study of the Synergy between Support Surface Properties and Catalyst Deactivation for CO2 Reforming over Supported Ni Nanoparticles. Appl. Catal. A Gen. 2017, 545, 113–126. [Google Scholar] [CrossRef]
- Pakhare, D.; Schwartz, V.; Abdelsayed, V.; Haynes, D.; Shekhawat, D.; Poston, J.; Spivey, J. Kinetic and Mechanistic Study of Dry (CO2) Reforming of Methane over Rh-Substituted La2Zr2O7 Pyrochlores. J. Catal. 2014, 316, 78–92. [Google Scholar] [CrossRef]
- Corthals, S.; Van Nederkassel, J.; Geboers, J.; De Winne, H.; Van Noyen, J.; Moens, B.; Sels, B.; Jacobs, P. Influence of Composition of MgAl2O4 Supported NiCeO2ZrO2 Catalysts on Coke Formation and Catalyst Stability for Dry Reforming of Methane. Catal. Today 2008, 138, 28–32. [Google Scholar] [CrossRef]
- Darujati, A.R.S.; Thomson, W.J. Kinetic Study of a Ceria-Promoted Mo2c/γ-Al2O3 Catalyst in Dry-Methane Reforming. Chem. Eng. Sci. 2006, 61, 4309–4315. [Google Scholar] [CrossRef]
- Daza, C.E.; Gallego, J.; Mondragón, F.; Moreno, S.; Molina, R. High Stability of Ce-Promoted Ni/Mg–Al Catalysts Derived from Hydrotalcites in Dry Reforming of Methane. Fuel 2010, 89, 592–603. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Douvartzides, S.L.; Siakavelas, G.I.; Tzounis, L.; Polychronopoulou, K.; Goula, M.A. The Relationship between Reaction Temperature and Carbon Deposition on Nickel Catalysts Based on Nickel Catalysts Based on Al2O3, ZrO2 or SiO2 Supports during the Biogas Dry Reforming Reaction. Catalysts 2019, 9, 676. [Google Scholar] [CrossRef]
- Charisiou, N.; Siakavelas, G.; Sebastian, V.; Hinder, S.; Baker, M.; Papadakis, V.; Wang, W.; Polychronopoulou, K.; Goula, M. Structural Investigation of the Carbon Deposits on Ni/Al2O3 Catalyst Modified by CaO-MgO for the Biogas Dry Reforming Reaction. Chem. Proc. 2020, 2, 15. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Sebastian, V.; Monzon, A.; Baker, M.A.; Hinder, S.J.; Polychronopoulou, K.; Yentekakis, I.V.; Goula, M.A. An in Depth Investigation of Deactivation through Carbon Formation during the Biogas Dry Reforming Reaction for Ni Supported on Modified with CeO2 and La2O3 Zirconia Catalysts. Int. J. Hydrogen Energy 2018, 43, 18955–18976. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Tzounis, L.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Investigating the Correlation between Deactivation and the Carbon Deposited on the Surface of Ni/Al2O3 and Ni/La2O3-Al2O3 Catalysts during the Biogas Reforming Reaction. Appl. Surf. Sci. 2019, 474, 42–56. [Google Scholar] [CrossRef]
- García, V.; Fernández, J.J.; Ruíz, W.; Mondragón, F.; Moreno, A. Effect of MgO Addition on the Basicity of Ni/ZrO2 and on Its Catalytic Activity in Carbon Dioxide Reforming of Methane. Catal. Commun. 2009, 11, 240–246. [Google Scholar] [CrossRef]
- Tauster, S.J.; Fung, S.C.; Garten, R.L. Strong Metal-Support Interactions. Group 8 Noble Metals Supported on Titanium Dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. [Google Scholar] [CrossRef]
- Antolini, E. Composite Materials: An Emerging Class of Fuel Cell Catalyst Supports. Appl. Catal. B Environ. 2010, 100, 413–426. [Google Scholar] [CrossRef]
- Ledesma, C.; Yang, J.; Chen, D.; Holmen, A. Recent Approaches in Mechanistic and Kinetic Studies of Catalytic Reactions Using SSITKA Technique. ACS Catal. 2014, 4, 4527–4547. [Google Scholar] [CrossRef]
- Mirodatos, C. Use of Isotopic Transient Kinetics in Heterogeneous Catalysis. Catal. Today 1991, 9, 83–95. [Google Scholar] [CrossRef]
- Todic, B.; Ma, W.; Jacobs, G.; Davis, B.H.; Bukur, D.B. CO-Insertion Mechanism Based Kinetic Model of the Fischer–Tropsch Synthesis Reaction over Re-Promoted Co Catalyst. Catal. Today 2014, 228, 32–39. [Google Scholar] [CrossRef]
- Berger, R.J.; Kapteijn, F.; Moulijn, J.A.; Marin, G.B.; De Wilde, J.; Olea, M.; Chen, D.; Holmen, A.; Lietti, L.; Tronconi, E.; et al. Dynamic Methods for Catalytic Kinetics. Appl. Catal. A Gen. 2008, 342, 3–28. [Google Scholar] [CrossRef]
- Le Cardinal, G. Transient Tracing. Chem. Eng. Sci. 1978, 33, 1568–1569. [Google Scholar] [CrossRef]
- Biloen, P. Transient Kinetic Methods. J. Mol. Catal. 1983, 21, 17–24. [Google Scholar] [CrossRef]
- Szedlacsek, P.; Efstathiou, A.M.; Verykios, X.E. A Novel Analysis of Equilibrium Adsorption and Desorption Using Transient Tracing Methods. Appl. Catal. A Gen. 1997, 151, 59–96. [Google Scholar] [CrossRef]
- Fakeeha, A.H.; Khan, W.U.; Fatesh, A.S.A.L.; Abasaeed, A.E. Stabilities of Zeolite-Supported Ni Catalysts for Dry Reforming of Methane. Chin. J. Catal. 2013, 34, 764–768. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Damaskinos, C.M.; Djinović, P.; Pintar, A.; Efstathiou, A.M. Dry Reforming of CH4 over NiCo/Ce0.75Zr0.25O2-δ: The Effect of Co on the Site Activity and Carbon Pathways Studied by Transient Techniques. Catal. Commun. 2021, 149, 106237. [Google Scholar] [CrossRef]
- Damaskinos, C.M.; Zavašnik, J.; Djinović, P.; Efstathiou, A.M. Dry Reforming of Methane over Ni/Ce0.8Ti0.2O2-δ: The Effect of Ni Particle Size on the Carbon Pathways Studied by Transient and Isotopic Techniques. Appl. Catal. B Environ. 2021, 296, 120321. [Google Scholar] [CrossRef]
- Vasiliades, M.A.; Damaskinos, C.M.; Kyprianou, K.K.; Kollia, M.; Efstathiou, A.M. The Effect of Pt on the Carbon Pathways in the Dry Reforming of Methane over Ni-Pt/Ce0.8Pr0.2O2-δ Catalyst. Catal. Today 2020, 355, 788–803. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Marin, G.B. Enhanced Carbon-Resistant Dry Reforming Fe-Ni Catalyst: Role of Fe. ACS Catal. 2015, 5, 3028–3039. [Google Scholar] [CrossRef]
- Marafi, M.; Stanislaus, A. Options and Processes for Spent Catalyst Handling and Utilization. J. Hazard. Mater. 2003, 101, 123–132. [Google Scholar] [CrossRef]
- Trimm, D. The Regeneration or Disposal of Deactivated Heterogeneous Catalysts. Appl. Catal. A Gen. 2001, 212, 153–160. [Google Scholar] [CrossRef]
- Chen, C.; Wang, X.; Zhang, L.; Zou, X.; Ding, W.; Lu, X. Synthesis of Mesoporous Ni–La2O3/SiO2 by Ploy(Ethylene Glycol)-Assisted Sol-Gel Route as Highly Efficient Catalysts for Dry Reforming of Methane with a H2/CO Ratio of Unity. Catal. Commun. 2017, 94, 38–41. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Sabbe, M.; Poelman, H.; Detavernier, C.; Marin, G.B. Controlling the Stability of a Fe–Ni Reforming Catalyst: Structural Organization of the Active Components. Appl. Catal. B Environ. 2017, 209, 405–416. [Google Scholar] [CrossRef]
- Rego de Vasconcelos, B.; Pham Minh, D.; Sharrock, P.; Nzihou, A. Regeneration Study of Ni/Hydroxyapatite Spent Catalyst from Dry Reforming. Catal. Today 2018, 310, 107–115. [Google Scholar] [CrossRef]
- Adesina, A.A. The Role of CO2 in Hydrocarbon Reforming Catalysis: Friend or Foe? Curr. Opin. Chem. Eng. 2012, 1, 272–280. [Google Scholar] [CrossRef]
- Izquierdo, U.; García-García, I.; Gutierrez, Á.; Arraibi, J.; Barrio, V.; Cambra, J.; Arias, P. Catalyst Deactivation and Regeneration Processes in Biogas Tri-Reforming Process. The Effect of Hydrogen Sulfide Addition. Catalysts 2018, 8, 12. [Google Scholar] [CrossRef]
- Abdelsadek, Z.; Sehailia, M.; Halliche, D.; Gonzalez-Delacruz, V.M.; Holgado, J.P.; Bachari, K.; Caballero, A.; Cherifi, O. In-Situ Hydrogasification/Regeneration of NiAl-Hydrotalcite Derived Catalyst in the Reaction of CO2 Reforming of Methane: A Versatile Approach to Catalyst Recycling. J. CO2 Util. 2016, 14, 98–105. [Google Scholar] [CrossRef]
- Wu, J.C.S.; Chou, H.-C. Bimetallic Rh–Ni/BN Catalyst for Methane Reforming with CO2. Chem. Eng. J. 2009, 148, 539–545. [Google Scholar] [CrossRef]
- Zhang, Z.; Verykios, X.E.; MacDonald, S.M.; Affrossman, S. Comparative Study of Carbon Dioxide Reforming of Methane to Synthesis Gas over Ni/La2O3 and Conventional Nickel-Based Catalysts. J. Phys. Chem. 1996, 100, 744–754. [Google Scholar] [CrossRef]
- Kehres, J.; Jakobsen, J.G.; Andreasen, J.W.; Wagner, J.B.; Liu, H.; Molenbroek, A.; Sehested, J.; Chorkendorff, I.; Vegge, T. Dynamical Properties of a Ru/MgAl2O4 Catalyst during Reduction and Dry Methane Reforming. J. Phys. Chem. C 2012, 116, 21407–21415. [Google Scholar] [CrossRef]
- Carrara, C.; Múnera, J.; Lombardo, E.A.; Cornaglia, L.M. Kinetic and Stability Studies of Ru/La2O3 Used in the Dry Reforming of Methane. Top. Catal. 2008, 51, 98–106. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Márquez-Alvarez, C.; Rodríguez-Ramos, I.; Schuurman, Y.; Guerrero-Ruiz, A.; Mirodatos, C. A Transient Kinetic Study of the Carbon Dioxide Reforming of Methane over Supported Ru Catalysts. J. Catal. 1999, 184, 202–212. [Google Scholar] [CrossRef]
- Gao, M.; Jiang, N.; Zhao, Y.; Xu, C.; Su, H.; Zeng, S. Copper-Cerium Oxides Supported on Carbon Nanomaterial for Preferential Oxidation of Carbon Monoxide. J. Rare Earths 2016, 34, 55–60. [Google Scholar] [CrossRef]
- Foppa, L.; Silaghi, M.-C.; Larmier, K.; Comas-Vives, A. Intrinsic Reactivity of Ni, Pd and Pt Surfaces in Dry Reforming and Competitive Reactions: Insights from First Principles Calculations and Microkinetic Modeling Simulations. J. Catal. 2016, 343, 196–207. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Comparative Study at Low and Medium Reaction Temperatures of Syngas Production by Methane Reforming with Carbon Dioxide over Silica and Alumina Supported Catalysts. Appl. Catal. A Gen. 1998, 170, 177–187. [Google Scholar] [CrossRef]
- Jones, G.; Jakobsen, J.; Shim, S.; Kleis, J.; Andersson, M.; Rossmeisl, J.; Abildpedersen, F.; Bligaard, T.; Helveg, S.; Hinnemann, B. First Principles Calculations and Experimental Insight into Methane Steam Reforming over Transition Metal Catalysts. J. Catal. 2008, 259, 147–160. [Google Scholar] [CrossRef]
- Hou, Z.; Yashima, T. Small Amounts of Rh-Promoted Ni Catalysts for Methane Reforming with CO2. Catal. Lett. 2003, 89, 193–197. [Google Scholar] [CrossRef]
- García-Diéguez, M.; Pieta, I.S.; Herrera, M.C.; Larrubia, M.A.; Alemany, L.J. RhNi Nanocatalysts for the CO2 and CO2 + H2O Reforming of Methane. Catal. Today 2011, 172, 136–142. [Google Scholar] [CrossRef]
- Pawelec, B.; Damyanova, S.; Arishtirova, K.; Fierro, J.L.G.; Petrov, L. Structural and Surface Features of PtNi Catalysts for Reforming of Methane with CO2. Appl. Catal. A Gen. 2007, 323, 188–201. [Google Scholar] [CrossRef]
- Steinhauer, B.; Kasireddy, M.R.; Radnik, J.; Martin, A. Development of Ni-Pd Bimetallic Catalysts for the Utilization of Carbon Dioxide and Methane by Dry Reforming. Appl. Catal. A Gen. 2009, 366, 333–341. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Pinna, F.; Canton, P.; Pernicone, N. Optimization of Bimetallic Dry Reforming Catalysts by Temperature Programmed Reaction. Appl. Catal. A Gen. 2012, 439–440, 80–87. [Google Scholar] [CrossRef]
- Nagaoka, K.; Takanabe, K.; Aika, K.I. Modification of Co/TiO2 for Dry Reforming of Methane at 2 MPa by Pt, Ru or Ni. Appl. Catal. A Gen. 2004, 268, 151–158. [Google Scholar] [CrossRef]
- Budiman, A.W.; Song, S.H.; Chang, T.S.; Shin, C.H.; Choi, M.J. Dry Reforming of Methane Over Cobalt Catalysts: A Literature Review of Catalyst Development. Catal. Surv. Asia 2012, 16, 183–197. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Fernández-García, M.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. Support Effect over Rhodium Catalysts during the Reforming of Methane by Carbon Dioxide. Stud. Surf. Sci. Catal. 2000, 130, 3675–3680. [Google Scholar]
- Bian, Z.; Das, S.; Wai, M.H.; Hongmanorom, P.; Kawi, S. A Review on Bimetallic Nickel-Based Catalysts for CO2 Reforming of Methane. ChemPhysChem 2017, 18, 3117–3134. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.-S.; Abdullah, A.Z.; Bhatia, S. Catalytic Technology for Carbon Dioxide Reforming of Methane to Synthesis Gas. ChemCatChem 2009, 1, 192–208. [Google Scholar] [CrossRef]
- Tomishige, K.; Chen, Y.; Fujimoto, K. Studies on Carbon Deposition in CO2 Reforming of CH4over Nickel–Magnesia Solid Solution Catalysts. J. Catal. 1999, 181, 91–103. [Google Scholar] [CrossRef]
- Chen, Y.-G.; Tomishige, K.; Yokoyama, K.; Fujimoto, K. Catalytic Performance and Catalyst Structure of Nickel–Magnesia Catalysts for CO2 Reforming of Methane. J. Catal. 1999, 184, 479–490. [Google Scholar] [CrossRef]
- Tomishige, K.; Himeno, Y.; Matsuo, Y.; Yoshinaga, Y.; Fujimoto, K. Catalytic Performance and Carbon Deposition Behavior of a NiO−MgO Solid Solution in Methane Reforming with Carbon Dioxide under Pressurized Conditions. Ind. Eng. Chem. Res. 2000, 39, 1891–1897. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, X.-M.; Zhu, J.; Hu, P. Activity and Coke Formation of Nickel and Nickel Carbide in Dry Reforming: A Deactivation Scheme from Density Functional Theory. J. Catal. 2014, 311, 469–480. [Google Scholar] [CrossRef]
- Kuntaiah, K.; Sudarsanam, P.; Reddy, B.M.; Vinu, A. Nanocrystalline Ce1−xSmxO2−δ (x = 0.4) Solid Solutions: Structural Characterization versus CO Oxidation. RSC Adv. 2013, 3, 7953. [Google Scholar] [CrossRef]
- Gonzalez-delaCruz, V.M.; Pereñiguez, R.; Ternero, F.; Holgado, J.P.; Caballero, A. In Situ XAS Study of Synergic Effects on Ni–Co/ZrO 2 Methane Reforming Catalysts. J. Phys. Chem. C 2012, 116, 2919–2926. [Google Scholar] [CrossRef]
- De Lima, S.M.; Assaf, J.M. Ni-Fe Catalysts Based on Perovskite-Type Oxides for Dry Reforming of Methane to Syngas. Catal. Lett. 2006, 108, 63–70. [Google Scholar] [CrossRef]
- Sutthiumporn, K.; Maneerung, T.; Kathiraser, Y.; Kawi, S. CO2 Dry-Reforming of Methane over La0.8Sr0.2Ni0.8M0.2O3 Perovskite (M = Bi, Co, Cr, Cu, Fe): Roles of Lattice Oxygen on C–H Activation and Carbon Suppression. Int. J. Hydrogen Energy 2012, 37, 11195–11207. [Google Scholar] [CrossRef]
- Pan, C.-J.; Tsai, M.-C.; Su, W.-N.; Rick, J.; Akalework, N.G.; Agegnehu, A.K.; Cheng, S.-Y.; Hwang, B.-J. Tuning/Exploiting Strong Metal-Support Interaction (SMSI) in Heterogeneous Catalysis. J. Taiwan Inst. Chem. Eng. 2017, 74, 154–186. [Google Scholar] [CrossRef]
- Schwab, G.-M. Electronics of Supported Catalysts. Adv. Catal. 1979, 27, 1–22. [Google Scholar]
- Haller, G.L.; Resasco, D.E. Metal–Support Interaction: Group VIII Metals and Reducible Oxides. Adv. Catal. 1989, 36, 173–235. [Google Scholar]
- Nicole, J.; Tsiplakides, D.; Pliangos, C.; Verykios, X.; Comninellis, C.; Vayenas, C. Electrochemical Promotion and Metal–Support Interactions. J. Catal. 2001, 204, 23–34. [Google Scholar] [CrossRef]
- Tauster, S.J. Strong Metal-Support Interactions. Acc. Chem. Res. 1987, 20, 389–394. [Google Scholar] [CrossRef]
- Resasco, D.E.; Weber, R.S.; Sakellson, S.; McMillan, M.; Haller, G.L. X-ray Absorption near-Edge Structure Evidence for Direct Metal-Metal Bonding and Electron Transfer in Reduced Rhodium/Titania Catalysts. J. Phys. Chem. 1988, 92, 189–193. [Google Scholar] [CrossRef]
- Beard, B.C.; Ross, P.N. Platinum-Titanium Alloy Formation from High-Temperature Reduction of a Titania-Impregnated Platinum Catalyst: Implications for Strong Metal-Support Interaction. J. Phys. Chem. 1986, 90, 6811–6817. [Google Scholar] [CrossRef]
- Solymosi, F. Importance of the Electric Properties of Supports in the Carrier Effect. Catal. Rev. 1968, 1, 233–255. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Hu, J.; Li, J.; Richards, R. TiO2 Nanoflakes Modified with Gold Nanoparticles as Photocatalysts with High Activity and Durability under near UV Irradiation. J. Phys. Chem. C 2010, 114, 1641–1645. [Google Scholar] [CrossRef]
- Neyman, K.M.; Vent, S.; Rösch, N.; Pacchioni, G. Transition Metals Supported on Oxides: Density Functional Cluster Studies of Palladium Deposition on MgO(001). Top. Catal. 1999, 9, 153–161. [Google Scholar] [CrossRef]
- Horsley, J.A. A Molecular Orbital Study of Strong Metal-Support Interaction between Platinum and Titanium Dioxide. J. Am. Chem. Soc. 1979, 101, 2870–2874. [Google Scholar] [CrossRef]
- Tung, R.T. Recent Advances in Schottky Barrier Concepts. Mater. Sci. Eng. R Rep. 2001, 35, 1–138. [Google Scholar] [CrossRef]
- Wagner, T.; Marien, J.; Duscher, G. Cu, Nb and V on (110) TiO2 (Rutile): Epitaxy and Chemical Reactions. Thin Solid Films 2001, 398–399, 419–426. [Google Scholar] [CrossRef]
- Ioannides, T.; Verykios, X.E. Charge Transfer in Metal Catalysts Supported on Doped TiO2: A Theoretical Approach Based on Metal–Semiconductor Contact Theory. J. Catal. 1996, 161, 560–569. [Google Scholar] [CrossRef]
- Fu, Q.; Wagner, T. Interaction of Nanostructured Metal Overlayers with Oxide Surfaces. Surf. Sci. Rep. 2007, 62, 431–498. [Google Scholar] [CrossRef]
- Mott, N.F. Note on the Contact between a Metal and an Insulator or Semi-Conductor. Math. Proc. Camb. Philos. Soc. 1938, 34, 568–572. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef]
- Pierret, R.F. MS Contacts and Schottky Diodes. In Semiconductor Device Fundamentals; Addison-Wesley Publishing Company: Boston, MA, USA, 1996; pp. 477–491. ISBN 9780201543933. [Google Scholar]
- Komaya, T.; Bell, A.T.; Wengsieh, Z.; Gronsky, R.; Engelke, F.; King, T.S.; Pruski, M. The Influence of Metal-Support Interactions on the Accurate Determination of Ru Dispersion for Ru/TiO2. J. Catal. 1994, 149, 142–148. [Google Scholar] [CrossRef]
- Kliewer, C.; Miseo, S.; Baumgartner, J.; Stach, E.; Zakharov, D. Early Stage Strong Metal Support Interaction (SMSI) Effects in an Experimental Titania-Supported Platinum Catalyst An Environmental TEM Study. Microsc. Microanal. 2009, 15, 1066–1067. [Google Scholar] [CrossRef]
- Fu, Q.; Wagner, T.; Olliges, S.; Carstanjen, H.-D. Metal−Oxide Interfacial Reactions: Encapsulation of Pd on TiO2 (110). J. Phys. Chem. B 2005, 109, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, M.; Jin, Z.; Wang, J.; Zhang, Z. Study of High-Temperature Hydrogen Reduced Pt0/TiO2 by X-ray Photoelectron Spectroscopy Combined with Argon Ion Sputtering—Diffusion-Encapsulation Effect in Relation to Strong Metal–Support Interaction. Appl. Surf. Sci. 2012, 258, 3991–3999. [Google Scholar] [CrossRef]
- Sá, J.; Bernardi, J.; Anderson, J.A. Imaging of Low Temperature Induced SMSI on Pd/TiO2 Catalysts. Catal. Lett. 2007, 114, 91–95. [Google Scholar] [CrossRef]
- Gao, Y.; Liang, Y.; Chambers, S.A. Thermal Stability and the Role of Oxygen Vacancy Defects in Strong Metal Support Interaction—Pt on Nb-Doped TiO2(100). Surf. Sci. 1996, 365, 638–648. [Google Scholar] [CrossRef]
- Van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of Metal-Support Interactions in Heterogeneous Catalysts to Enhance Activity and Selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Ghods, B.; Rezaei, M.; Meshkani, F. Ni Catalysts Supported on Mesoporous Nanocrystalline Magnesium Silicate in Dry and Steam Reforming Reactions. Chem. Eng. Technol. 2017, 40, 760–768. [Google Scholar] [CrossRef]
- Labich, S.; Taglauer, E.; Knözinger, H. Metal-Support Interactions on Rhodium Model Catalysts. Top. Catal. 2000, 14, 153–161. [Google Scholar] [CrossRef]
- Hayek, K.; Kramer, R.; Paál, Z. Metal-Support Boundary Sites in Catalysis. Appl. Catal. A Gen. 1997, 162, 1–15. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Panagiotopoulou, P.; Kondarides, D.I.; Efstathiou, A.M. Kinetic and Mechanistic Studies of the Water–Gas Shift Reaction on Pt/TiO2 Catalyst. J. Catal. 2009, 264, 117–129. [Google Scholar] [CrossRef]
- Raimondi, F.; Scherer, G.G.; Kötz, R.; Wokaun, A. Nanoparticles in Energy Technology: Examples from Electrochemistry and Catalysis. Angew. Chem. Int. Ed. 2005, 44, 2190–2209. [Google Scholar] [CrossRef]
- Munera, J.; Irusta, S.; Cornaglia, L.; Lombardo, E.; Vargascesar, D.; Schmal, M. Kinetics and Reaction Pathway of the CO2 Reforming of Methane on Rh Supported on Lanthanum-Based Solid. J. Catal. 2007, 245, 25–34. [Google Scholar] [CrossRef]
- Solymosi, F.; Tolmacsov, P.; Kedves, K. CO2 Reforming of Propane over Supported Rh. J. Catal. 2003, 216, 377–385. [Google Scholar] [CrossRef]
- Bradford, M.C.J.; Vannice, M.A. CO2 Reforming of CH4 over Supported Ru Catalysts. J. Catal. 1999, 183, 69–75. [Google Scholar] [CrossRef]
- O’Connor, A.M.; Schuurman, Y.; Ross, J.R.H.; Mirodatos, C. Transient Studies of Carbon Dioxide Reforming of Methane over Pt/ZrO2 and Pt/Al2O3. Catal. Today 2006, 115, 191–198. [Google Scholar] [CrossRef]
- Hu, Y.H.; Ruckenstein, E. An Optimum NiO Content in the CO2 Reforming of CH4 with NiO/MgO Solid Solution Catalysts. Catal. Lett. 1996, 36, 145–149. [Google Scholar] [CrossRef]
- Olsbye, U.; Wurzel, T.; Mleczko, L. Kinetic and Reaction Engineering Studies of Dry Reforming of Methane over a Ni/La/Al2O3 Catalyst. Ind. Eng. Chem. Res. 1997, 36, 5180–5188. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Rodríguez-Ramos, I.; Anderson, J.; Guerrero-Ruiz, A. Mechanistic Aspects of the Dry Reforming of Methane over Ruthenium Catalysts. Appl. Catal. A Gen. 2000, 202, 183–196. [Google Scholar] [CrossRef]
- Bitter, J.H.; Seshan, K.; Lercher, J.A. Mono and Bifunctional Pathways of CO2/CH4 Reforming over Pt and Rh Based Catalysts. J. Catal. 1998, 176, 93–101. [Google Scholar] [CrossRef]
- Gronchi, P.; Mazzocchia, C.; Del Rosso, R. Carbon Dioxide Reaction with Methane on La2O3 Supported Rh Catalysts. Energy Convers. Manag. 1995, 36, 605–608. [Google Scholar] [CrossRef]
- Djinović, P.; Batista, J.; Pintar, A. Efficient Catalytic Abatement of Greenhouse Gases: Methane Reforming with CO2 Using a Novel and Thermally Stable Rh–CeO2 Catalyst. Int. J. Hydrogen Energy 2012, 37, 2699–2707. [Google Scholar] [CrossRef]
- De Miguel, S.R.; Vilella, I.M.J.; Maina, S.P.; San José-Alonso, D.; Román-Martínez, M.C.; Illán-Gómez, M.J. Influence of Pt Addition to Ni Catalysts on the Catalytic Performance for Long Term Dry Reforming of Methane. Appl. Catal. A Gen. 2012, 435–436, 10–18. [Google Scholar] [CrossRef]
- Osojnik Črnivec, I.G.; Djinović, P.; Erjavec, B.; Pintar, A. Effect of Synthesis Parameters on Morphology and Activity of Bimetallic Catalysts in CO2-CH4 Reforming. Chem. Eng. J. 2012, 207–208, 299–307. [Google Scholar] [CrossRef]
- Gohier, A.; Ewels, C.P.; Minea, T.M.; Djouadi, M.A. Carbon Nanotube Growth Mechanism Switches from Tip- to Base-Growth with Decreasing Catalyst Particle Size. Carbon N. Y. 2008, 46, 1331–1338. [Google Scholar] [CrossRef]
- Kim, J.-H.; Suh, D.J.; Park, T.-J.; Kim, K.-L. Effect of Metal Particle Size on Coking during CO2 Reforming of CH4 over Ni–Alumina Aerogel Catalysts. Appl. Catal. A Gen. 2000, 197, 191–200. [Google Scholar] [CrossRef]
- Chen, D.; Christensen, K.; Ochoafernandez, E.; Yu, Z.; Totdal, B.; Latorre, N.; Monzon, A.; Holmen, A. Synthesis of Carbon Nanofibers: Effects of Ni Crystal Size during Methane Decomposition. J. Catal. 2005, 229, 82–96. [Google Scholar] [CrossRef]
- Tang, S.; Ji, L.; Lin, J.; Zeng, H.C.; Tan, K.L.; Li, K. CO2 Reforming of Methane to Synthesis Gas over Sol–Gel-Made Ni/γ-Al2O3 Catalysts from Organometallic Precursors. J. Catal. 2000, 194, 424–430. [Google Scholar] [CrossRef]
- Wang, S. CO2 Reforming of Methane on Ni Catalysts: Effects of the Support Phase and Preparation Technique. Appl. Catal. B Environ. 1998, 16, 269–277. [Google Scholar] [CrossRef]
- Gonzalez-Delacruz, V.M.; Ternero, F.; Pereñíguez, R.; Caballero, A.; Holgado, J.P. Study of Nanostructured Ni/CeO2 Catalysts Prepared by Combustion Synthesis in Dry Reforming of Methane. Appl. Catal. A Gen. 2010, 384, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, H.-M.; Xu, B.-Q. Durable Ni/MgO Catalysts for CO2 Reforming of Methane: Activity and Metal–Support Interaction. J. Mol. Catal. A Chem. 2009, 299, 44–52. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, T.; Zhang, P.; Qi, R.; Huang, R.; Song, X.; Gao, L. Facile Synthesis of Hollow Hierarchical Ni/γ-Al2O3 Nanocomposites for Methane Dry Reforming Catalysis. RSC Adv. 2014, 4, 51184–51193. [Google Scholar] [CrossRef]
- Naeem, M.A.; Al-Fatesh, A.S.; Abasaeed, A.E.; Fakeeha, A.H. Activities of Ni-Based Nano Catalysts for CO2–CH4 Reforming Prepared by Polyol Process. Fuel Process. Technol. 2014, 122, 141–152. [Google Scholar] [CrossRef]
- Gonzalezdelacruz, V.; Holgado, J.; Pereniguez, R.; Caballero, A. Morphology Changes Induced by Strong Metal–Support Interaction on a Ni–Ceria Catalytic System. J. Catal. 2008, 257, 307–314. [Google Scholar] [CrossRef]
- Hannora, A.E.; Ataya, S. Structure and Compression Strength of Hydroxyapatite/Titania Nanocomposites Formed by High Energy Ball Milling. J. Alloys Compd. 2016, 658, 222–233. [Google Scholar] [CrossRef]
- Koo, K.Y.; Roh, H.-S.; Jung, U.H.; Yoon, W.L. CeO2 Promoted Ni/Al2O3 Catalyst in Combined Steam and Carbon Dioxide Reforming of Methane for Gas to Liquid (GTL) Process. Catal. Lett. 2009, 130, 217–221. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Liu, H.; Wang, X. One-Pot Synthesis of Ni-Nanoparticle-Embedded Mesoporous Titania/Silica Catalyst and Its Application for CO2-Reforming of Methane. Catal. Commun. 2008, 9, 995–1000. [Google Scholar] [CrossRef]
- Raberg, L.; Jensen, M.; Olsbye, U.; Daniel, C.; Haag, S.; Mirodatos, C.; Sjastad, A. Propane Dry Reforming to Synthesis Gas over Ni-Based Catalysts: Influence of Support and Operating Parameters on Catalyst Activity and Stability. J. Catal. 2007, 249, 250–260. [Google Scholar] [CrossRef]
- Zhang, Q.-H.; Li, Y.; Xu, B.-Q. Reforming of Methane and Coalbed Methane over Nanocomposite Ni/ZrO2 Catalyst. Catal. Today 2004, 98, 601–605. [Google Scholar] [CrossRef]
- Wang, N.; Xu, Z.; Deng, J.; Shen, K.; Yu, X.; Qian, W.; Chu, W.; Wei, F. One-Pot Synthesis of Ordered Mesoporous NiCeAl Oxide Catalysts and a Study of Their Performance in Methane Dry Reforming. ChemCatChem 2014, 6, 1470–1480. [Google Scholar] [CrossRef]
- Han, J.W.; Park, J.S.; Choi, M.S.; Lee, H. Uncoupling the Size and Support Effects of Ni Catalysts for Dry Reforming of Methane. Appl. Catal. B Environ. 2017, 203, 625–632. [Google Scholar] [CrossRef]
- Van Hardeveld, R.; Hartog, F. The Statistics of Surface Atoms and Surface Sites on Metal Crystals. Surf. Sci. 1969, 15, 189–230. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst Design for Dry Reforming of Methane: Analysis Review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Ruiz Puigdollers, A.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef]
- Wang, H.; Ruckenstein, E. Carbon Dioxide Reforming of Methane to Synthesis Gas over Supported Rhodium Catalysts: The Effect of Support. Appl. Catal. A Gen. 2000, 204, 143–152. [Google Scholar] [CrossRef]
- Nakagawa, K.; Anzai, K.; Matsui, N.; Ikenaga, N.; Suzuki, T.; Teng, Y. 99/00322 Effect of Support on the Conversion of Methane to Synthesis Gas over Supported Iridium Catalysts. Fuel Energy Abstr. 1999, 40, 32. [Google Scholar] [CrossRef]
- Zhang, R.; Xia, G.; Li, M.; Wu, Y.; Nie, H.; Li, D. Effect of Support on the Performance of Ni-Based Catalyst in Methane Dry Reforming. J. Fuel Chem. Technol. 2015, 43, 1359–1365. [Google Scholar] [CrossRef]
- Naeem, M.A.; Al-Fatesh, A.S.; Khan, W.U.; Abasaeed, A.E.; Fakeeha, A.H. Syngas Production from Dry Reforming of Methane over Nano Ni Polyol Catalysts. Int. J. Chem. Eng. Appl. 2013, 315–320. [Google Scholar] [CrossRef]
- Xue, Y.; Xu, L.; Chen, M.; Wu, C.; Cheng, G.; Wang, N.; Hu, X. Constructing Ni-Based Confinement Catalysts with Advanced Performances toward the CO2 Reforming of CH4: State-of-the-Art Review and Perspectives. Catal. Sci. Technol. 2021, 11, 6344–6368. [Google Scholar] [CrossRef]
- De Jesus, A.S.; Maloncy, M.L.; Batista, M.S. Effect of MgO Loading over Zeolite-Supported Ni Catalysts in Methane Reforming with Carbon Dioxide for Synthesis Gas Production. React. Kinet. Mech. Catal. 2017, 122, 501–511. [Google Scholar] [CrossRef]
- Najfach, A.J.; Almquist, C.B.; Edelmann, R.E. Effect of Manganese and Zeolite Composition on Zeolite-Supported Ni-Catalysts for Dry Reforming of Methane. Catal. Today 2021, 369, 31–47. [Google Scholar] [CrossRef]
- Luengnaruemitchai, A.; Kaengsilalai, A. Activity of Different Zeolite-Supported Ni Catalysts for Methane Reforming with Carbon Dioxide. Chem. Eng. J. 2008, 144, 96–102. [Google Scholar] [CrossRef]
- Alotaibi, R.; Alenazey, F.; Alotaibi, F.; Wei, N. Ni Catalysts with Different Promoters Supported on Zeolite for Dry Reforming of Methane. Appl. Petrochem. Res. 2015, 5, 329–337. [Google Scholar] [CrossRef]
- Shan, B.; Cho, K. First-Principles Study of Work Functions of Double-Wall Carbon Nanotubes. Phys. Rev. B 2006, 73, 081401. [Google Scholar] [CrossRef]
- Xu, L.; Song, H.; Chou, L. Mesoporous Nanocrystalline Ceria–Zirconia Solid Solutions Supported Nickel Based Catalysts for CO2 Reforming of CH4. Int. J. Hydrogen Energy 2012, 37, 18001–18020. [Google Scholar] [CrossRef]
- Xia, D.; Chen, Y.; Li, C.; Liu, C.; Zhou, G. Carbon Dioxide Reforming of Methane to Syngas over Ordered Mesoporous Ni/KIT-6 Catalysts. Int. J. Hydrogen Energy 2018, 43, 20488–20499. [Google Scholar] [CrossRef]
- Lv, Y.; Xin, Z.; Meng, X.; Tao, M.; Bian, Z. Ni Based Catalyst Supported on KIT-6 Silica for CO Methanation: Confinement Effect of Three Dimensional Channel on NiO and Ni Particles. Microporous Mesoporous Mater. 2018, 262, 89–97. [Google Scholar] [CrossRef]
- Sidik, S.M.; Triwahyono, S.; Jalil, A.A.; Majid, Z.A.; Salamun, N.; Talib, N.B.; Abdullah, T.A.T. CO2 Reforming of CH4 over Ni–Co/MSN for Syngas Production: Role of Co as a Binder and Optimization Using RSM. Chem. Eng. J. 2016, 295, 1–10. [Google Scholar] [CrossRef]
- Sidik, S.M.; Jalil, A.A.; Triwahyono, S.; Abdullah, T.A.T.; Ripin, A. CO2 Reforming of CH 4 over Ni/Mesostructured Silica Nanoparticles (Ni/MSN). RSC Adv. 2015, 5, 37405–37414. [Google Scholar] [CrossRef]
- Dai, Y.-M.; Lu, C.-Y.; Chang, C.-J. Catalytic Activity of Mesoporous Ni/CNT, Ni/SBA-15 and (Cu, Ca, Mg, Mn, Co)–Ni/SBA-15 Catalysts for CO2 Reforming of CH4. RSC Adv. 2016, 6, 73887–73896. [Google Scholar] [CrossRef]
- Dou, J.; Zeng, H.C. Targeted Synthesis of Silicomolybdic Acid (Keggin Acid) inside Mesoporous Silica Hollow Spheres for Friedel-Crafts Alkylation. J. Am. Chem. Soc. 2012, 134, 16235–16246. [Google Scholar] [CrossRef]
- Okutan, C.; Arbag, H.; Yasyerli, N.; Yasyerli, S. Catalytic Activity of SBA-15 Supported Ni Catalyst in CH4 Dry Reforming: Effect of Al, Zr, and Ti Co-Impregnation and Al Incorporation to SBA-15. Int. J. Hydrogen Energy 2020, 45, 13911–13928. [Google Scholar] [CrossRef]
- Abdullah, N.; Ainirazali, N.; Chong, C.C.; Razak, H.A.; Setiabudi, H.D.; Jalil, A.A.; Vo, D.-V.N. Influence of Impregnation Assisted Methods of Ni/SBA-15 for Production of Hydrogen via Dry Reforming of Methane. Int. J. Hydrogen Energy 2020, 45, 18426–18439. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, M.; Ning, P.; Long, K.; Wang, J.; Tang, T.; Fan, J.; Sun, H.; Yin, L.; Lin, Q. Effect of Thermal Induction Temperature on Re-Dispersion Behavior of Ni Nanoparticles over Ni/SBA-15 for Dry Reforming of Methane. Appl. Surf. Sci. 2019, 469, 368–377. [Google Scholar] [CrossRef]
- El Hassan, N.; Kaydouh, M.N.; Geagea, H.; El Zein, H.; Jabbour, K.; Casale, S.; El Zakhem, H.; Massiani, P. Low Temperature Dry Reforming of Methane on Rhodium and Cobalt Based Catalysts: Active Phase Stabilization by Confinement in Mesoporous SBA-15. Appl. Catal. A Gen. 2016, 520, 114–121. [Google Scholar] [CrossRef]
- Kaydouh, M.N.; El Hassan, N.; Davidson, A.; Massiani, P. Optimization of Synthesis Conditions of Ni/SbA-15 Catalysts: Confined Nanoparticles and Improved Stability in Dry Reforming of Methane. Catalysts 2021, 11, 44. [Google Scholar] [CrossRef]
- Rodriguez-Gomez, A.; Pereñiguez, R.; Caballero, A. Nickel Particles Selectively Confined in the Mesoporous Channels of SBA-15 Yielding a Very Stable Catalyst for DRM Reaction. J. Phys. Chem. B 2018, 122, 500–510. [Google Scholar] [CrossRef]
- Baktash, E.; Littlewood, P.; Schomäcker, R.; Thomas, A.; Stair, P.C. Alumina Coated Nickel Nanoparticles as a Highly Active Catalyst for Dry Reforming of Methane. Appl. Catal. B Environ. 2015, 179, 122–127. [Google Scholar] [CrossRef]
- Qiu, S.; Zhang, Q.; Lv, W.; Wang, T.; Zhang, Q.; Ma, L. Simply Packaging Ni Nanoparticles inside SBA-15 Channels by Co-Impregnation for Dry Reforming of Methane. RSC Adv. 2017, 7, 24551–24560. [Google Scholar] [CrossRef]
- Tian, J.; Li, H.; Zeng, X.; Wang, Z.; Huang, J.; Zhao, C. Facile Immobilization of Ni Nanoparticles into Mesoporous MCM-41 Channels for Efficient Methane Dry Reforming. Chin. J. Catal. 2019, 40, 1395–1404. [Google Scholar] [CrossRef]
- Hoyos, L.S.; Faroldi, B.M.; Cornaglia, L.M. A coke-resistant catalyst for the dry reforming of methane based on Ni nanoparticles confined within rice husk-derived mesoporous materials. Catal. Commun. 2019, 105898. [Google Scholar] [CrossRef]
- Alves, R.H.; Reis, T.V.d.S.; Rovani, S.; Fungaro, D.A. Green Synthesis and Characterization of Biosilica Produced from Sugarcane Waste Ash. J. Chem. 2017, 2017, 6129035. [Google Scholar] [CrossRef]
- Wang, J.; Jin, P.; Bishop, P.L.; Li, F. Upgrade of Three Municipal Wastewater Treatment Lagoons Using a High Surface Area Media. Front. Environ. Sci. Eng. 2012, 6, 288–293. [Google Scholar] [CrossRef]
- Aguiar, M.; Cazula, B.B.; Saragiotto Colpini, L.M.; Borba, C.E.; Alves da Silva, F.; Noronha, F.B.; Alves, H.J. Si-MCM-41 Obtained from Different Sources of Silica and Its Application as Support for Nickel Catalysts Used in Dry Reforming of Methane. Int. J. Hydrogen Energy 2019, 44, 32003–32018. [Google Scholar] [CrossRef]
- Wang, X.; Yang, M.; Zhu, L.; Zhu, X.; Wang, S. CO2 Methanation over Ni/Mg@MCM-41 Prepared by In-Situ Synthesis Method. J. Fuel Chem. Technol. 2020, 48, 456–465. [Google Scholar] [CrossRef]
- Taherian, Z.; Khataee, A.; Orooji, Y. Facile Synthesis of Yttria-Promoted Nickel Catalysts Supported on MgO-MCM-41 for Syngas Production from Greenhouse Gases. Renew. Sustain. Energy Rev. 2020, 134, 110130. [Google Scholar] [CrossRef]
- Ibrahim, A.A.; Al-Fatesh, A.A.; Atia, H.; Fakeeha, A.H.; Kasim, S.O.; Abasaeed, A.E. Influence of Promoted 5%Ni/MCM-41 Catalysts on Hydrogen Yield in CO2 Reforming of CH4. Int. J. Energy Res. 2018, 42, 4120–4130. [Google Scholar] [CrossRef]
- Xu, J.; Xia, P.; Guo, F.; Xie, J.; Xia, Y.; Tian, H. Enhancement of the Catalytic Performance and Coke Resistance of Alcohol-Promoted Ni/MCM-41 Catalysts for CH4/CO2 Reforming. Int. J. Hydrogen Energy 2020, 45, 30484–30495. [Google Scholar] [CrossRef]
- Amin, M.H. Relationship Between the Pore Structure of Mesoporous Silica Supports and the Activity of Nickel Nanocatalysts in the CO2 Reforming of Methane. Catalysts 2020, 10, 51. [Google Scholar] [CrossRef]
- He, S.; An, Z.; Wei, M.; Evans, D.G.; Duan, X. Layered Double Hydroxide-Based Catalysts: Nanostructure Design and Catalytic Performance. Chem. Commun. 2013, 49, 5912. [Google Scholar] [CrossRef]
- Wang, Q.; O’Hare, D. Recent Advances in the Synthesis and Application of Layered Double Hydroxide (LDH) Nanosheets. Chem. Rev. 2012, 112, 4124–4155. [Google Scholar] [CrossRef]
- Ashok, J.; Kathiraser, Y.; Ang, M.L.; Kawi, S. Bi-Functional Hydrotalcite-Derived NiO–CaO–Al2O3 Catalysts for Steam Reforming of Biomass and/or Tar Model Compound at Low Steam-to-Carbon Conditions. Appl. Catal. B Environ. 2015, 172–173, 116–128. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Yakovenchuk, V.N.; Zolotarev, A.A.; Ivanyuk, G.N.; Pakhomovsky, Y.A. Cation Ordering and Superstructures in Natural Layered Double Hydroxides. Chim. Int. J. Chem. 2010, 64, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Ashok, J.; Kawi, S. Steam Reforming of Toluene as a Biomass Tar Model Compound over CeO2 Promoted Ni/CaO–Al2O3 Catalytic Systems. Int. J. Hydrogen Energy 2013, 38, 13938–13949. [Google Scholar] [CrossRef]
- Bel Hadjltaief, H.; Da Costa, P.; Beaunier, P.; Gálvez, M.E.; Ben Zina, M. Fe-Clay-Plate as a Heterogeneous Catalyst in Photo-Fenton Oxidation of Phenol as Probe Molecule for Water Treatment. Appl. Clay Sci. 2014, 91–92, 46–54. [Google Scholar] [CrossRef]
- Gil, A.; Korili, S.A.; Vicente, M.A. Recent Advances in the Control and Characterization of the Porous Structure of Pillared Clay Catalysts. Catal. Rev. 2008, 50, 153–221. [Google Scholar] [CrossRef]
- Zhou, C.H. An Overview on Strategies towards Clay-Based Designer Catalysts for Green and Sustainable Catalysis. Appl. Clay Sci. 2011, 53, 87–96. [Google Scholar] [CrossRef]
- Dewangan, N.; Hui, W.M.; Jayaprakash, S.; Bawah, A.-R.; Poerjoto, A.J.; Jie, T.; Jangam, A.; Hidajat, K.; Kawi, S. Recent Progress on Layered Double Hydroxide (LDH) Derived Metal-Based Catalysts for CO2 Conversion to Valuable Chemicals. Catal. Today 2020, 356, 490–513. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, F.; Evans, D.G.; Duan, X. Layered Double Hydroxide Films: Synthesis, Properties and Applications. Chem. Commun. 2010, 46, 5197. [Google Scholar] [CrossRef]
- Aider, N.; Touahra, F.; Bali, F.; Djebarri, B.; Lerari, D.; Bachari, K.; Halliche, D. Improvement of Catalytic Stability and Carbon Resistance in the Process of CO2 Reforming of Methane by CoAl and CoFe Hydrotalcite-Derived Catalysts. Int. J. Hydrogen Energy 2018, 43, 8256–8266. [Google Scholar] [CrossRef]
- Feng, J.; He, Y.; Liu, Y.; Du, Y.; Li, D. Supported Catalysts Based on Layered Double Hydroxides for Catalytic Oxidation and Hydrogenation: General Functionality and Promising Application Prospects. Chem. Soc. Rev. 2015, 44, 5291–5319. [Google Scholar] [CrossRef]
- Jiang, Z.; Yu, J.; Cheng, J.; Xiao, T.; Jones, M.O.; Hao, Z.; Edwards, P.P. Catalytic Combustion of Methane over Mixed Oxides Derived from Co–Mg/Al Ternary Hydrotalcites. Fuel Process. Technol. 2010, 91, 97–102. [Google Scholar] [CrossRef]
- Dębek, R.; Motak, M.; Duraczyska, D.; Launay, F.; Galvez, M.E.; Grzybek, T.; Da Costa, P. Methane Dry Reforming over Hydrotalcite-Derived Ni–Mg–Al Mixed Oxides: The Influence of Ni Content on Catalytic Activity, Selectivity and Stability. Catal. Sci. Technol. 2016, 6, 6705–6715. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, S.; Chen, B.; Zhang, Z.; Shi, C. Effect of Mg/Al Ratio of NiMgAl Mixed Oxide Catalyst Derived from Hydrotalcite for Carbon Dioxide Reforming of Methane. Catal. Today 2016, 264, 163–170. [Google Scholar] [CrossRef]
- Huang, J.; Yan, Y.; Saqline, S.; Liu, W.; Liu, B. High Performance Ni Catalysts Prepared by Freeze Drying for Efficient Dry Reforming of Methane. Appl. Catal. B Environ. 2020, 275, 119109. [Google Scholar] [CrossRef]
- Zhao, M.-Q.; Zhang, Q.; Zhang, W.; Huang, J.-Q.; Zhang, Y.; Su, D.S.; Wei, F. Embedded High Density Metal Nanoparticles with Extraordinary Thermal Stability Derived from Guest−Host Mediated Layered Double Hydroxides. J. Am. Chem. Soc. 2010, 132, 14739–14741. [Google Scholar] [CrossRef]
- Bu, K.; Kuboon, S.; Deng, J.; Li, H.; Yan, T.; Chen, G.; Shi, L.; Zhang, D. Methane Dry Reforming over Boron Nitride Interface-Confined and LDHs-Derived Ni Catalysts. Appl. Catal. B Environ. 2019, 252, 86–97. [Google Scholar] [CrossRef]
- Bu, K.; Deng, J.; Zhang, X.; Kuboon, S.; Yan, T.; Li, H.; Shi, L.; Zhang, D. Promotional Effects of B-Terminated Defective Edges of Ni/Boron Nitride Catalysts for Coking- and Sintering-Resistant Dry Reforming of Methane. Appl. Catal. B Environ. 2020, 267, 118692. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, C.; Zhang, Y.; Xu, Y.; Shang, S.; Yin, Y. Ni–Co Catalyst Derived from Layered Double Hydroxides for Dry Reforming of Methane. Int. J. Hydrogen Energy 2015, 40, 16115–16126. [Google Scholar] [CrossRef]
- Gandara-Loe, J.; Pastor-Perez, L.; Bobadilla, L.F.; Odriozola, J.A.; Reina, T.R. Understanding the Opportunities of Metal-Organic Frameworks (MOFs) for CO2 capture and Gas-Phase CO2 conversion Processes: A Comprehensive Overview. React. Chem. Eng. 2021, 6, 787–814. [Google Scholar] [CrossRef]
- Jiao, L.; Seow, J.Y.R.; Skinner, W.S.; Wang, Z.U.; Jiang, H.-L. Metal–Organic Frameworks: Structures and Functional Applications. Mater. Today 2019, 27, 43–68. [Google Scholar] [CrossRef]
- Xu, W.; Thapa, K.B.; Ju, Q.; Fang, Z.; Huang, W. Heterogeneous Catalysts Based on Mesoporous Metal–Organic Frameworks. Coord. Chem. Rev. 2018, 373, 199–232. [Google Scholar] [CrossRef]
- Kitao, T.; Zhang, Y.; Kitagawa, S.; Wang, B.; Uemura, T. Hybridization of MOFs and Polymers. Chem. Soc. Rev. 2017, 46, 3108–3133. [Google Scholar] [CrossRef] [PubMed]
- Karam, L.; Reboul, J.; Casale, S.; Massiani, P.; El Hassan, N. Porous Nickel-Alumina Derived from Metal-Organic Framework (MIL-53): A New Approach to Achieve Active and Stable Catalysts in Methane Dry Reforming. ChemCatChem 2020, 12, 373–385. [Google Scholar] [CrossRef]
- Price, C.-A.H.; Pastor-Pérez, L.; Ramirez Reina, T.; Liu, J. Robust Mesoporous Bimetallic Yolk–Shell Catalysts for Chemical CO2 Upgrading via Dry Reforming of Methane. React. Chem. Eng. 2018, 3, 433–436. [Google Scholar] [CrossRef]
- Chin, K.C.; Leong, L.K.; Lu, S.-Y.; Tsai, D.-H.; Sethupathi, S. Preparation of Metal Organic Framework (MOF) Derived Bimetallic Catalyst for Dry Reforming of Methane. Int. J. Technol. 2019, 10, 1437. [Google Scholar] [CrossRef]
- Vakili, R.; Gholami, R.; Stere, C.E.; Chansai, S.; Chen, H.; Holmes, S.M.; Jiao, Y.; Hardacre, C.; Fan, X. Plasma-Assisted Catalytic Dry Reforming of Methane (DRM) over Metal-Organic Frameworks (MOFs)-Based Catalysts. Appl. Catal. B Environ. 2020, 260, 118195. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Studt, F.; Abild-Pedersen, F.; Bligaard, T. Fundamental Concepts in Heterogeneous Catalysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; ISBN 9781118892114. [Google Scholar]
- Montemore, M.M.; Medlin, J.W. Scaling Relations between Adsorption Energies for Computational Screening and Design of Catalysts. Catal. Sci. Technol. 2014, 4, 3748–3761. [Google Scholar] [CrossRef]
- Medlin, J.W.; Montemore, M.M. Scaling the Rough Heights. Nat. Chem. 2015, 7, 378–380. [Google Scholar] [CrossRef]
- Chowdhury, A.J.; Yang, W.; Walker, E.; Mamun, O.; Heyden, A.; Terejanu, G.A. Prediction of Adsorption Energies for Chemical Species on Metal Catalyst Surfaces Using Machine Learning. J. Phys. Chem. C 2018, 122, 28142–28150. [Google Scholar] [CrossRef]
- Chowdhury, A.J.; Yang, W.; Abdelfatah, K.E.; Zare, M.; Heyden, A.; Terejanu, G.A. A Multiple Filter Based Neural Network Approach to the Extrapolation of Adsorption Energies on Metal Surfaces for Catalysis Applications. J. Chem. Theory Comput. 2020, 16, 1105–1114. [Google Scholar] [CrossRef]
- Medford, A.J.; Kunz, M.R.; Ewing, S.M.; Borders, T.; Fushimi, R. Extracting Knowledge from Data through Catalysis Informatics. ACS Catal. 2018, 8, 7403–7429. [Google Scholar] [CrossRef]
- Ulissi, Z.W.; Medford, A.J.; Bligaard, T.; Nørskov, J.K. To Address Surface Reaction Network Complexity Using Scaling Relations Machine Learning and DFT Calculations. Nat. Commun. 2017, 8, 14621. [Google Scholar] [CrossRef] [PubMed]
- Mamun, O.; Winther, K.T.; Boes, J.R.; Bligaard, T. High-Throughput Calculations of Catalytic Properties of Bimetallic Alloy Surfaces. Sci. Data 2019, 6, 76. [Google Scholar] [CrossRef] [PubMed]
- Winther, K.T.; Hoffmann, M.J.; Boes, J.R.; Mamun, O.; Bajdich, M.; Bligaard, T. Catalysis-Hub.Org, an Open Electronic Structure Database for Surface Reactions. Sci. Data 2019, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.; Ammal, S.C.; Terejanu, G.A.; Heyden, A. Uncertainty Quantification Framework Applied to the Water–Gas Shift Reaction over Pt-Based Catalysts. J. Phys. Chem. C 2016, 120, 10328–10339. [Google Scholar] [CrossRef]
- Sutton, J.E.; Guo, W.; Katsoulakis, M.A.; Vlachos, D.G. Effects of Correlated Parameters and Uncertainty in Electronic-Structure-Based Chemical Kinetic Modelling. Nat. Chem. 2016, 8, 331–337. [Google Scholar] [CrossRef]
- Walker, E.A.; Mitchell, D.; Terejanu, G.A.; Heyden, A. Identifying Active Sites of the Water–Gas Shift Reaction over Titania Supported Platinum Catalysts under Uncertainty. ACS Catal. 2018, 8, 3990–3998. [Google Scholar] [CrossRef]
- Watwe, R.M.; Bengaard, H.S.; Rostrup-Nielsen, J.R.; Dumesic, J.A.; Nørskov, J.K. Theoretical Studies of Stability and Reactivity of CHx Species on Ni(111). J. Catal. 2000, 189, 16–30. [Google Scholar] [CrossRef]
- Michaelides, A.; Hu, P. Methyl Chemisorption on Ni(111) and C H M Multicentre Bonding: A Density Functional Theory Study. Surf. Sci. 1999, 437, 362–376. [Google Scholar] [CrossRef]
- Michaelides, A.; Hu, P. A First Principles Study of CH3 Dehydrogenation on Ni(111). J. Chem. Phys. 2000, 112, 8120–8125. [Google Scholar] [CrossRef]
- Michaelides, A.; Hu, P. A Density Functional Theory Study of CH2 and H Adsorption on Ni(111). J. Chem. Phys. 2000, 112, 6006–6014. [Google Scholar] [CrossRef][Green Version]
- Zhu, Y.A.; Chen, D.; Zhou, X.G.; Yuan, W.K. DFT Studies of Dry Reforming of Methane on Ni Catalyst. Catal. Today 2009, 148, 260–267. [Google Scholar] [CrossRef]
- Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. CO2 Dissociation on Ni(211). Surf. Sci. 2009, 603, 2991–2998. [Google Scholar] [CrossRef]
- Wang, S.-G.; Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. Chemisorption of CO2 on Nickel Surfaces. J. Phys. Chem. B 2005, 109, 18956–18963. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-G.; Liao, X.-Y.; Hu, J.; Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. Kinetic Aspect of CO2 Reforming of CH4 on Ni(111): A Density Functional Theory Calculation. Surf. Sci. 2007, 601, 1271–1284. [Google Scholar] [CrossRef]
- Wang, S.-G.; Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. CO2 Reforming of CH4 on Ni(111): A Density Functional Theory Calculation. J. Phys. Chem. B 2006, 110, 9976–9983. [Google Scholar] [CrossRef]
- Ko, J.; Kim, B.-K.; Han, J.W. Density Functional Theory Study for Catalytic Activation and Dissociation of CO2 on Bimetallic Alloy Surfaces. J. Phys. Chem. C 2016, 120, 3438–3447. [Google Scholar] [CrossRef]
- Schlexer, P.; Chen, H.-Y.T.; Pacchioni, G. CO2 Activation and Hydrogenation: A Comparative DFT Study of Ru10/TiO2 and Cu10/TiO2 Model Catalysts. Catal. Lett. 2017, 147, 1871–1881. [Google Scholar] [CrossRef]
- Takanabe, K.; Basset, J.-M.; Park, J.-H.; Samal, A.; Alsabban, B. Boron-Containing Catalysts for Dry Reforming of Methane to Synthesis Gas. U.S. Patent 20190330059A1, 31 October 2019. [Google Scholar]
- Ko, C.H.; Jeon, J.Y.; Yun, J.W.; Park, E.G. Perovskite Metal Oxide Catalyst, in Which Metal Ion Is Substituted, for Reducing Carbon Deposition, Preparation Method Therefor, and Methane Reforming Reaction Method Using Same. U.S. Patent 20200269217A1, 27 August 2020. [Google Scholar]
- Methane Dry Reforming Reaction, Catalyst Containing Nickel and Cerium and with Core-Shell Structure for Methane Dry Reforming Reaction and Preparation Method of Catalyst. Patent CN 107921427B, 3 August 2021.
- Ying, J.Y.; Weiss, S.E. Method of Dry Reforming a Reactant Gas with Intermetallic Catalyst. U.S. Patent 8173010B2, 8 May 2012. [Google Scholar]
- Wu, S.; Xu, J.; Liu, H.; Lin, Q.; Xiao, H. Method of Coupling Methane Dry-Reforming and Composite Catalyst Regeneration. U.S. Patent 20200368728A1, 26 November 2020. [Google Scholar]
- Nickel-Molybdenum Carbide Composite Catalyst for Preparing Synthesis Gas through Dry Reforming of Methane. Patent CN 105107515B, 19 April 2017.
- The Preparation Method of the Ni-Based Methane Dry Reforming Catalyst of Core Shell Structure. Patent CN 104998649B, 25 July 2017.
- Preparation Method of Anti-Carbon Accumulation and Anti-Sintering Methane Dry Reforming Ni-Based Catalyst. Patent CN 105964261A, 28 September 2016.
- Ravon, U.; D’Souza, L.; Viswanath, V. Use of Hollow Zeolites Doped with Bimetallic or Trimetallic Particles for Hydrocarbon Reforming Reactions. Patent WO 2017072698A1, 1 November 2018. [Google Scholar]
- Siriwardane, R.V. Metal Ferrite Catalyst for Conversion of CO2 and Methane to Synthesis Gas via Reforming. U.S. Patent 10427138B1, 1 October 2019. [Google Scholar]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussien, A.G.S.; Polychronopoulou, K. A Review on the Different Aspects and Challenges of the Dry Reforming of Methane (DRM) Reaction. Nanomaterials 2022, 12, 3400. https://doi.org/10.3390/nano12193400
Hussien AGS, Polychronopoulou K. A Review on the Different Aspects and Challenges of the Dry Reforming of Methane (DRM) Reaction. Nanomaterials. 2022; 12(19):3400. https://doi.org/10.3390/nano12193400
Chicago/Turabian StyleHussien, Aseel G. S., and Kyriaki Polychronopoulou. 2022. "A Review on the Different Aspects and Challenges of the Dry Reforming of Methane (DRM) Reaction" Nanomaterials 12, no. 19: 3400. https://doi.org/10.3390/nano12193400
APA StyleHussien, A. G. S., & Polychronopoulou, K. (2022). A Review on the Different Aspects and Challenges of the Dry Reforming of Methane (DRM) Reaction. Nanomaterials, 12(19), 3400. https://doi.org/10.3390/nano12193400