
An Atomistic Model Describing the Structure and Morphology of Cu-Doped C-S-H Hardening Accelerator Nanoparticles

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
1.1. A Brief Overview on Available C-S-H Structural Models
2. Materials and Methods
2.1. Synthesis of Me-S-H/PCE Nanocomposite—Mapefast Ultra
2.2. Small-Angle X-ray Scattering (SAXS)
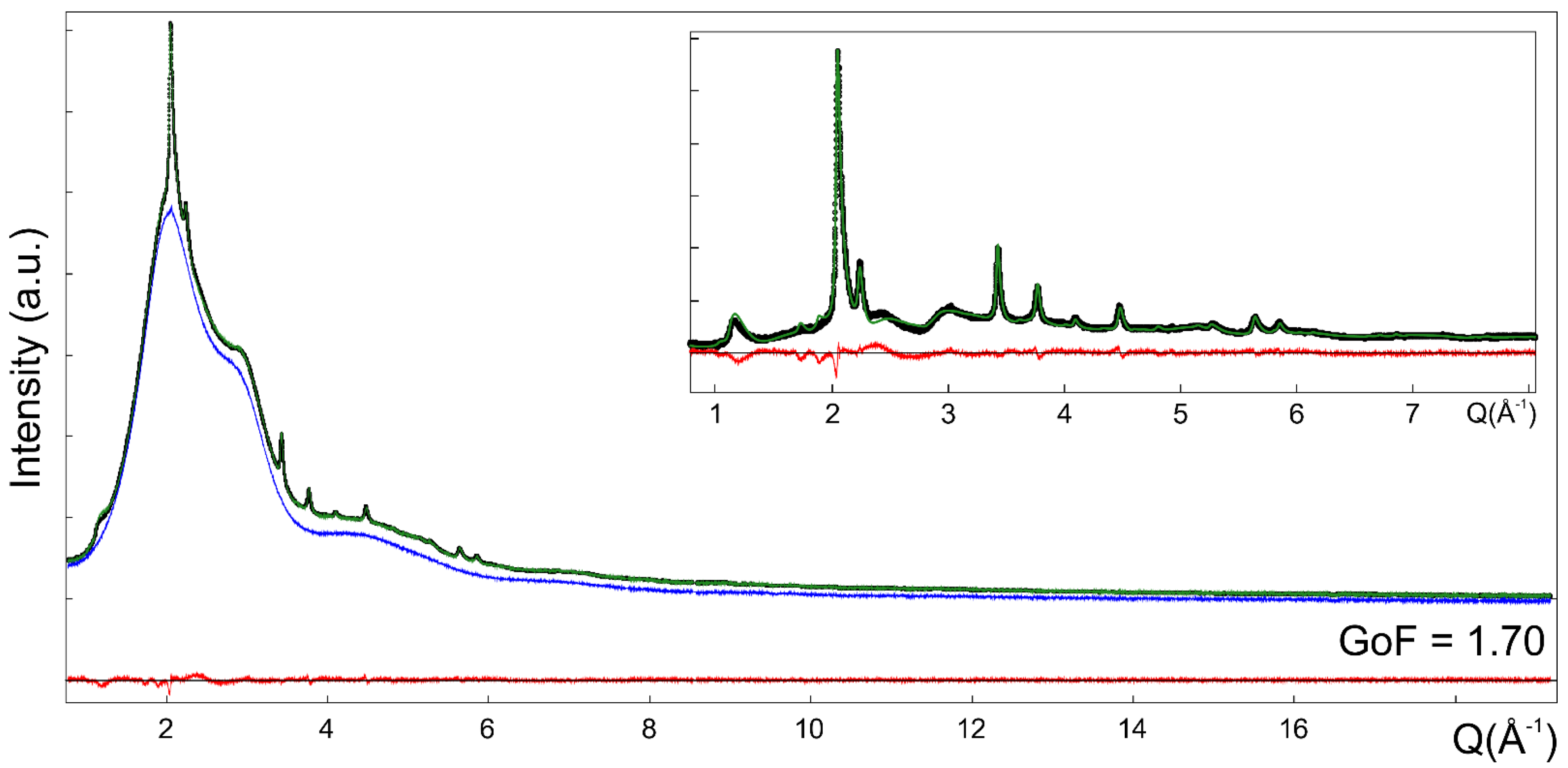
2.3. Synchrotron Wide Angle X-ray Total Scattering (WAXTS)
2.4. The Debye Scattering Equation (DSE) Method
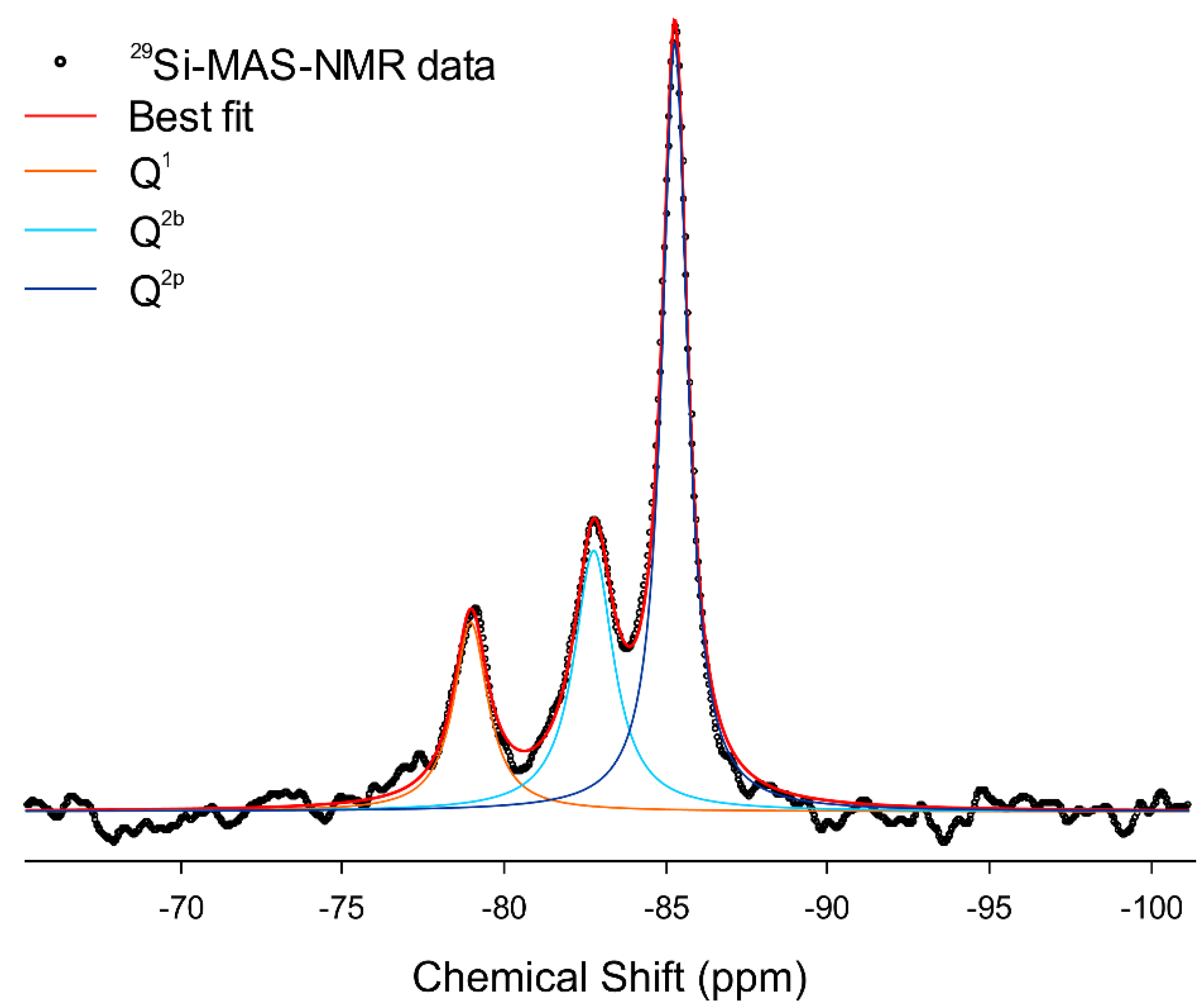
2.5. 29Si Magic-Angle-Spinning Nuclear Magnetic Resonance (NMR)
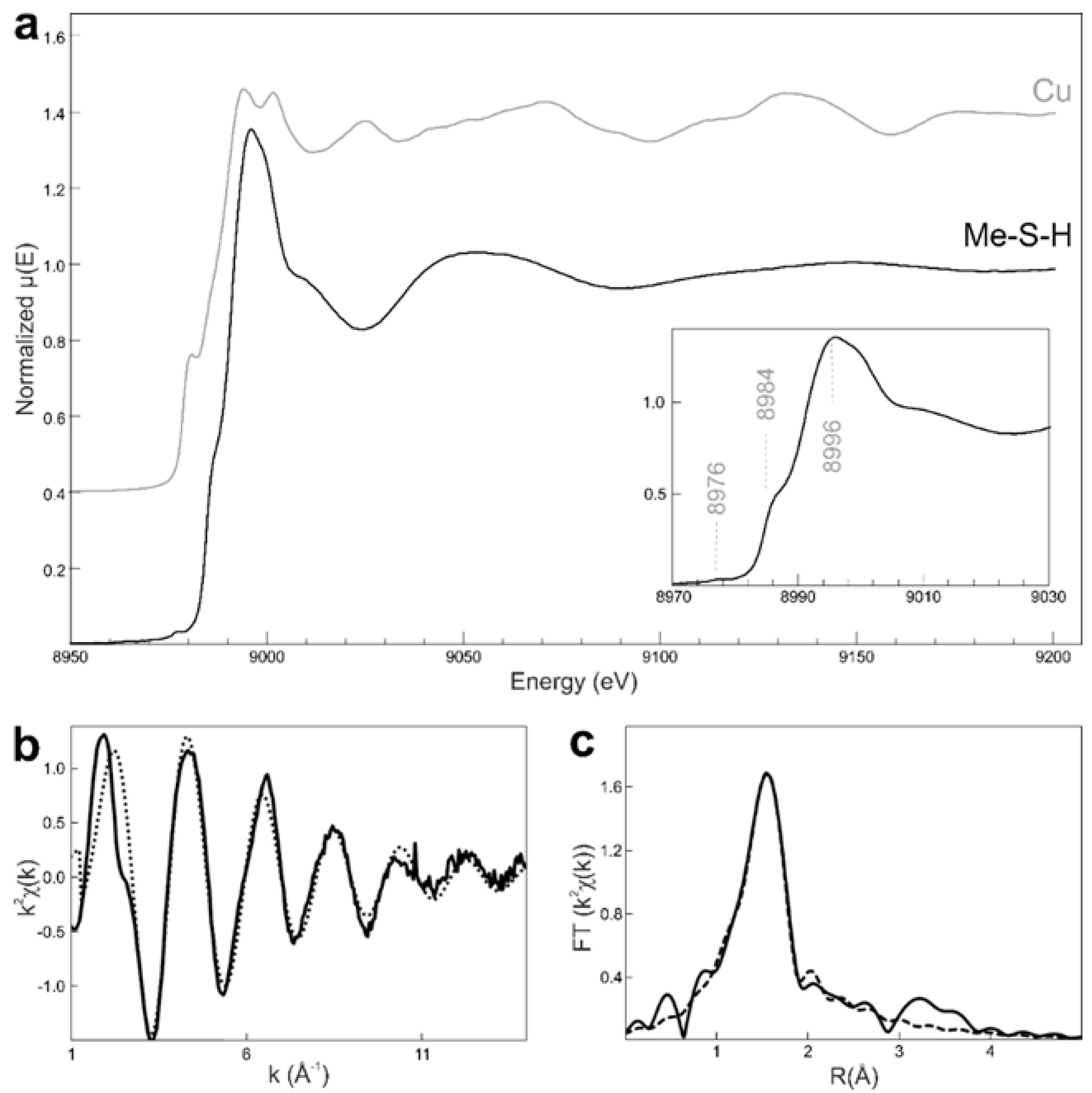
2.6. Cu K-Edge X-ray Absorption Spectroscopy (XAS)
3. Results and Discussion
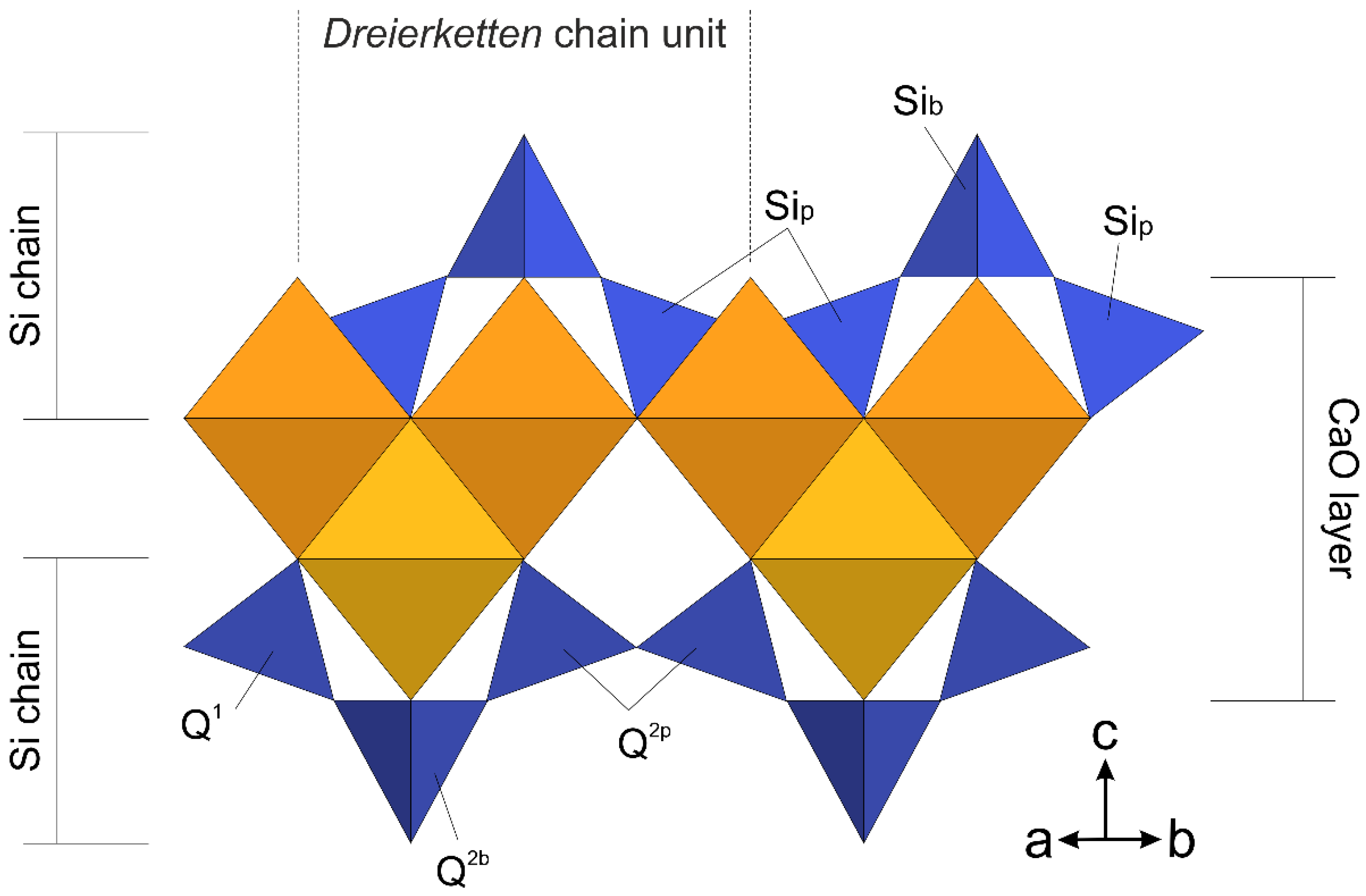
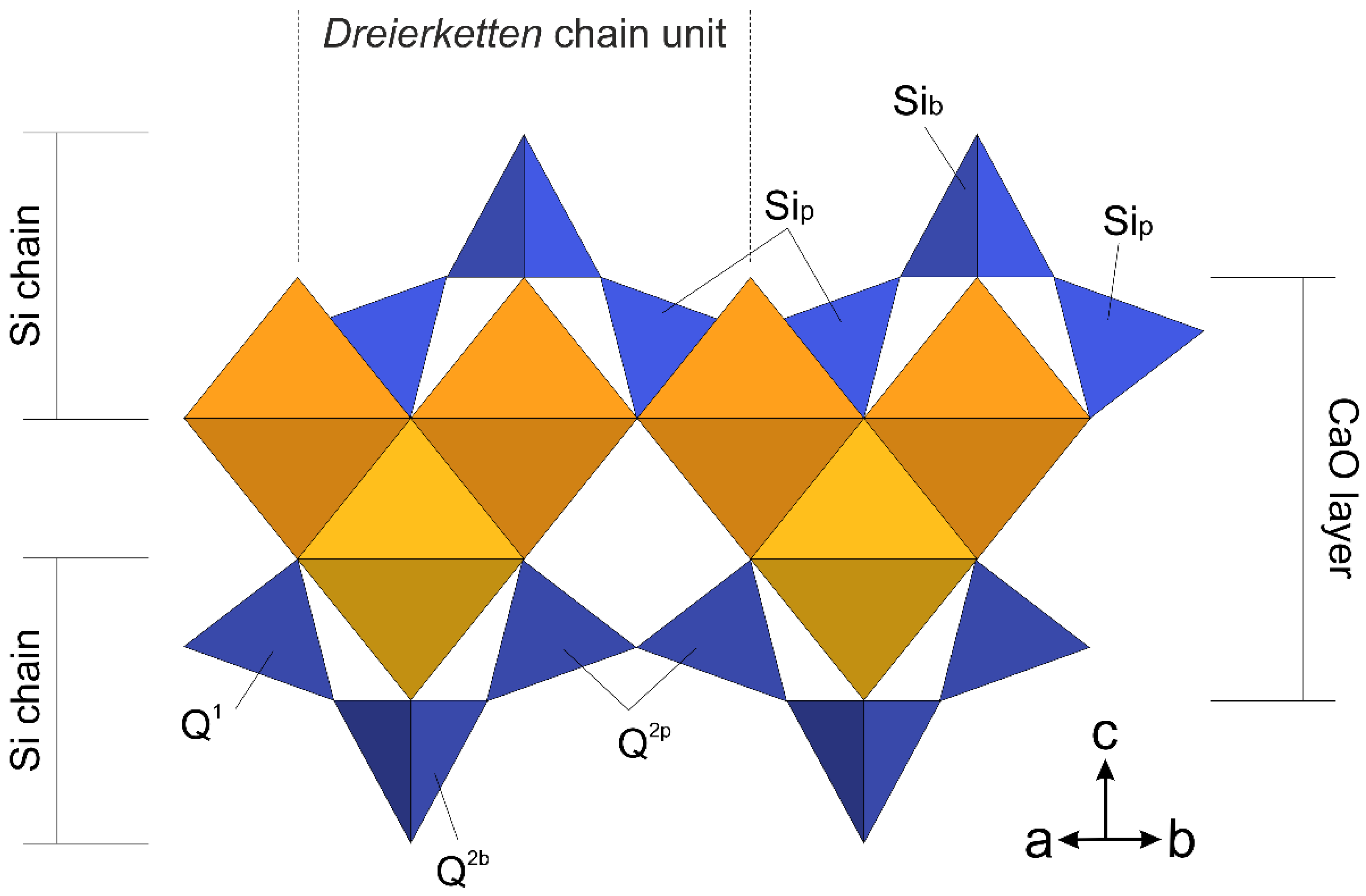
3.1. Defining a Suitable Atomistic Model for Me-S-H Nanoparticles
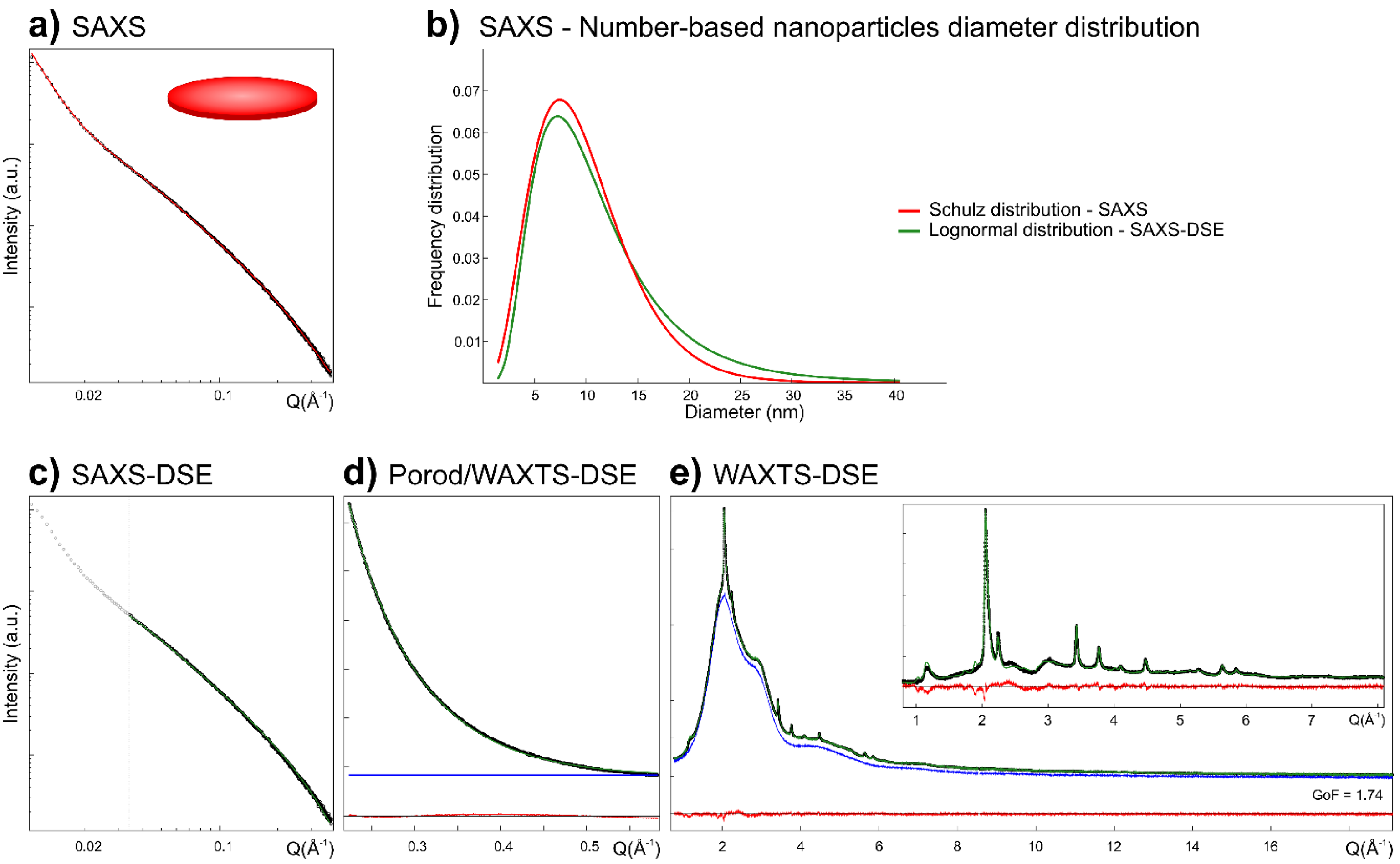
3.2. Me-S-H Nanoparticles Size and Morphology by SAXS Analysis
3.2.1. Conventional SAXS Modeling
3.2.2. SAXS Modeling Based on the DSE
3.3. WAXTS-DSE Analysis of Me-S-H Nanoparticles
3.4. Combining 29Si MAS-NMR and WAXTS-DSE Analyses to Describe the Silicate Tetrahedra Connectivity
3.5. On the Effective Copper Incorporation into the Me-S-H Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scrivener, K.L.; Juilland, P.; Monteiro, P.J.M. Advances in understanding hydration of Portland cement. Cem. Concr. Res. 2015, 78, 38–56. [Google Scholar] [CrossRef]
- Valentini, L.; Favero, M.; Dalconi, M.C.; Russo, V.; Ferrari, G.; Artioli, G. Kinetic Model of Calcium-Silicate Hydrate Nucleation and Growth in the Presence of PCE Superplasticizers. Cryst. Growth Des. 2016, 16, 646–654. [Google Scholar] [CrossRef]
- Valentini, L.; Ferrari, G.; Russo, V.; Štefančič, M.; Zalar Serjun, V.; Artioli, G. Use of nanocomposites as permeability reducing admixtures. J. Am. Ceram. Soc. 2018, 101, 4275–4284. [Google Scholar] [CrossRef] [Green Version]
- Artioli, G.; Valentini, L.; Dalconi, M.C.; Parisatto, M.; Voltolini, M.; Russo, V.; Ferrari, G. Imaging of nano-seeded nucleation in cement pastes by X-ray diffraction tomography. Int. J. Mater. Res. 2014, 105, 628–631. [Google Scholar] [CrossRef]
- Provis, J.L. Alkali-activated materials. Cem. Concr. Res. 2018, 114, 40–48. [Google Scholar] [CrossRef]
- Lothenbach, B.; Scrivener, K.; Hooton, R.D. Supplementary cementitious materials. Cem. Concr. Res. 2011, 41, 1244–1256. [Google Scholar] [CrossRef]
- Ez-zaki, H.; Marangu, J.M.; Bellotto, M.; Dalconi, M.C.; Artioli, G.; Valentini, L. A Fresh View on Limestone Calcined Clay Cement (LC3) Pastes. Materials 2021, 14, 3037. [Google Scholar] [CrossRef]
- White, C.E.; Daemen, L.L.; Hartl, M.; Page, K. Intrinsic differences in atomic ordering of calcium (alumino)silicate hydrates in conventional and alkali-activated cements. Cem. Concr. Res. 2015, 67, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Grangeon, S.; Fernandez-Martinez, A.; Baronnet, A.; Marty, N.; Poulain, A.; Elkaïm, E.; Roosz, C.; Gaboreau, S.; Henocq, P.; Claret, F. Quantitative X-ray pair distribution function analysis of nanocrystalline calcium silicate hydrates: A contribution to the understanding of cement chemistry. J. Appl. Crystallogr. 2017, 50, 14–21. [Google Scholar] [CrossRef]
- Meral, C.; Benmore, C.J.; Monteiro, P.J.M. The study of disorder and nanocrystallinity in C–S–H, supplementary cementitious materials and geopolymers using pair distribution function analysis. Cem. Concr. Res. 2011, 41, 696–710. [Google Scholar] [CrossRef]
- Theobald, M.; Plank, J. β-Naphthalene sulfonate formaldehyde-based nanocomposites as new seeding materials for Portland cement. Constr. Build. Mater. 2020, 264, 120240. [Google Scholar] [CrossRef]
- John, E.; Matschei, T.; Stephan, D. Nucleation seeding with calcium silicate hydrate—A review. Cem. Concr. Res. 2018, 113, 74–85. [Google Scholar] [CrossRef]
- Artioli, G.; Ferrari, G.; Dalconi, M.C.; Valentini, L. Nanoseeds as modifiers of the cement hydration kinetics. In Smart Nanoconcretes and Cement-Based Materials; Elsevier: Amsterdam, The Netherlands, 2020; pp. 257–269. ISBN 9780128178553. [Google Scholar]
- Zou, F.; Zhang, M.; Hu, C.; Wang, F.; Hu, S. Novel C-A-S-H/PCE nanocomposites: Design, characterization and the effect on cement hydration. Chem. Eng. J. 2021, 412, 128569. [Google Scholar] [CrossRef]
- Pedrosa, H.C.; Reales, O.M.; Reis, V.D.; das Dores Paiva, M.; Fairbairn, E.M.R. Hydration of Portland cement accelerated by C-S-H seeds at different temperatures. Cem. Concr. Res. 2020, 129, 105978. [Google Scholar] [CrossRef]
- Wang, F.; Kong, X.; Jiang, L.; Wang, D. The acceleration mechanism of nano-C-S-H particles on OPC hydration. Constr. Build. Mater. 2020, 249, 118734. [Google Scholar] [CrossRef]
- Sun, J.; Shi, H.; Qian, B.; Xu, Z.; Li, W.; Shen, X. Effects of synthetic C-S-H/PCE nanocomposites on early cement hydration. Constr. Build. Mater. 2017, 140, 282–292. [Google Scholar] [CrossRef]
- Provis, J.L.; Hajimohammadi, A.; White, C.E.; Bernal, S.A.; Myers, R.J.; Winarski, R.P.; Rose, V.; Proffen, T.E.; Llobet, A.; Van Deventer, J.S.J. Nanostructural characterization of geopolymers by advanced beamline techniques. Cem. Concr. Compos. 2013, 36, 56–64. [Google Scholar] [CrossRef]
- Grangeon, S.; Claret, F.; Roosz, C.; Sato, T.; Gaboreau, S.; Linard, Y. Structure of nanocrystalline calcium silicate hydrates: Insights from X-ray diffraction, synchrotron X-ray absorption and nuclear magnetic resonance. J. Appl. Crystallogr. 2016, 49, 771–783. [Google Scholar] [CrossRef]
- Kunhi Mohamed, A.; Parker, S.C.; Bowen, P.; Galmarini, S. An atomistic building block description of C-S-H—Towards a realistic C-S-H model. Cem. Concr. Res. 2018, 107, 221–235. [Google Scholar] [CrossRef]
- Li, J.; Geng, G.; Myers, R.; Yu, Y.S.; Shapiro, D.; Carraro, C.; Maboudian, R.; Monteiro, P.J.M. The chemistry and structure of calcium (alumino) silicate hydrate: A study by XANES, ptychographic imaging, and wide- and small-angle scattering. Cem. Concr. Res. 2019, 115, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Nonat, A. The structure and stoichiometry of C-S-H. Cem. Concr. Res. 2004, 34, 1521–1528. [Google Scholar] [CrossRef]
- Kumar, A.; Walder, B.J.; Kunhi Mohamed, A.; Hofstetter, A.; Srinivasan, B.; Rossini, A.J.; Scrivener, K.; Emsley, L.; Bowen, P. The Atomic-Level Structure of Cementitious Calcium Silicate Hydrate. J. Phys. Chem. C 2017, 121, 17188–17196. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.; Santacruz, I.; De la Torre, A.G.; Dapiaggi, M.; Zea-Garcia, J.D.; Aranda, M.A.G. Local structure and Ca/Si ratio in C-S-H gels from hydration of blends of tricalcium silicate and silica fume. Cem. Concr. Res. 2021, 143, 106405. [Google Scholar] [CrossRef]
- Walkley, B.; Provis, J.L. Solid-state nuclear magnetic resonance spectroscopy of cements. Mater. Today Adv. 2019, 1, 100007. [Google Scholar] [CrossRef]
- Richardson, I.G. The calcium silicate hydrates. Cem. Concr. Res. 2008, 38, 137–158. [Google Scholar] [CrossRef]
- Kirkpatrick, R.J.; Yarger, J.L.; McMillan, P.F.; Ping, Y.; Cong, X. Raman spectroscopy of C-S-H, tobermorite, and jennite. Adv. Cem. Based Mater. 1997, 5, 93–99. [Google Scholar] [CrossRef]
- Lolli, F.; Manzano, H.; Provis, J.L.; Bignozzi, M.C.; Masoero, E. Atomistic Simulations of Geopolymer Models: The Impact of Disorder on Structure and Mechanics. ACS Appl. Mater. Interfaces 2018, 10, 22809–22820. [Google Scholar] [CrossRef] [Green Version]
- Grangeon, S.; Claret, F.; Linard, Y.; Chiaberge, C. X-ray diffraction: A powerful tool to probe and understand the structure of nanocrystalline calcium silicate hydrates. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Grangeon, S.; Claret, F.; Lerouge, C.; Warmont, F.; Sato, T.; Anraku, S.; Numako, C.; Linard, Y.; Lanson, B. On the nature of structural disorder in calcium silicate hydrates with a calcium/silicon ratio similar to tobermorite. Cem. Concr. Res. 2013, 52, 31–37. [Google Scholar] [CrossRef]
- Renaudin, G.; Russias, J.; Leroux, F.; Frizon, F.; Cau-dit-Coumes, C. Structural characterization of C–S–H and C–A–S–H samples—Part I: Long-range order investigated by Rietveld analyses. J. Solid State Chem. 2009, 182, 3312–3319. [Google Scholar] [CrossRef]
- Bertolotti, F.; Moscheni, D.; Guagliardi, A.; Masciocchi, N. When Crystals Go Nano—The Role of Advanced X-ray Total Scattering Methods in Nanotechnology. Eur. J. Inorg. Chem. 2018, 3789–3803. [Google Scholar] [CrossRef]
- Cervellino, A.; Frison, R.; Masciocchi, N.; Guagliardi, A. X-Ray Powder Diffraction Characterization of Nanomaterials. In X-ray and Neutron Techniques for Nanomaterials Characterization; Kumar, C.S.S.R., Ed.; Springer-Verlag GmbH Germany: Berlin/Heidelberg, Germany,, 2016; pp. 545–608. ISBN 978-3-662-48606-1. [Google Scholar]
- Debye, P. Zerstreuung von Röntgenstrahlen. Ann. Phys. 1915, 351, 809–823. [Google Scholar] [CrossRef] [Green Version]
- Cervellino, A.; Frison, R.; Bertolotti, F.; Guagliardi, A. DEBUSSY 2.0: The new release of a Debye user system for nanocrystalline and/or disordered materials. J. Appl. Crystallogr. 2015, 48, 2026–2032. [Google Scholar] [CrossRef] [Green Version]
- Egami, T.; Billinge, S.J.L. Underneath the Bragg Peaks; Pergamon Press: New York, NY, USA, 2003. [Google Scholar]
- Cuesta, A.; Zea-Garcia, J.D.; Londono-Zuluaga, D.; De la Torre, A.G.; Santacruz, I.; Vallcorba, O.; Aranda, M. Synchrotron Radiation Pair Distribution Function Analysis of Gels in Cements. Crystals 2017, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- White, C.E.; Provis, J.L.; Bloomer, B.; Henson, N.J.; Page, K. In situ X-ray pair distribution function analysis of geopolymer gel nanostructure formation kinetics. Phys. Chem. Chem. Phys. 2013, 15, 8573–8582. [Google Scholar] [CrossRef]
- White, C.E.; Provis, J.L.; Llobet, A.; Proffen, T.; Van Deventer, J.S.J. Evolution of local structure in geopolymer gels: An in situ neutron pair distribution function analysis. J. Am. Ceram. Soc. 2011, 94, 3532–3539. [Google Scholar] [CrossRef]
- Bertolotti, F.; Carmona, F.J.; Dal Sasso, G.; Ramírez-Rodríguez, G.B.; Delgado-López, J.M.; Pedersen, J.S.; Ferri, F.; Masciocchi, N.; Guagliardi, A. On the amorphous layer in bone mineral and biomimetic apatite: A combined small- and wide-angle X-ray scattering analysis. Acta Biomater. 2021, 120, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Carmona, F.J.; Dal Sasso, G.; Bertolotti, F.; Ramírez-Rodríguez, G.B.; Delgado-López, J.M.; Pedersen, J.S.; Masciocchi, N.; Guagliardi, A. The role of nanoparticle structure and morphology in the dissolution kinetics and nutrient release of nitrate-doped calcium phosphate nanofertilizers. Sci. Rep. 2020, 10, 12396. [Google Scholar] [CrossRef]
- Moscheni, D.; Bertolotti, F.; Piveteau, L.; Protesescu, L.; Dirin, D.N.; Kovalenko, M.V.; Cervellino, A.; Pedersen, J.S.; Masciocchi, N.; Guagliardi, A. Size-Dependent Fault-Driven Relaxation and Faceting in Zincblende CdSe Colloidal Quantum Dots. ACS Nano 2018, 12, 12558–12570. [Google Scholar] [CrossRef]
- Cervellino, A.; Giannini, C.; Guagliardi, A. On the efficient evaluation of Fourier patterns for nanoparticles and clusters. J. Comput. Chem. 2006, 27, 995–1008. [Google Scholar] [CrossRef]
- TAYLOR, H.F.W. Proposed Structure for Calcium Silicate Hydrate Gel. J. Am. Ceram. Soc. 1986, 69, 464–467. [Google Scholar] [CrossRef]
- Richardson, I. The nature of C-S-H in hardened cements. Cem. Concr. Res. 1999, 29, 1131–1147. [Google Scholar] [CrossRef]
- Allen, A.J.; Thomas, J.J.; Jennings, H.M. Composition and density of nanoscale calcium-silicate-hydrate in cement. Nat. Mater. 2007, 6, 311–316. [Google Scholar] [CrossRef]
- Lothenbach, B.; Kulik, D.A.; Matschei, T.; Balonis, M.; Baquerizo, L.; Dilnesa, B.; Miron, G.D.; Myers, R.J. Cemdata18: A chemical thermodynamic database for hydrated Portland cements and alkali-activated materials. Cem. Concr. Res. 2019, 115, 472–506. [Google Scholar] [CrossRef] [Green Version]
- Roosz, C.; Vieillard, P.; Blanc, P.; Gaboreau, S.; Gailhanou, H.; Braithwaite, D.; Montouillout, V.; Denoyel, R.; Henocq, P.; Madé, B. Thermodynamic properties of C-S-H, C-A-S-H and M-S-H phases: Results from direct measurements and predictive modelling. Appl. Geochem. 2018, 92, 140–156. [Google Scholar] [CrossRef]
- Merlino, S.; Bonaccorsi, E.; Armbruster, T. The real structure of tobermorite 11A: Normal and anomalous forms, OD character and polytypic modifications. Eur. J. Mineral. 2001, 13, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Merlino, S.; Bonaccorsi, E.; Armbruster, T. Tobermorites; their real structure and order-disorder (OD) character. Am. Mineral. 1999, 84, 1613–1621. [Google Scholar] [CrossRef]
- Bonaccorsi, E.; Merlino, S.; Kampf, A.R. The Crystal Structure of Tobermorite 14 A (Plombierite), a C-S-H Phase. J. Am. Ceram. Soc. 2005, 88, 505–512. [Google Scholar] [CrossRef]
- Merlino, S.; Bonaccorsi, E.; Armbruster, T. The real structures of clinotobermorite and tobermorite 9 Å: OD character, polytypes, and structural relationships. Eur. J. Mineral. 2000, 12, 411–429. [Google Scholar] [CrossRef]
- Hamid, S.A. The crystal structure of the 11 Ä natural tobermorite Ca2.25[Si3O7.5(OH)1.5]·1H2O. Z. Für Krist.-Cryst. Mater. 1981, 154, 189–198. [Google Scholar] [CrossRef]
- Gaboreau, S.; Grangeon, S.; Claret, F.; Ihiawakrim, D.; Ersen, O.; Montouillout, V.; Maubec, N.; Roosz, C.; Henocq, P.; Carteret, C. Hydration Properties and Interlayer Organization in Synthetic C-S-H. Langmuir 2020, 36, 9449–9464. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Zea-Garcia, J.D.; Londono-Zuluaga, D.; De La Torre, A.G.; Santacruz, I.; Vallcorba, O.; Dapiaggi, M.; Sanfélix, S.G.; Aranda, M.A.G. Multiscale understanding of tricalcium silicate hydration reactions. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Basquiroto de Souza, F.; Sagoe-Crentsil, K.; Duan, W. Determining the disordered nanostructure of calcium silicate hydrate (C-S-H) from broad X-ray diffractograms. J. Am. Ceram. Soc. 2022, 105, 1491–1502. [Google Scholar] [CrossRef]
- Pedersen, J.S. A flux- and background-optimized version of the NanoSTAR small-angle X-ray scattering camera for solution scattering. J. Appl. Crystallogr. 2004, 37, 369–380. [Google Scholar] [CrossRef]
- Pedersen, J.S. X-Ray Analyzing System for X-Ray Scattering Analysis. U.S. Patent 9,958,404, 1 May 2018. [Google Scholar]
- Lyngsø, J.; Pedersen, J.S. A high-flux automated laboratory small-angle X-ray scattering instrument optimized for solution scattering. J. Appl. Crystallogr. 2021, 54, 295–305. [Google Scholar] [CrossRef]
- Fournet, G. Étude théorique et expérimentale de la diffusion des rayons X par les ensembles denses de particules. Bull. Société Française Minéralogie Cristallogr. 1951, 74, 37–172. [Google Scholar] [CrossRef]
- Schulz, G.V. Über die Kinetik der Kettenpolymerisationen. V. Z. Für Phys. Chem. 1939, 43B, 25–46. [Google Scholar] [CrossRef]
- Pedersen, J.S. Analysis of small-angle scattering data from colloids and polymer solutions: Modeling and least-squares fitting. Adv. Colloid Interface Sci. 1997, 70, 171–210. [Google Scholar] [CrossRef]
- Willmott, P.R.; Meister, D.; Leake, S.J.; Lange, M.; Bergamaschi, A.; Böge, M.; Calvi, M.; Cancellieri, C.; Casati, N.; Cervellino, A.; et al. The Materials Science beamline upgrade at the Swiss Light Source. J. Synchrotron Radiat. 2013, 20, 667–682. [Google Scholar] [CrossRef]
- Gordon, T.R.; Diroll, B.T.; Paik, T.; Doan-Nguyen, V.V.T.; Gaulding, E.A.; Murray, C.B. Characterization of shape and monodispersity of anisotropic Nanocrystals through atomistic x-ray scattering simulation. Chem. Mater. 2015, 27, 2502–2506. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Di Cicco, A.; Aquilanti, G.; Minicucci, M.; Principi, E.; Novello, N.; Cognigni, A.; Olivi, L. Novel XAFS capabilities at ELETTRA synchrotron light source. J. Phys. Conf. Ser. 2009, 190, 012043. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, F.; Dirin, D.N.; Ibáñez, M.; Krumeich, F.; Cervellino, A.; Frison, R.; Voznyy, O.; Sargent, E.H.; Kovalenko, M.V.; Guagliardi, A.; et al. Crystal symmetry breaking and vacancies in colloidal lead chalcogenide quantum dots. Nat. Mater. 2016, 15, 987–994. [Google Scholar] [CrossRef]
- Larsen, A.H.; Pedersen, J.S.; Arleth, L. Assessment of structure factors for analysis of small-angle scattering data from desired or undesired aggregates. J. Appl. Crystallogr. 2020, 53, 991–1005. [Google Scholar] [CrossRef]
- Chiang, Y.; Chang, S.-W. Bridging the gap between NMR measured mean silicate chain length and nano-scale silicate polymorphism of calcium silicate hydrates. Cem. Concr. Res. 2021, 140, 106268. [Google Scholar] [CrossRef]
- Kau, L.S.; Spira-Solomon, D.J.; Penner-Hahn, J.E.; Hodgson, K.O.; Solomon, E.I. X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc. 1987, 109, 6433–6442. [Google Scholar] [CrossRef]
- Persson, I.; Lundberg, D.; Bajnóczi, É.G.; Klementiev, K.; Just, J.; Sigfridsson Clauss, K.G.V. EXAFS Study on the Coordination Chemistry of the Solvated Copper(II) Ion in a Series of Oxygen Donor Solvents. Inorg. Chem. 2020, 59, 9538–9550. [Google Scholar] [CrossRef]
- Persson, I.; Persson, P.; Sandström, M.; Ullström, A.-S. Structure of Jahn–Teller distorted solvated copper(ii) ions in solution, and in solids with apparently regular octahedral coordination geometry. J. Chem. Soc. Dalt. Trans. 2002, 1256. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Shell | N | S02 | R(Å) | σ2(Å2) | ΔE (eV) | R-Factor |
|---|---|---|---|---|---|---|
| Cu-O | 4 | 0.90 (4) | 1.945 (5) | 0.0039(6) | 5.9 (6) | 0.002 |
| k-range: 3.2–13.0; R-range: 1.1–2.3; k-weight: 1,2 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Sasso, G.; Dalconi, M.C.; Ferrari, G.; Pedersen, J.S.; Tamburini, S.; Bertolotti, F.; Guagliardi, A.; Bruno, M.; Valentini, L.; Artioli, G. An Atomistic Model Describing the Structure and Morphology of Cu-Doped C-S-H Hardening Accelerator Nanoparticles. Nanomaterials 2022, 12, 342. https://doi.org/10.3390/nano12030342
Dal Sasso G, Dalconi MC, Ferrari G, Pedersen JS, Tamburini S, Bertolotti F, Guagliardi A, Bruno M, Valentini L, Artioli G. An Atomistic Model Describing the Structure and Morphology of Cu-Doped C-S-H Hardening Accelerator Nanoparticles. Nanomaterials. 2022; 12(3):342. https://doi.org/10.3390/nano12030342
Chicago/Turabian StyleDal Sasso, Gregorio, Maria Chiara Dalconi, Giorgio Ferrari, Jan Skov Pedersen, Sergio Tamburini, Federica Bertolotti, Antonietta Guagliardi, Marco Bruno, Luca Valentini, and Gilberto Artioli. 2022. "An Atomistic Model Describing the Structure and Morphology of Cu-Doped C-S-H Hardening Accelerator Nanoparticles" Nanomaterials 12, no. 3: 342. https://doi.org/10.3390/nano12030342
APA StyleDal Sasso, G., Dalconi, M. C., Ferrari, G., Pedersen, J. S., Tamburini, S., Bertolotti, F., Guagliardi, A., Bruno, M., Valentini, L., & Artioli, G. (2022). An Atomistic Model Describing the Structure and Morphology of Cu-Doped C-S-H Hardening Accelerator Nanoparticles. Nanomaterials, 12(3), 342. https://doi.org/10.3390/nano12030342