
Fabrication and Characterisation of Calcium Sulphate Hemihydrate Enhanced with Zn- or B-Doped Hydroxyapatite Nanoparticles for Hard Tissue Restoration

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Composite Preparation
2.2. Samples Characterisation
3. Results
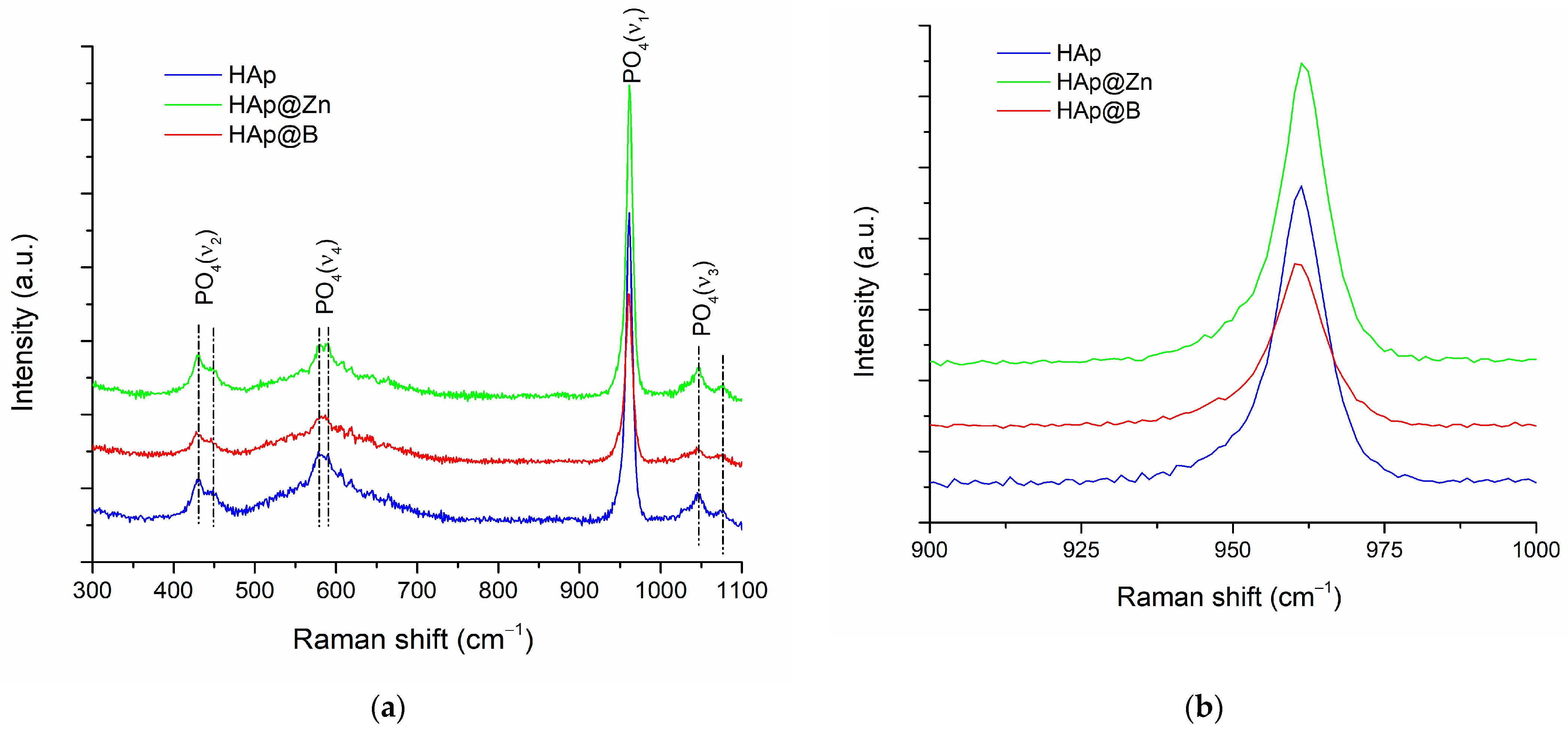
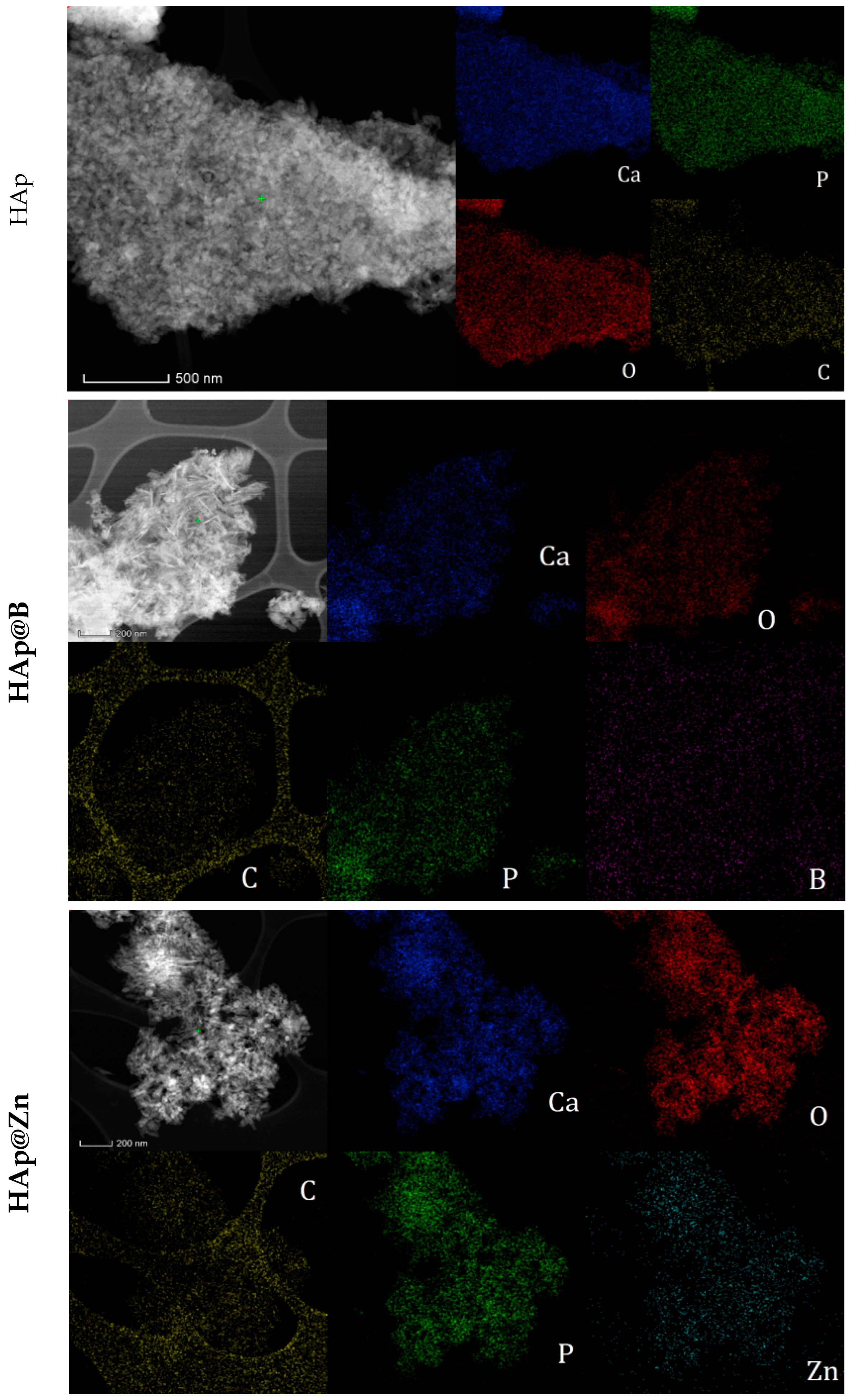
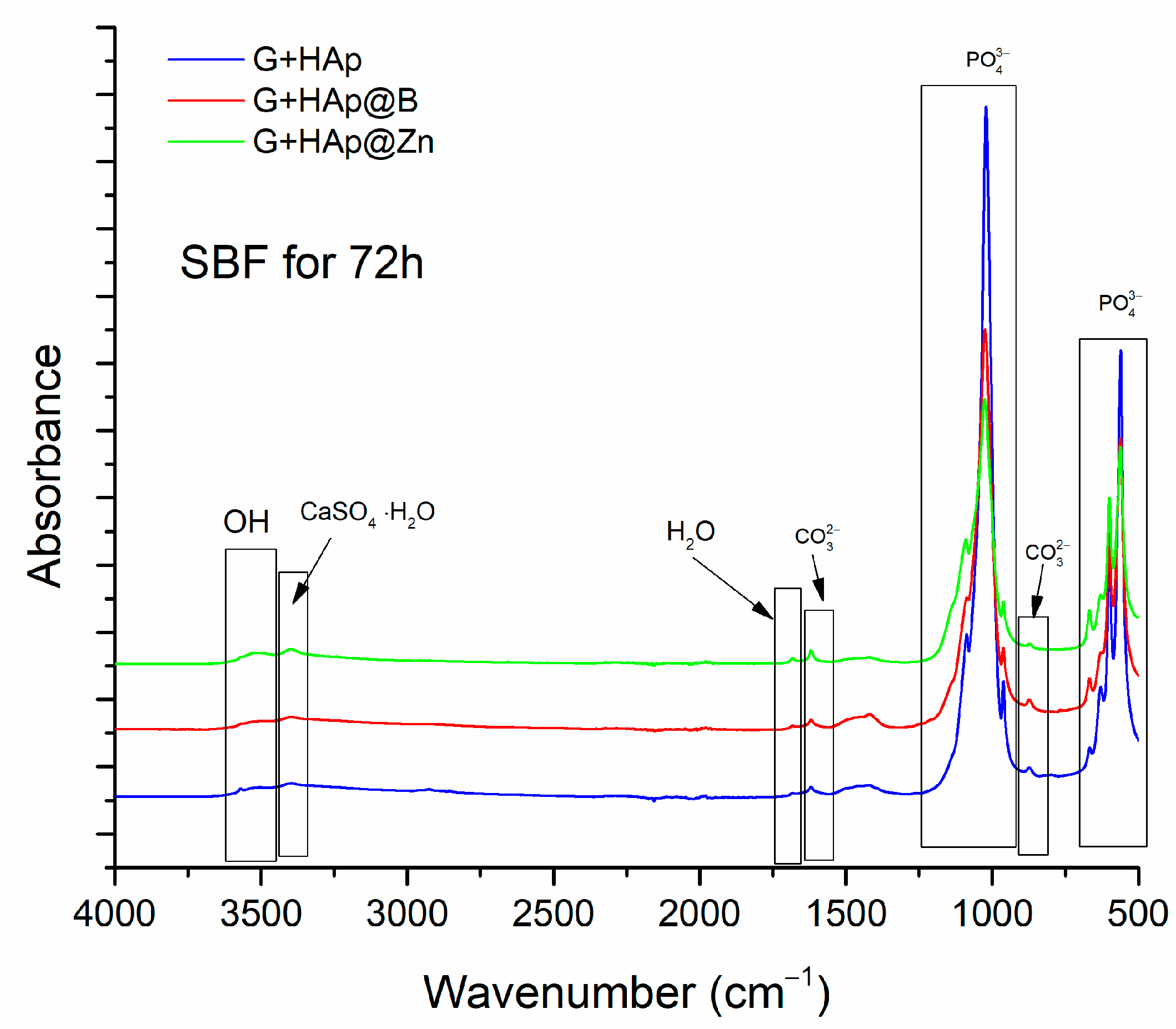
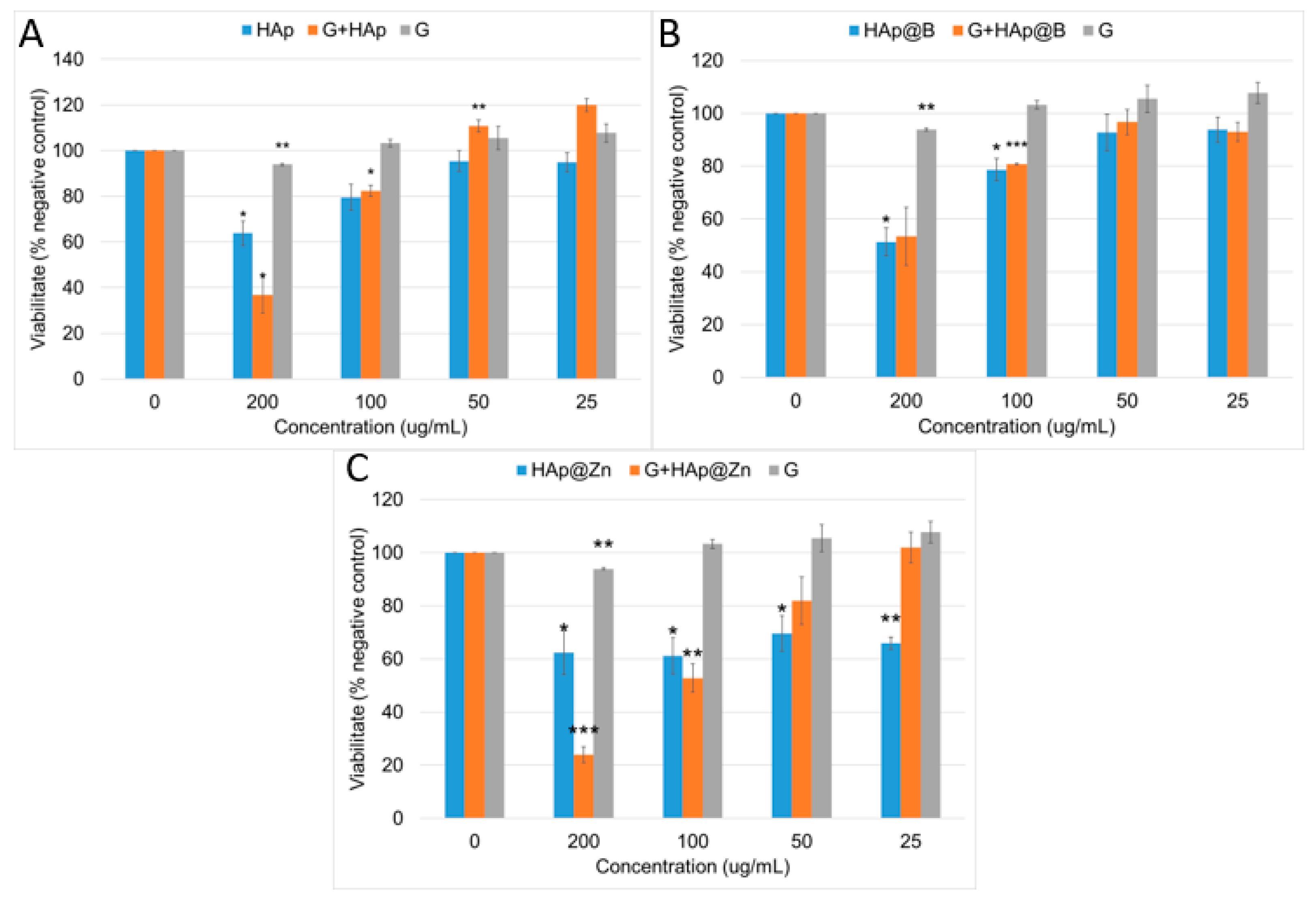
3.1. HAp, HAp@Zn and HAp@B Powder Characterisation
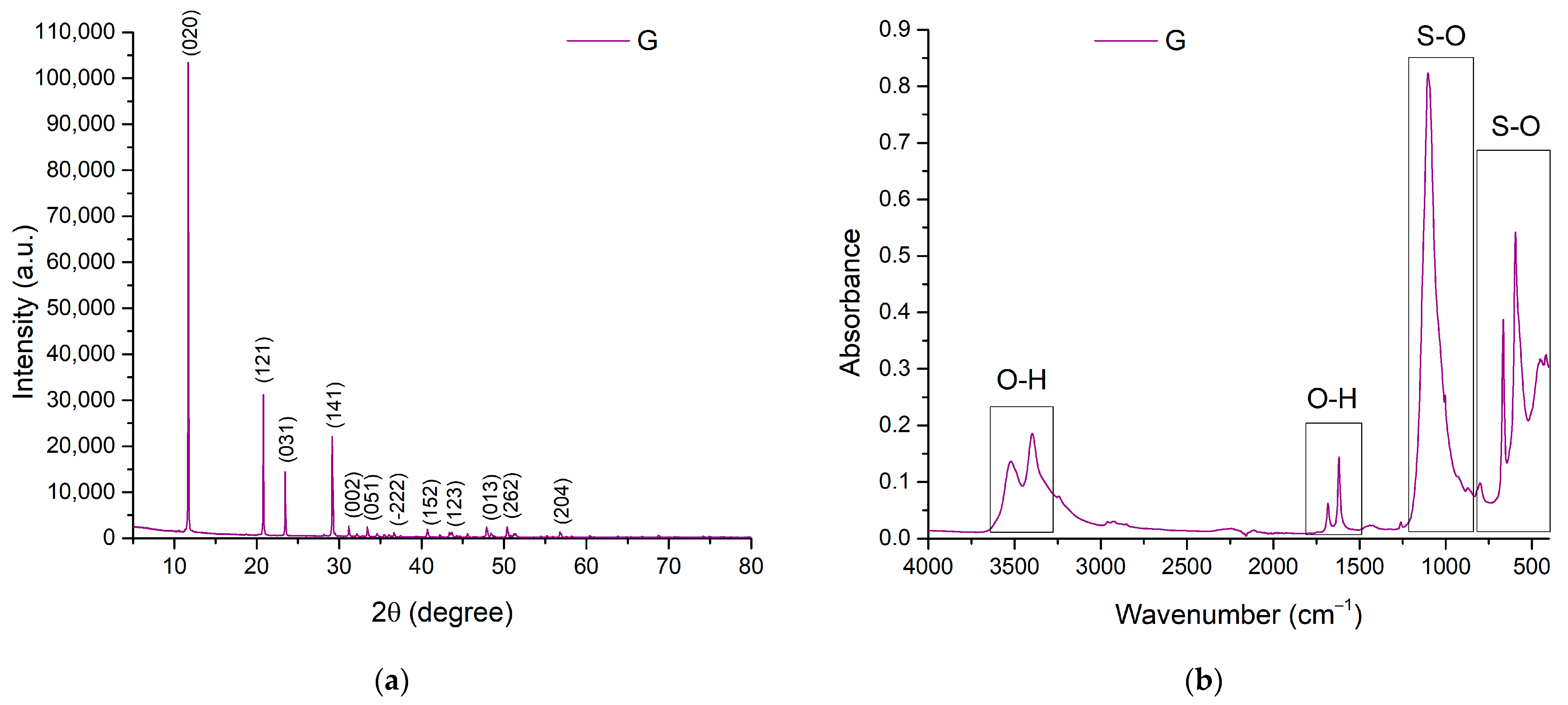
3.2. Gypsum Analysis
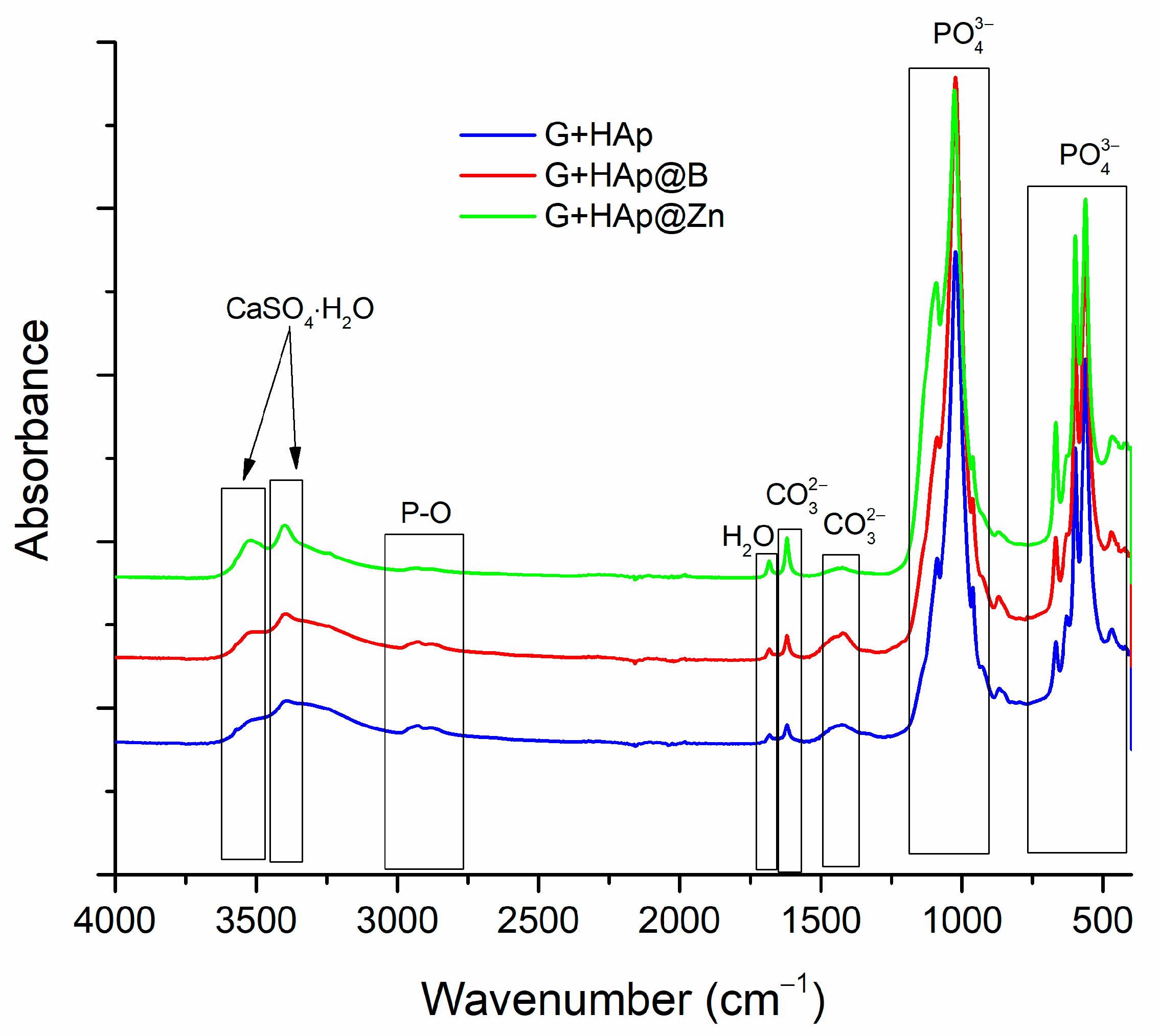
3.3. Composites Material Characterisation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fuchs, R.K.; Thompson, W.R.; Warden, S.J. Bone Biology. In Bone Repair Biomaterials; Elsevier: Amsterdam, The Netherlands, 2019; pp. 15–52. [Google Scholar]
- Pawelec, K.M.; White, A.A.; Best, S.M. Properties and Characterization of Bone Repair Materials. In Bone Repair Biomaterials; Elsevier: Amsterdam, The Netherlands, 2019; pp. 65–102. [Google Scholar]
- Florencio-Silva, R.; Sasso, G.R.d.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [Green Version]
- Langdahl, B.; Ferrari, S.; Dempster, D.W. Bone Modeling and Remodeling: Potential as Therapeutic Targets for the Treatment of Osteoporosis. Ther. Adv. Musculoskelet. Dis. 2016, 8, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.V.; Puleo, D.A. Calcium Sulfate: Properties and Clinical Applications. J. Biomed. Mater. Res. Part. B Appl. Biomater. 2009, 88B, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Crespi, R.; Capparè, P.; Gherlone, E. Magnesium-Enriched Hydroxyapatite Compared to Calcium Sulfate in the Healing of Human Extraction Sockets: Radiographic and Histomorphometric Evaluation at 3 Months. J. Periodontol. 2009, 80, 210–218. [Google Scholar] [CrossRef]
- Tran, D.L.; Hong, A.P.N.; Nguyen, N.H.; Huynh, N.T.; Le Tran, B.H.; Tran, C.T.; Truong, M.D.; Nguyen, Q.D.; Park, K.D.; Nguyen, D.H. α-Calcium Sulfate Hemihydrate Bioceramic Prepared via Salt Solution Method to Enhance Bone Regenerative Efficiency. J. Ind. Eng. Chem. 2023, 120, 293–301. [Google Scholar] [CrossRef]
- Lewis, K.N.; Thomas, M.V.; Puleo, D.A. Mechanical and Degradation Behavior of Polymer-Calcium Sulfate Composites. J. Mater. Sci. Mater. Med. 2006, 17, 531–537. [Google Scholar] [CrossRef]
- Yahav, A.; Kurtzman, G.M.; Katzap, M.; Dudek, D.; Baranes, D. Bone Regeneration. Dent. Clin. N. Am. 2020, 64, 453–472. [Google Scholar] [CrossRef]
- Tan, V.; Evaniew, N.; Finlay, K.; Jurriaans, E.; Ghert, M.; Deheshi, B.; Parasu, N. Chronology of the Radiographic Appearances of the Calcium Sulfate-Calcium Phosphate Synthetic Bone Graft Composite Following Resection of Bone Tumors: A Follow-up Study of Postoperative Appearances. Can. Assoc. Radiol. J. 2016, 67, 21–27. [Google Scholar] [CrossRef]
- Evaniew, N.; Tan, V.; Parasu, N.; Jurriaans, E.; Finlay, K.; Deheshi, B.; Ghert, M. Use of a Calcium Sulfate–Calcium Phosphate Synthetic Bone Graft Composite in the Surgical Management of Primary Bone Tumors. Orthopedics 2013, 36, e216–e222. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Liu, H.; Liu, X.; Lian, X.; Guo, Z.; Jiang, H.-J.; Cui, F.-Z. Improved Workability of Injectable Calcium Sulfate Bone Cement by Regulation of Self-Setting Properties. Mater. Sci. Eng. C 2013, 33, 1048–1053. [Google Scholar] [CrossRef]
- Wang, J.-S.; Tägil, M.; Isaksson, H.; Boström, M.; Lidgren, L. Tissue Reaction and Material Biodegradation of a Calcium Sulfate/Apatite Biphasic Bone Substitute in Rat Muscle. J. Orthop. Transl. 2016, 6, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.; Zheng, M.H.; Tägil, M. The Composite of Hydroxyapatite and Calcium Sulphate: A Review of Preclinical Evaluation and Clinical Applications. Expert. Rev. Med. Devices 2013, 10, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Abramo, A.; Geijer, M.; Kopylov, P.; Tägil, M. Osteotomy of Distal Radius Fracture Malunion Using a Fast Remodeling Bone Substitute Consisting of Calcium Sulphate and Calcium Phosphate. J. Biomed. Mater. Res. Part. B Appl. Biomater. 2010, 92B, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, E.; Iaquinta, M.R.; Lanzillotti, C.; Mazziotta, C.; Maritati, M.; Montesi, M.; Sprio, S.; Tampieri, A.; Tognon, M.; Martini, F. Bioactive Materials for Soft Tissue Repair. Front. Bioeng. Biotechnol. 2021, 9, 613787. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhou, Z.; Li, W.; Fan, Y.; Li, Z.; Wei, J. Hydroxyapatite Based Materials for Bone Tissue Engineering: A Brief and Comprehensive Introduction. Crystals 2021, 11, 149. [Google Scholar] [CrossRef]
- Panda, S.; Biswas, C.K.; Paul, S. A Comprehensive Review on the Preparation and Application of Calcium Hydroxyapatite: A Special Focus on Atomic Doping Methods for Bone Tissue Engineering. Ceram. Int. 2021, 47, 28122–28144. [Google Scholar] [CrossRef]
- Kim, H.-L.; Jung, G.-Y.; Yoon, J.-H.; Han, J.-S.; Park, Y.-J.; Kim, D.-G.; Zhang, M.; Kim, D.-J. Preparation and Characterization of Nano-Sized Hydroxyapatite/Alginate/Chitosan Composite Scaffolds for Bone Tissue Engineering. Mater. Sci. Eng. C 2015, 54, 20–25. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.-T.; Jamshidi, A. A New Strategy for Fabrication of Bone Scaffolds Using Electrospun Nano-HAp/PHB Fibers and Protein Hydrogels. Chem. Eng. J. 2016, 289, 38–47. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis Methods for Nanosized Hydroxyapatite with Diverse Structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef]
- Soraya Shahnaz Tadjoedin, E.; Sunarso. Fabrication and Mechanical Properties of Newly Developed Triphasic Blocks Composed of Gypsum-Brushite-Monetite for Bone Graft Applications. Saudi Dent. J. 2022, 34, 757–762. [Google Scholar] [CrossRef]
- Ielo, I.; Calabrese, G.; De Luca, G.; Conoci, S. Recent Advances in Hydroxyapatite-Based Biocomposites for Bone Tissue Regeneration in Orthopedics. Int. J. Mol. Sci. 2022, 23, 9721. [Google Scholar] [CrossRef] [PubMed]
- Uysal, I.; Yilmaz, B.; Evis, Z. Zn-Doped Hydroxyapatite in Biomedical Applications. J. Aust. Ceram. Soc. 2021, 57, 869–897. [Google Scholar] [CrossRef]
- Negrila, C.C.; Predoi, M.V.; Iconaru, S.L.; Predoi, D. Development of Zinc-Doped Hydroxyapatite by Sol-Gel Method for Medical Applications. Molecules 2018, 23, 2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Predoi, D.; Iconaru, S.L.; Predoi, M.V.; Motelica-Heino, M.; Guegan, R.; Buton, N. Evaluation of Antibacterial Activity of Zinc-Doped Hydroxyapatite Colloids and Dispersion Stability Using Ultrasounds. Nanomaterials 2019, 9, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begam, H.; Kundu, B.; Chanda, A.; Nandi, S.K. MG63 Osteoblast Cell Response on Zn Doped Hydroxyapatite (HAp) with Various Surface Features. Ceram. Int. 2017, 43, 3752–3760. [Google Scholar] [CrossRef]
- Beaufils, S.; Rouillon, T.; Millet, P.; Le Bideau, J.; Weiss, P.; Chopart, J.-P.; Daltin, A.-L. Synthesis of Calcium-Deficient Hydroxyapatite Nanowires and Nanotubes Performed by Template-Assisted Electrodeposition. Mater. Sci. Eng. C 2019, 98, 333–346. [Google Scholar] [CrossRef]
- Toledano, M.; Osorio, R.; Vallecillo-Rivas, M.; Osorio, E.; Lynch, C.D.; Aguilera, F.S.; Toledano, R.; Sauro, S. Zn-Doping of Silicate and Hydroxyapatite-Based Cements: Dentin Mechanobiology and Bioactivity. J. Mech. Behav. Biomed. Mater. 2021, 114, 104232. [Google Scholar] [CrossRef]
- Martinez-Zelaya, V.R.; Zarranz, L.; Herrera, E.Z.; Alves, A.T.; Uzeda, M.J.; Mavropoulos, E.; Rossi, A.L.; Mello, A.; Granjeiro, J.M.; Calasans-Maia, M.D.; et al. In Vitro and in Vivo Evaluations of Nanocrystalline Zn-Doped Carbonated Hydroxyapatite/Alginate Microspheres: Zinc and Calcium Bioavailability and Bone Regeneration. Int. J. Nanomedicine 2019, 14, 3471–3490. [Google Scholar] [CrossRef] [Green Version]
- Gümüşderelioğlu, M.; Tunçay, E.Ö.; Kaynak, G.; Demirtaş, T.T.; Aydın, S.T.; Hakkı, S.S. Encapsulated Boron as an Osteoinductive Agent for Bone Scaffolds. J. Trace Elem. Med. Biol. 2015, 31, 120–128. [Google Scholar] [CrossRef]
- Tunçay, E.Ö.; Demirtaş, T.T.; Gümüşderelioğlu, M. Microwave-Induced Production of Boron-Doped HAp (B-HAp) and B-HAp Coated Composite Scaffolds. J. Trace Elem. Med. Biol. 2017, 40, 72–81. [Google Scholar] [CrossRef]
- Kaygili, O.; Keser, S.; Kom, M.; Bulut, N.; Dorozhkin, S.V. The Effect of Simulating Body Fluid on the Structural Properties of Hydroxyapatite Synthesized in the Presence of Citric Acid. Prog. Biomater. 2016, 5, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokubo, T.; Kushitani, H.; Sakka, S.; Kitsugi, T.; Yamamuro, T. Solutions Able to Reproduce in Vivo Surface-structure Changes in Bioactive Glass-ceramic A-W3. J. Biomed. Mater. Res. 1990, 24, 721–734. [Google Scholar] [CrossRef] [PubMed]
- ISO 3051:1974; Gypsum Plasters—Determination of Mechanical Properties. International Organization for Standardization: Geneva, Switzerland, 1974.
- Yoo, C.-K.; Jeon, J.-Y.; Kim, Y.-J.; Kim, S.-G.; Hwang, K.-G. Cell Attachment and Proliferation of Osteoblast-like MG63 Cells on Silk Fibroin Membrane for Guided Bone Regeneration. Maxillofac. Plast. Reconstr. Surg. 2016, 38, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C.D.; Brett, E.A.; Moore, A.L.; Wan, D.C.; Longaker, M.T. In Vitro and In Vivo Osteogenic Differentiation of Human Adipose-Derived Stromal Cells. In Bone Morphogenetic Proteins. Methods in Molecular Biology; Rogers, M., Ed.; Humana Press: New York, NY, USA, 2019; pp. 9–18. [Google Scholar]
- Predoi, D.; Iconaru, S.; Deniaud, A.; Chevallet, M.; Michaud-Soret, I.; Buton, N.; Prodan, A. Textural, Structural and Biological Evaluation of Hydroxyapatite Doped with Zinc at Low Concentrations. Materials 2017, 10, 229. [Google Scholar] [CrossRef] [Green Version]
- Waghmare, P.G.; Narwade, V.N.; Naik, K.B.; Kutte, V.D.; Bogle, K.A.; Mahabole, M.P. CuO/HAp Composites: Excellent Dielectric Materials. Mater. Today Proc. 2023, 72, 2681–2686. [Google Scholar] [CrossRef]
- Miyaji, F.; Kono, Y.; Suyama, Y. Formation and Structure of Zinc-Substituted Calcium Hydroxyapatite. Mater. Res. Bull. 2005, 40, 209–220. [Google Scholar] [CrossRef]
- Popa, C.L.; Deniaud, A.; Michaud-Soret, I.; Guégan, R.; Motelica-Heino, M.; Predoi, D. Structural and Biological Assessment of Zinc Doped Hydroxyapatite Nanoparticles. J. Nanomater. 2016, 2016, 1062878. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Prieto, S.J.; Fonseca, L.F.; Sequeda-Castañeda, L.G.; Díaz, K.J.; Castañeda, L.Y.; Leyva-Rojas, J.A.; Salcedo-Reyes, J.C.; Acosta, A.P. Elaboration and Biocompatibility of an Eggshell-Derived Hydroxyapatite Material Modified with Si/PLGA for Bone Regeneration in Dentistry. Int. J. Dent. 2019, 2019, 5949232. [Google Scholar] [CrossRef] [Green Version]
- Venkatasubbu, G.D.; Ramasamy, S.; Ramakrishnan, V.; Avadhani, G.S.; Thangavel, R.; Kumar, J. Investigations on Zinc Doped Nanocrystalline Hydroxyapatite. Int. J. Nanosci. Nanotechnol. 2011, 2, 1–23. [Google Scholar]
- El-Maghraby, H.F.; Greish, Y.E. Preparation, Structural Characterization, and Biomedical Applications of Gypsum-Based Nanocomposite Bone Cements. In Novel Nanomaterials; IntechOpen: London, UK, 2021. [Google Scholar]
- Prasad, P.S.R. Direct Formation of the -CaSO4 Phase in Dehydration Process of Gypsum: In Situ FTIR Study. Am. Mineral. 2005, 90, 672–678. [Google Scholar] [CrossRef]
- Böke, H.; Akkurt, S.; Özdemir, S.; Göktürk, E.H.; Caner Saltik, E.N. Quantification of CaCO3–CaSO3·0.5H2O–CaSO4·2H2O Mixtures by FTIR Analysis and Its ANN Model. Mater. Lett. 2004, 58, 723–726. [Google Scholar] [CrossRef] [Green Version]
- Bueno, V.B.; Bentini, R.; Catalani, L.H.; Barbosa, L.R.S.; Petri, D.F.S. Synthesis and Characterization of Xanthan–Hydroxyapatite Nanocomposites for Cellular Uptake. Mater. Sci. Eng. C 2014, 37, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Okulus, Z.; Buchwald, T.; Szybowicz, M.; Voelkel, A. Study of a New Resin-Based Composites Containing Hydroxyapatite Filler Using Raman and Infrared Spectroscopy. Mater. Chem. Phys. 2014, 145, 304–312. [Google Scholar] [CrossRef]
- Kaygili, O.; Keser, S.; Al Orainy, R.H.; Ates, T.; Yakuphanoglu, F. In Vitro Characterization of Polyvinyl Alcohol Assisted Hydroxyapatite Derived by Sol—Gel Method. Mater. Sci. Eng. C 2014, 35, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Wakamura, M.; Kondo, S. Surface Characterization of Calcium Hydroxylapatite by Fourier Transform Infrared Spectroscopy. Langmuir 1989, 5, 140–144. [Google Scholar] [CrossRef]
- Kaygili, O.; Tatar, C.; Yakuphanoglu, F. Structural and Dielectrical Properties of Mg3–Ca3(PO4)2 Bioceramics Obtained from Hydroxyapatite by Sol–Gel Method. Ceram. Int. 2012, 38, 5713–5722. [Google Scholar] [CrossRef]
- Trommer, R.M.; Santos, L.A.; Bergmann, C.P. Nanostructured Hydroxyapatite Powders Produced by a Fl Ame-Based Technique. Mater. Sci. Eng. C 2009, 29, 1770–1775. [Google Scholar] [CrossRef]
- Borovanský, J.; Riley, P.A. Cytotoxicity of Zinc in Vitro. Chem. Biol. Interact. 1989, 69, 279–291. [Google Scholar] [CrossRef]
- Song, W.; Zhang, J.; Guo, J.; Zhang, J.; Ding, F.; Li, L.; Sun, Z. Role of the Dissolved Zinc Ion and Reactive Oxygen Species in Cytotoxicity of ZnO Nanoparticles. Toxicol. Lett. 2010, 199, 389–397. [Google Scholar] [CrossRef]
- Grue, B.H.; Veres, S.P. Effect of Increasing Mineralization on Pre-Osteoblast Response to Native Collagen Fibril Scaffolds for Bone Tissue Repair and Regeneration. J. Appl. Biomater. Funct. Mater. 2022, 20, 228080002211040. [Google Scholar] [CrossRef]
- Noda, M. Cellular and Molecular Biology of Bone; Academic Press: London, UK, 1993; ISBN 0-12-520225-3. [Google Scholar]
- Zhang, N.; Ying, M.-D.; Wu, Y.-P.; Zhou, Z.-H.; Ye, Z.-M.; Li, H.; Lin, D.-S. Hyperoside, a Flavonoid Compound, Inhibits Proliferation and Stimulates Osteogenic Differentiation of Human Osteosarcoma Cells. PLoS ONE 2014, 9, e98973. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Initial pH | Final pH |
|---|---|---|
| HAp | 5.11 | 9.25 |
| HAp@B | 4.92 | 9.15 |
| HAp@Zn | 4.87 | 9.62 |
| Notation | Gypsum (%) | HAp (%) | HAp@B (%) | HAp@Zn (%) | Gly (%) | H2O (%) |
|---|---|---|---|---|---|---|
| G+HAp | 26.60 | 26.60 | - | - | 4.68 | 42.12 |
| G+HAp@B | 27.18 | - | 27.18 | - | 4.56 | 41.08 |
| G+HAp@Zn | 27.18 | - | - | 27.18 | 4.56 | 41.08 |
| G | 62.50 | - | - | - | 3.75 | 33.75 |
| Elementary Cell Parameters | Sample | ||
|---|---|---|---|
| HAp | HAp@Zn | HAp@B | |
| a (Å) | 9.4240 ± 0.0008 | 9.4245 ± 0.0011 | 9.4116 ± 0.0019 |
| b (Å) | 9.4240 ± 0.0008 | 9.4245 ± 0.0011 | 9.4116 ± 0.0020 |
| c (Å) | 6.8851 ± 0.0007 | 6.8844 ± 0.0009 | 6.8834 ± 0.0015 |
| V (Å3) | 529.56 | 529.57 | 528.04 |
| Crystallinity (%) | 31.11 | 33.38 | 36.72 |
| Average crystallite size (nm) | 19.44 ± 3.13 | 16.63 ± 1.83 | 10.69 ± 1.59 |
| Microstrain (%) | 0.47 | 0.55 | 0.86 |
| Elements | HAp | HAp@B | HAp@Zn | |||
|---|---|---|---|---|---|---|
| Mass % | Error % | Mass % | Error % | Mass % | Error % | |
| C K | 16.37 | 9.96 | 5.51 | 15.91 | 3.91 | 16.03 |
| O K | 53.74 | 9.82 | 50.41 | 10.07 | 57.15 | 9.75 |
| P K | 10.80 | 4.40 | 13.32 | 4.63 | 14.29 | 4.67 |
| Ca K | 19.09 | 1.41 | 27.04 | 1.66 | 24.24 | 1.77 |
| Zn K | - | - | - | - | 0.33 | 18.59 |
| B K | - | - | 3.72 | 34.24 | - | - |
| 3 Days | 7 Days | 28 Days | SBF | |
|---|---|---|---|---|
| Gypsum (G) | - | 8.5 | 10.43 | - |
| G+HAp | 0.46 | 1.04 | 1.15 | 1.08 |
| G+HAp@B | 0.63 | 0.69 | 0.83 | 0.74 |
| G+HAp@Zn | 1.16 | 1.91 | 2.06 | 1.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicoara, A.I.; Voineagu, T.G.; Alecu, A.E.; Vasile, B.S.; Maior, I.; Cojocaru, A.; Trusca, R.; Popescu, R.C. Fabrication and Characterisation of Calcium Sulphate Hemihydrate Enhanced with Zn- or B-Doped Hydroxyapatite Nanoparticles for Hard Tissue Restoration. Nanomaterials 2023, 13, 2219. https://doi.org/10.3390/nano13152219
Nicoara AI, Voineagu TG, Alecu AE, Vasile BS, Maior I, Cojocaru A, Trusca R, Popescu RC. Fabrication and Characterisation of Calcium Sulphate Hemihydrate Enhanced with Zn- or B-Doped Hydroxyapatite Nanoparticles for Hard Tissue Restoration. Nanomaterials. 2023; 13(15):2219. https://doi.org/10.3390/nano13152219
Chicago/Turabian StyleNicoara, Adrian Ionut, Teodor Gabriel Voineagu, Andrada Elena Alecu, Bogdan Stefan Vasile, Ioana Maior, Anca Cojocaru, Roxana Trusca, and Roxana Cristina Popescu. 2023. "Fabrication and Characterisation of Calcium Sulphate Hemihydrate Enhanced with Zn- or B-Doped Hydroxyapatite Nanoparticles for Hard Tissue Restoration" Nanomaterials 13, no. 15: 2219. https://doi.org/10.3390/nano13152219