
Electrocatalytic Oxidation of Benzaldehyde on Gold Nanoparticles Supported on Titanium Dioxide
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
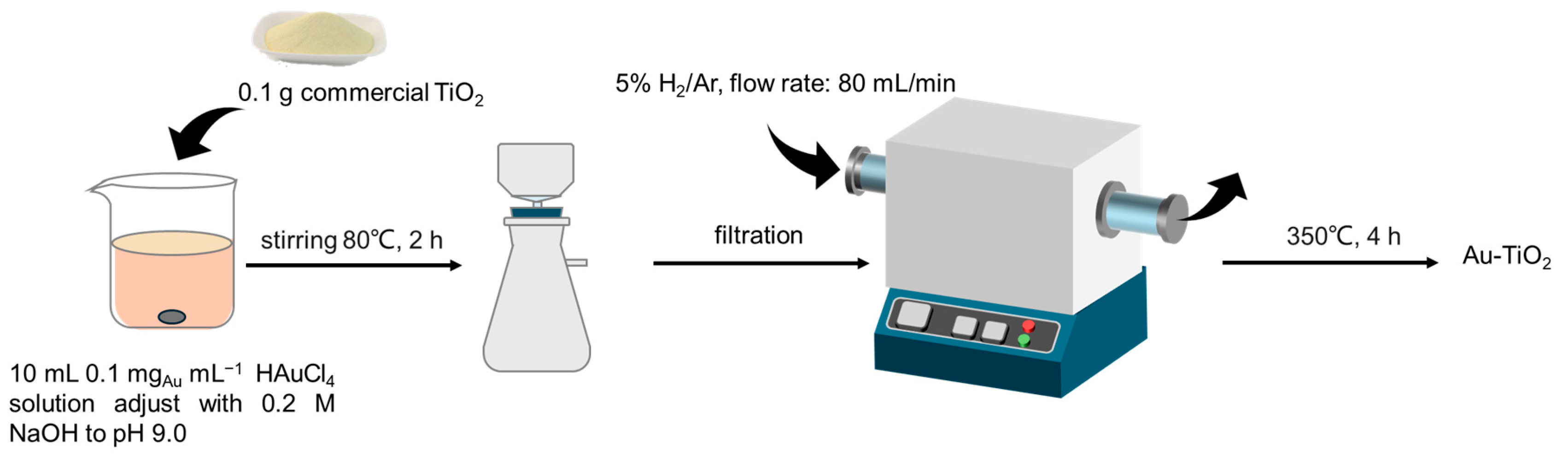
2.2. Synthesis of the Au-TiO2-P25, Au- TiO2-A, and Au-TiO2-R
2.3. Characterizations
2.4. Product Identification and Conversion and Selectivity Quantification
2.5. Electrochemistry Tests
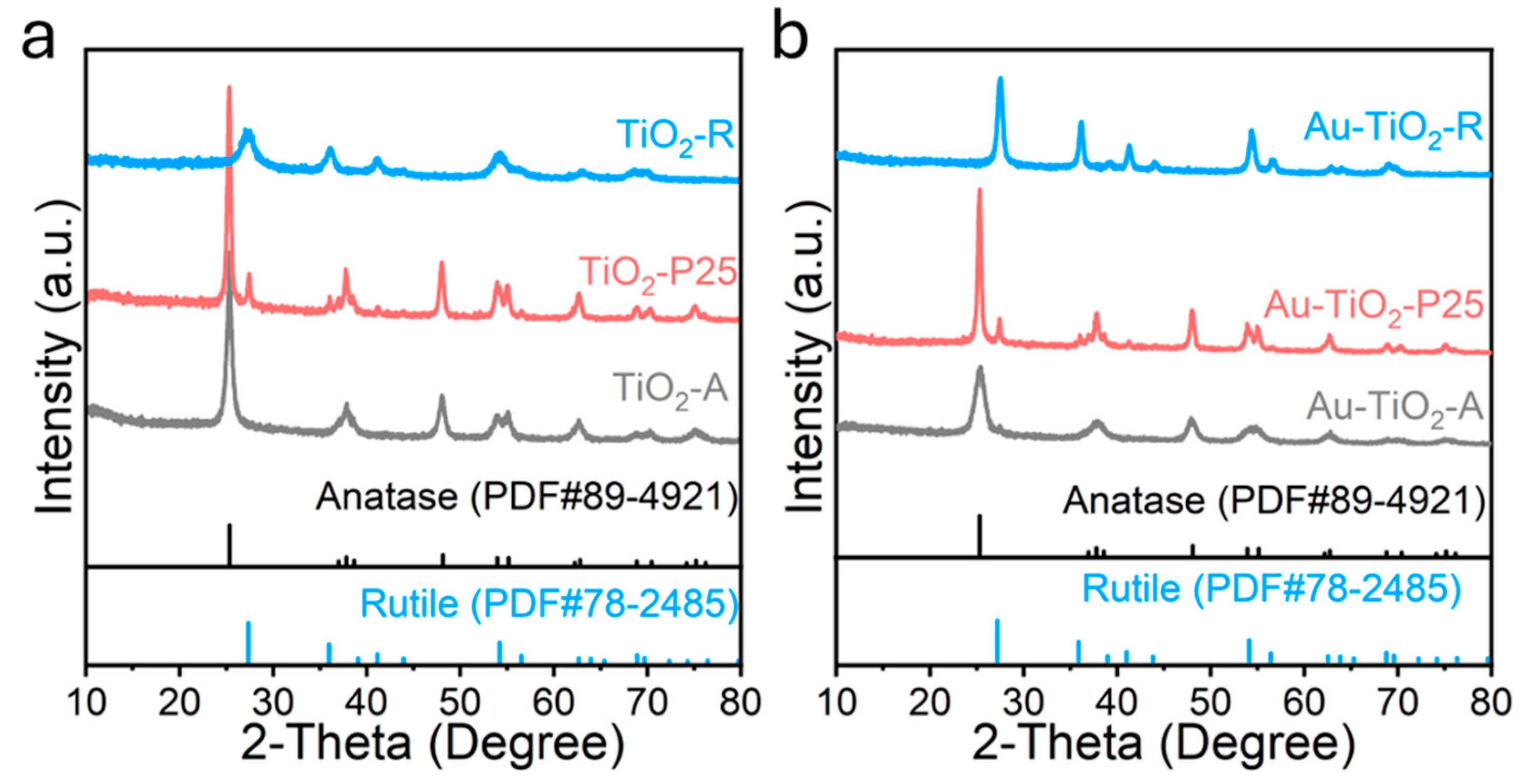
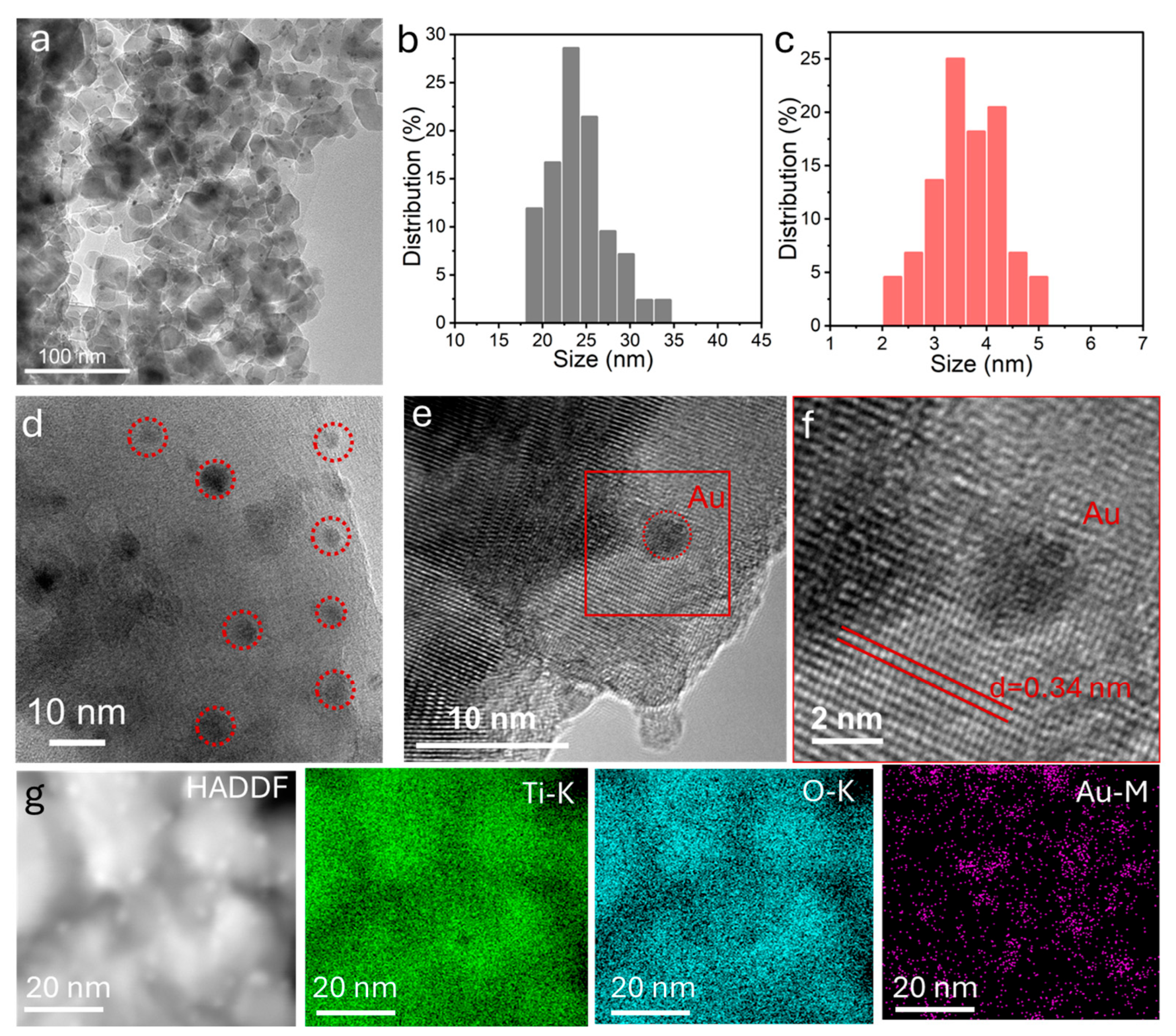
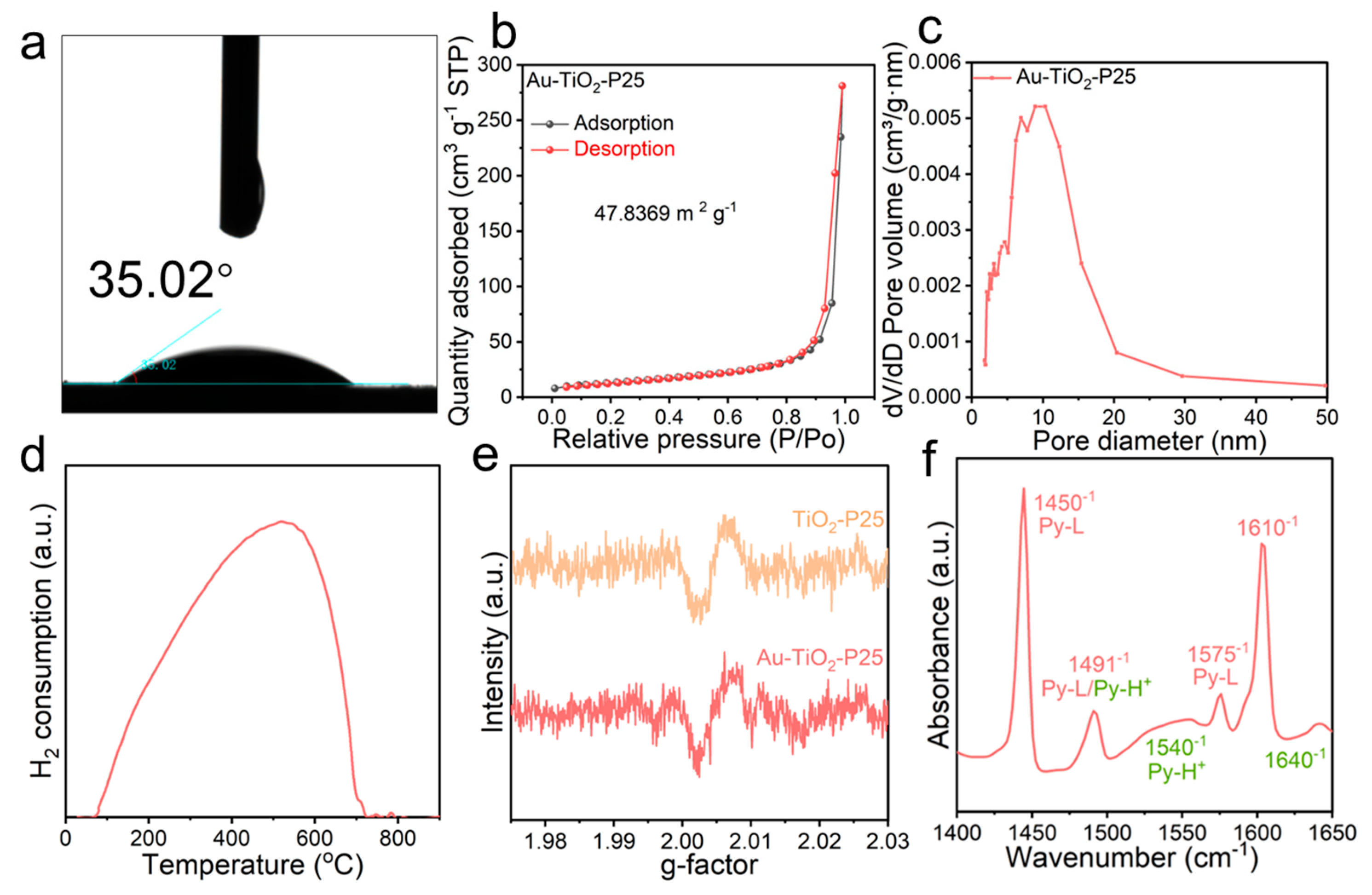
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zeng, G.; Sun, Q.; Horta, S.; Wang, S.; Lu, X.; Zhang, C.Y.; Li, J.; Li, J.; Ci, L.; Tian, Y.; et al. A Layered Bi2Te3@PPy Cathode for Aqueous Zinc-Ion Batteries: Mechanism and Application in Printed Flexible Batteries (Adv. Mater. 1/2024). Adv. Mater. 2024, 36, 2470004. [Google Scholar] [CrossRef]
- Zhong, W.; Huang, W.; Ruan, S.; Zhang, Q.; Wang, Y.; Xie, S. Electrocatalytic Reduction of CO2 Coupled with Organic Conversion to Selectively Synthesize High-Value Chemicals. Chem. Eur. J. 2023, 29, e202203228. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Cao, C.; Zhu, Q.-L. Paired electrosynthesis design strategy for sustainable CO2 conversion and product upgrading. EnergyChem 2023, 5, 100111. [Google Scholar] [CrossRef]
- Gao, G.; Chen, X.; Han, L.; Zhu, G.; Jia, J.; Cabot, A.; Sun, Z. Advances in MOFs and their derivatives for non-noble metal electrocatalysts in water splitting. Coord. Chem. Rev. 2024, 503, 215639. [Google Scholar] [CrossRef]
- Wang, X.; Li, D.; Dai, J.; Xue, Q.; Yang, C.; Xia, L.; Qi, X.; Bao, B.; Yang, S.; Xu, Y.; et al. Blocking Metal Nanocluster Growth through Ligand Coordination and Subsequent Polymerization: The Case of Ruthenium Nanoclusters as Robust Hydrogen Evolution Electrocatalysts. Small 2023, 20, 2309176. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, Y.; Wu, Z.-S. Design principle of electrocatalysts for the electrooxidation of organics. Chem 2022, 8, 2594–2629. [Google Scholar] [CrossRef]
- Chen, G.; Li, X.; Feng, X. Upgrading Organic Compounds through the Coupling of Electrooxidation with Hydrogen Evolution. Angew. Chem. Int. Ed. 2022, 61, e202209014. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cao, X.; Jiao, L. Progress in Hydrogen Production Coupled with Electrochemical Oxidation of Small Molecules. Angew. Chem. Int. Ed. 2022, 61, e202213328. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Ma, X.; Wang, J.; Zhao, J.; Zhang, Y.; He, R.; Yang, Y.; Cabot, A.; Zhu, Y. Unraveling the role of iron on Ni-Fe alloy nanoparticles during the electrocatalytic ethanol-to-acetate process. Nano Res. 2024, 17, 2328–2336. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Wang, J.; Cabot, A.; Zhu, Y. Boosting Hydrogen Evolution by Methanol Oxidation Reaction on Ni-Based Electrocatalysts: From Fundamental Electrochemistry to Perspectives. ACS Energy Lett. 2024, 9, 853–879. [Google Scholar] [CrossRef]
- Shrestha, N.K.; Patil, S.A.; Salunke, A.S.; Inamdar, A.I.; Im, H. Unprecedented urea oxidation on Zn@Ni-MOF with an ultra-high current density: Understanding the competition between UOR and OER, catalytic activity limitation and reaction selectivity. J. Mater. Chem. A 2023, 11, 14870–14877. [Google Scholar] [CrossRef]
- Shen, P.; Zhou, B.; Chen, Z.; Xiao, W.; Fu, Y.; Wan, J.; Wu, Z.; Wang, L. Ruthenium-doped 3D Cu2O nanochains as efficient electrocatalyst towards hydrogen evolution and hydrazine oxidation. Appl. Catal. B 2023, 325, 122305. [Google Scholar] [CrossRef]
- Cao, Y.; Yuan, S.; Meng, L.; Wang, Y.; Hai, Y.; Su, S.; Ding, W.; Liu, Z.; Li, X.; Luo, M. Recent Advances in Electrocatalytic Nitrate Reduction to Ammonia: Mechanism Insight and Catalyst Design. ACS Sustain. Chem. Eng. 2023, 11, 7965–7985. [Google Scholar] [CrossRef]
- Zhong, C.; Hu, W.B.; Cheng, Y.F. Recent advances in electrocatalysts for electro-oxidation of ammonia. J. Mater. Chem. A 2013, 1, 3216–3238. [Google Scholar] [CrossRef]
- Howard, E.E.; Allen, J.T.; Coleman, J.L.; Small, S.D.; Karl, J.P.; O’Fallon, K.S.; Margolis, L.M. Ketone Monoester Plus Carbohydrate Supplementation Does Not Alter Exogenous and Plasma Glucose Oxidation or Metabolic Clearance Rate During Exercise in Men Compared with Carbohydrate Alone. J. Nutr. 2023, 153, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Liu, X.; Dong, J.; You, B.; Liu, X.; Sun, Y. Electrocatalysis of Furfural Oxidation Coupled with H2 Evolution via Nickel-Based Electrocatalysts in Water. ChemNanoMat 2017, 3, 491–495. [Google Scholar] [CrossRef]
- Wang, T.; Tao, L.; Zhu, X.; Chen, C.; Chen, W.; Du, S.; Zhou, Y.; Zhou, B.; Wang, D.; Xie, C.; et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat. Catal. 2022, 5, 66–73. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Q.; Li, T.; Wang, Y.; Li, H.; Cabot, A. PdMoSb trimetallene as high-performance alcohol oxidation electrocatalyst. Nano Res. 2023, 16, 2041–2048. [Google Scholar] [CrossRef]
- Li, T.; Wang, Q.; Wu, J.; Sui, Y.; Tang, P.; Liu, H.; Zhang, W.; Li, H.; Wang, Y.; Cabot, A.; et al. Strain and Shell Thickness Engineering in Pd3Pb@Pt Bifunctional Electrocatalyst for Ethanol Upgrading Coupled with Hydrogen Production. Small 2024, 20, 2306178. [Google Scholar] [CrossRef]
- Sharma, R.K.; Bandichhor, R.; Mishra, V.; Sharma, S.; Yadav, S.; Mehta, S.; Arora, B.; Rana, P.; Dutta, S.; Solanki, K. Advanced metal oxide-based nanocatalysts for the oxidative synthesis of fine chemicals. Mater. Adv. 2023, 4, 1795–1830. [Google Scholar] [CrossRef]
- Van Thuan, D.; Ngo, H.L.; Thi, H.P.; Chu, T.T.H. Photodegradation of hazardous organic pollutants using titanium oxides -based photocatalytic: A review. Environ. Res. 2023, 229, 116000. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Ren, X.; Dai, Z.; Zhang, Y.; Qi, X.; Pan, C. Present Perspectives of Advanced Characterization Techniques in TiO2-Based Photocatalysts. ACS Appl. Mater. Interfaces 2017, 9, 23265–23286. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, C.Y.; Li, J.; Montaña-Mora, G.; Botifoll, M.; Guo, T.; Arbiol, J.; Zhou, J.Y.; Kallio, T.; Martأ nez-Alanis, P.R.; et al. Enhanced Electrochemical Hydrogenation of Benzaldehyde to Benzyl Alcohol on Pd@Ni-MOF by Modifying the Adsorption Configuration. ACS Appl. Mater. Interfaces 2024, 16, 6948–6957. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, C.Y.; Mu, X.; Han, X.; Li, J.; Arbiol, J.; Zhou, J.Y.; Kallio, T.; Martأ nez-Alanis, P.R.; Cabot, A. Semiconductor nanosheets for electrocatalytic self-coupling of benzaldehyde to hydrobenzoin. J. Chem. Eng. 2024, 479, 147612. [Google Scholar] [CrossRef]
- Dong, Y.; Dong, L.; Gu, X.; Wang, Y.; Liao, Y.; Luque, R.; Chen, Z. Sustainable production of active pharmaceutical ingredients from lignin-based benzoic acid derivatives via “demand orientation”. Green Chem. 2023, 25, 3791–3815. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, S.; Gao, B.; Bi, F.; Liu, Q.; Zhao, Q.; Xu, J.; Lu, G.; Yang, Y.; Wu, M. Effect of benzoic acid and dopamine hydrochloride as a modulator in the water resistance of Universitetet i Oslo-67: Adsorption performance and mechanism. J. Colloid Interface Sci. 2023, 651, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sun, Z.; Guo, W.; Chen, M.; Zhu, C.; Fei, J.; Liu, Y.; He, H.; Cao, Y.; Bao, X. Upgrading Waste Polylactide via Catalyst-Controlled Tandem Hydrolysis-Oxidation. ChemSusChem 2023, 16, e202301128. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zheng, P.; Song, S.; Wang, X.; Zhang, M.; Chi, K.; Xu, C.; Duan, A.; Zhao, Z. Synthesis of a novel micro/mesoporous composite material Beta-FDU-12 and its hydro-upgrading performance for FCC gasoline. RSC Adv. 2016, 6, 1018–1026. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Y.; Hao, Y.; Yin, L.; Kuo, C.-H.; Chen, H.-Y.; Li, L.; Peng, S. Lewis Acid Driving Asymmetric Interfacial Electron Distribution to Stabilize Active Species for Efficient Neutral Water Oxidation. Adv. Mater. 2024, 36, 2308925. [Google Scholar] [CrossRef]
- Qi, J.; Shen, X.; Chen, M.; Shangguan, E.; Zhang, W.; Cao, R. Lewis acid Mg2+-doped cobalt phosphate nanosheets for enhanced electrocatalytic oxygen evolution reaction. Chem. Commun. 2022, 58, 10801–10804. [Google Scholar] [CrossRef]
- Guo, J.; Wang, G.; Cui, S.; Xia, B.; Liu, Z.; Zang, S.-Q.; Wang, S. Enhanced adsorption with hydroxymethyl and aldehyde over the heterophase interface for efficient biomass electrooxidation. Sci. China Mater. 2023, 66, 2698–2707. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, Z.-Y.; Fang, H.; Jiao, H.; Gao, T.-T.; Yang, J.-T.; Bian, L.; Wang, Z.-L. Selective electrooxidation of methane to formic acid by atomically dispersed CuOx and its induced Lewis acid sites on V2O5 in a tubular electrode. Appl. Catal. B 2024, 351, 124001. [Google Scholar] [CrossRef]
- Cabot, A.; Arbiol, J.; Morante, J.R.; Weimar, U.; Bârsan, N.; Göpel, W. Analysis of the noble metal catalytic additives introduced by impregnation of as obtained SnO2 sol-gel nanocrystals for gas sensors. Sens. Actuators B Chem. 2000, 70, 87–100. [Google Scholar] [CrossRef]
- Tremiliosi-Filho, G.; Gonzalez, E.R.; Motheo, A.J.; Belgsir, E.M.; Léger, J.M.; Lamy, C. Electro-oxidation of ethanol on gold: Analysis of the reaction products and mechanism. J. Electroanal. Chem. 1998, 444, 31–39. [Google Scholar] [CrossRef]
- Suga, T.; Shida, N.; Atobe, M. Au-catalyzed electrochemical oxidation of alcohols using an electrochemical column flow cell. Electrochem. Commun. 2021, 124, 106944. [Google Scholar] [CrossRef]
- Bender, M.T.; Warburton, R.E.; Hammes-Schiffer, S.; Choi, K.-S. Understanding Hydrogen Atom and Hydride Transfer Processes during Electrochemical Alcohol and Aldehyde Oxidation. ACS Catal. 2021, 11, 15110–15124. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, L.; Jin, Y.; Zhao, S.; Wang, K.; Martínez-Alanis, P.R.; Cabot, A. Electrocatalytic Oxidation of Benzaldehyde on Gold Nanoparticles Supported on Titanium Dioxide. Nanomaterials 2024, 14, 1005. https://doi.org/10.3390/nano14121005
Gong L, Jin Y, Zhao S, Wang K, Martínez-Alanis PR, Cabot A. Electrocatalytic Oxidation of Benzaldehyde on Gold Nanoparticles Supported on Titanium Dioxide. Nanomaterials. 2024; 14(12):1005. https://doi.org/10.3390/nano14121005
Chicago/Turabian StyleGong, Li, Yu Jin, Shiling Zhao, Kaizhi Wang, Paulina R. Martínez-Alanis, and Andreu Cabot. 2024. "Electrocatalytic Oxidation of Benzaldehyde on Gold Nanoparticles Supported on Titanium Dioxide" Nanomaterials 14, no. 12: 1005. https://doi.org/10.3390/nano14121005
APA StyleGong, L., Jin, Y., Zhao, S., Wang, K., Martínez-Alanis, P. R., & Cabot, A. (2024). Electrocatalytic Oxidation of Benzaldehyde on Gold Nanoparticles Supported on Titanium Dioxide. Nanomaterials, 14(12), 1005. https://doi.org/10.3390/nano14121005