
Atomistic Insight on Effect of Silica Fume on Intermolecular Interactions between Poly(carboxylate) Superplasticizer and Calcium Ions in Concrete

 , ,
, ,
Abstract
:1. Introduction
1.1. Literature Review on the Application of Silica Fume in the Concrete Industry
1.2. Molecular Modeling Application for the Current Research
2. Materials and Methods
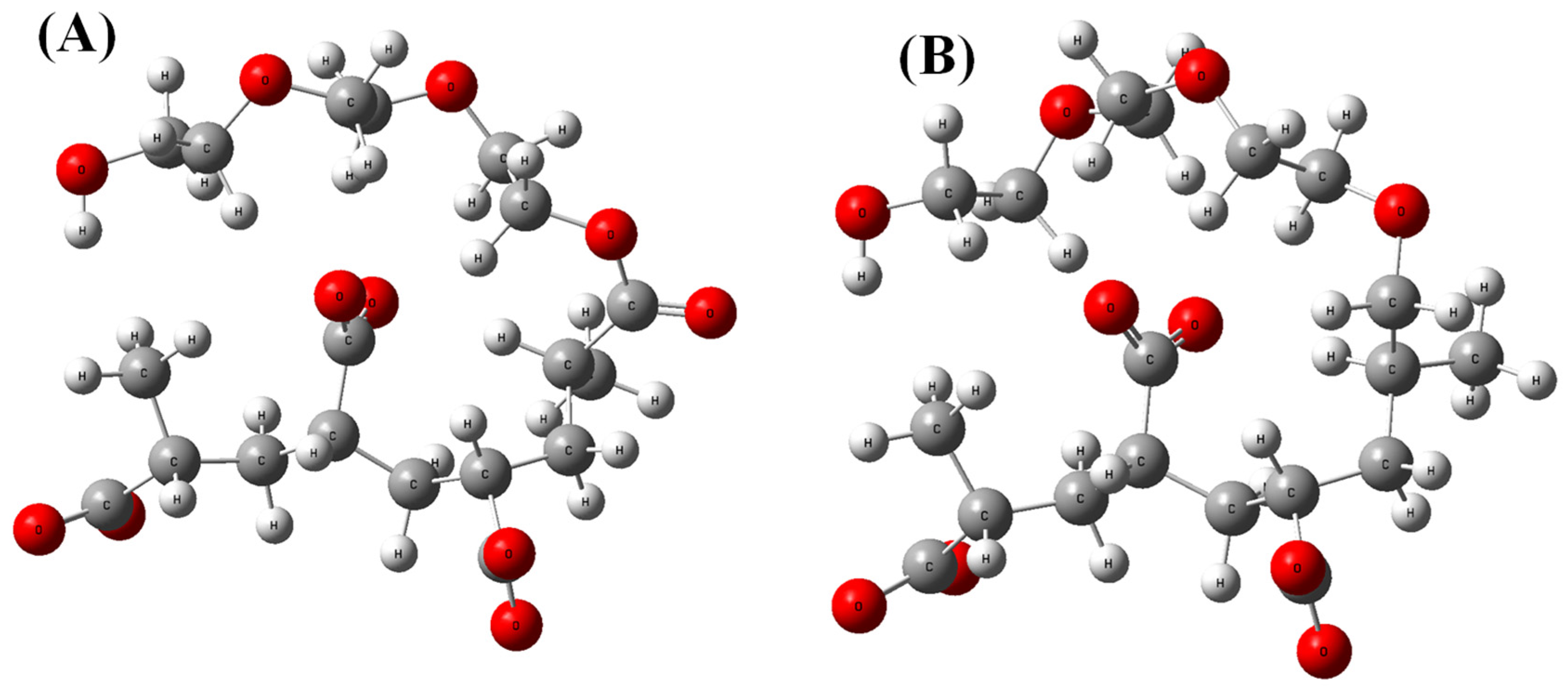
2.1. Theoretical Models
2.2. Classical All-Atom Molecular Dynamics Simulations
2.3. Density Functional Theory Calculations
3. Results and Discussion
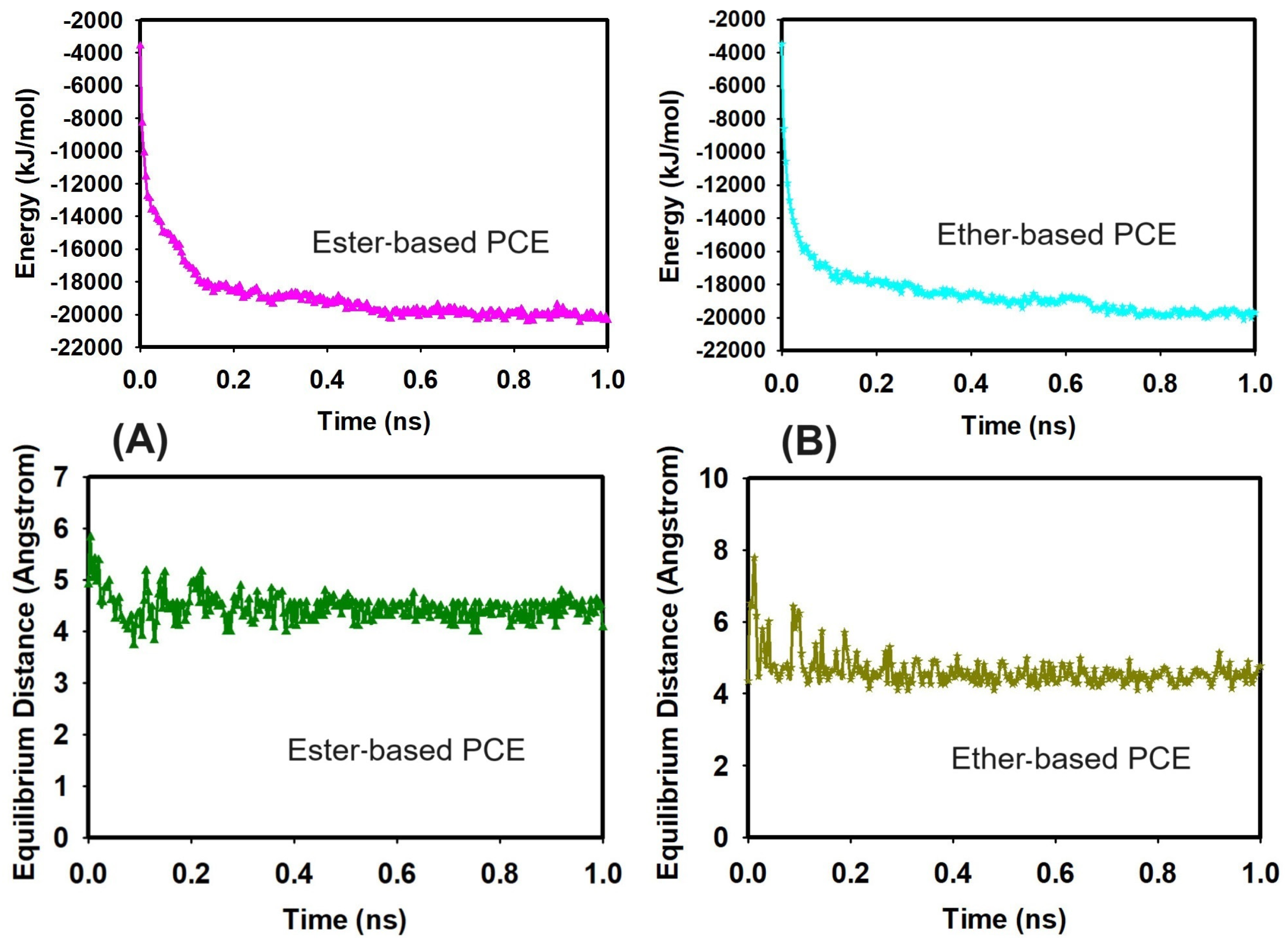
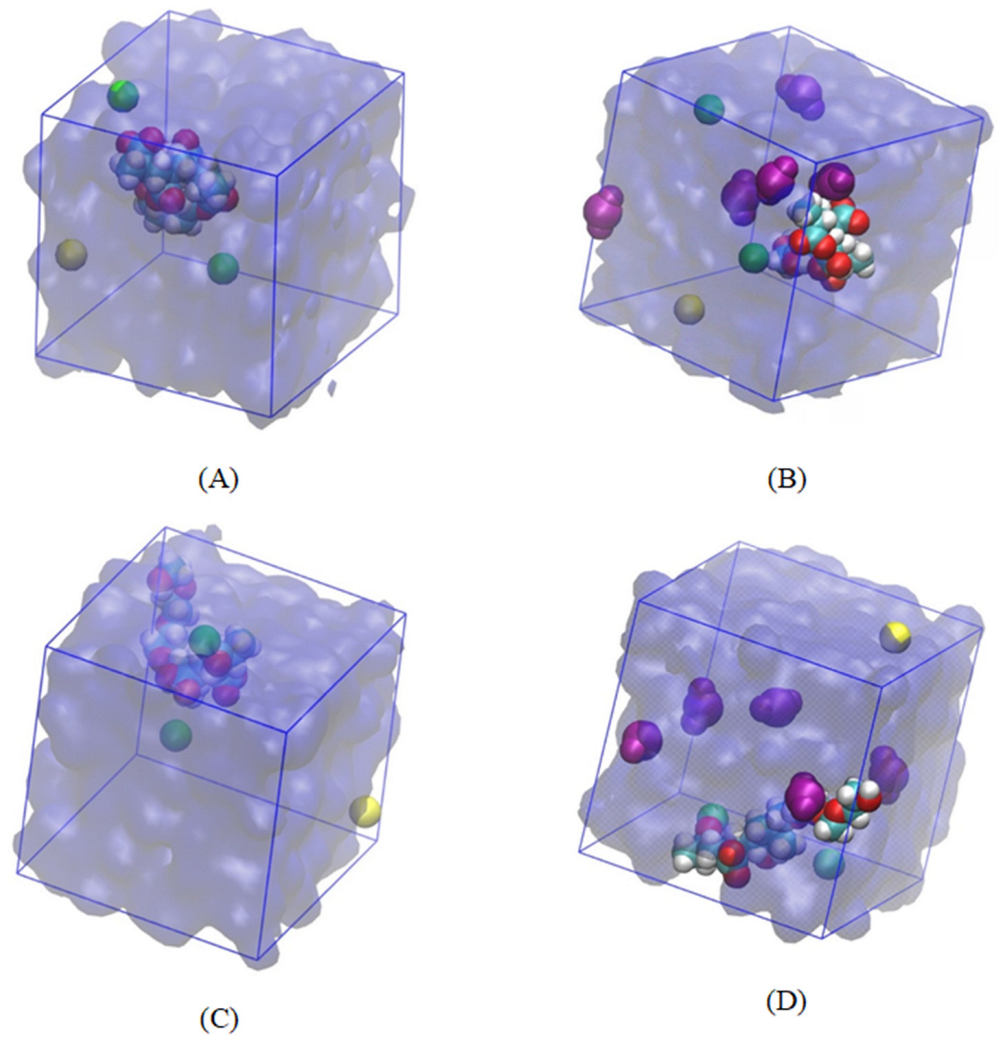
3.1. Validation of the Stability of the System before Classical All-Atom MD Production Run
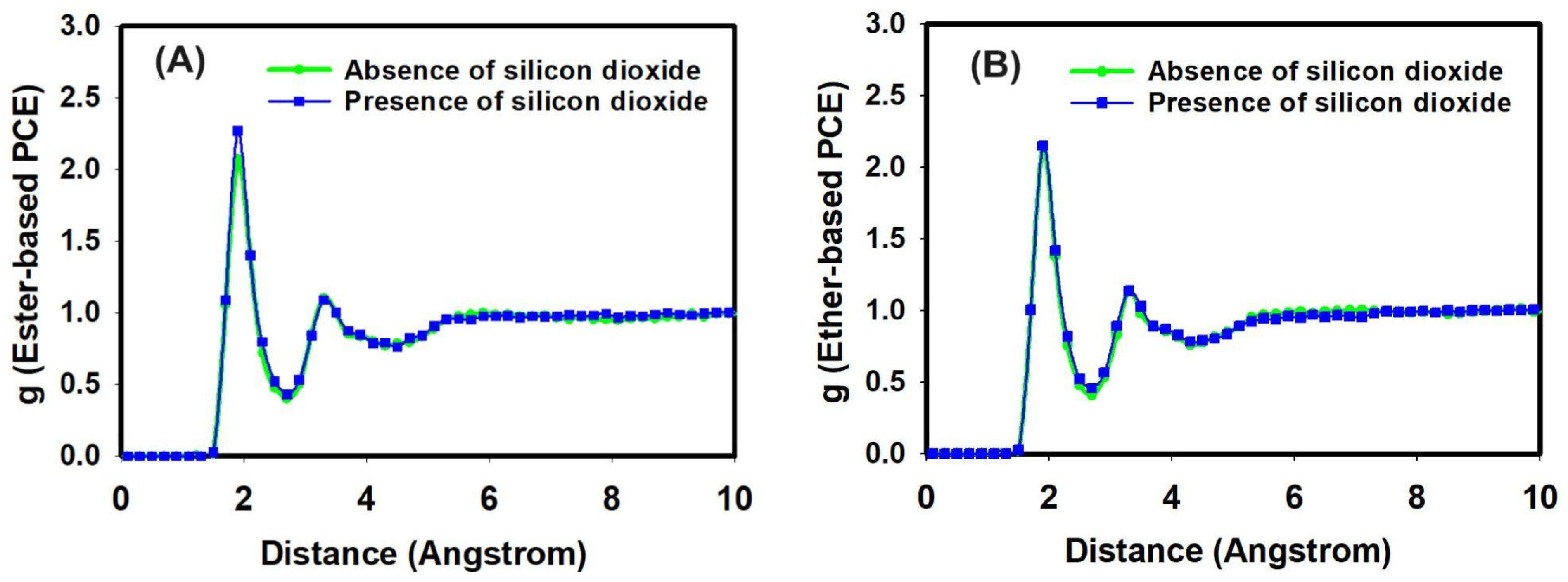
3.2. Classical All-Atom MD Simulation Results
3.3. DFT Calculation Results
3.4. Discussion of Classical All-Atom MD and DFT Calculation Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamil, T.; Javadi, A.; Heinz, H. Mechanism of molecular interaction of acrylate-polyethylene glycol acrylate copolymers with calcium silicate hydrate surfaces. Green Chem. 2020, 22, 1577–1593. [Google Scholar] [CrossRef]
- Scrivener, K.L.; Nonat, A. Hydration of cementitious materials, present and future. Cem. Concr. Res. 2011, 41, 651–665. [Google Scholar] [CrossRef]
- Cucek, L.; Klemes, J.J.; Kravanja, Z. A review of footprint tools for monitoring impacts on sustainability. J. Clean. Prod. 2012, 34, 9–20. [Google Scholar] [CrossRef]
- Torkmahalleh, A.M.; Karibayev, M.; Konakbayeva, D.; Fyrillas, M.M.; Rule, A.M. Aqueous chemistry of airborne hexavalent chromium during sampling. Aqueous chemistry of airborne hexavalent chromium during sampling. Air Qual. Atmos. Health. 2018, 11, 1059–1068. [Google Scholar] [CrossRef]
- McLellan, B.C.; Williams, R.P.; Lay, J.; Van Riessen, A.; Corder, G.D. Costs and carbon emissions for geopolymer pastes in comparison to ordinary portland cement. J. Clean. Prod. 2011, 19, 1080–1090. [Google Scholar] [CrossRef]
- Felekoglu, B.; Tosun, K.; Baradan, B.; Altun, A.; Uyulgan, B. The effect of fly ash and limestone fillers on the viscosity and compressive strength of self-compacting repair mortars. Cem. Concr. Res. 2006, 36, 1719–1726. [Google Scholar] [CrossRef]
- Sahmaran, M.; Christianto, H.A.; Yaman, I.O. The effect of chemical admixtures and mineral additives on the properties of self-compacting mortars. Cem. Concr. Compos. 2006, 28, 432–440. [Google Scholar] [CrossRef]
- Deilami, S.; Aslani, F.; Elchalakani, M. An experimental study on the durability and strength of SCC incorporating FA, GGBS and MS. Proc. Inst. Civ. Eng. Struct. Build. 2019, 172, 327–339. [Google Scholar] [CrossRef]
- Frhaan, W.K.M.; Bakar, H.B.A.; Hilal, N.; Al-Hadithi, A.I. Effect of Silica Fume and Super-Plasticizer on Mechanical Properties of Self-Compacting Concrete: A Revew. IOP Conf. Ser. Mater. Sci. Eng. 2020, 978, 012052. [Google Scholar] [CrossRef]
- Al-Hadithi, A.I.; Hilal, N.N. The possibility of enhancing some properties of self-compacting concrete by adding waste plastic fibers. J. Build. Eng. 2016, 8, 20–28. [Google Scholar] [CrossRef]
- Al-Hadithi, A.I.; Noaman, A.T.; Mosleh, W.K. Mechanical properties and impact behavior of PET fiber reinforced self-compacting concrete (SCC). Compos. Struct. 2019, 224, 111021. [Google Scholar] [CrossRef]
- Mohammed, M.K.; Al-Hadithi, A.I.; Mohammed, M.H. Production and optimization of eco-efficient self-compacting concrete SCC with limestone and PET. Constr. Build. Mater. 2019, 197, 734–746. [Google Scholar] [CrossRef]
- Faraj, R.H.; Ali, H.F.H.; Sherwani, A.F.H.; Hassan, B.R.; Karim, H. Use of recycled plastic in self-compacting concrete: A comprehensive review on fresh and mechanical properties. J. Build. Eng. 2020, 30, 101283. [Google Scholar] [CrossRef]
- Plank, J.; Zhimin, D.; Keller, H.; Hossle, F.V.; Seidl, W. Fundamental mechanisms for poly(carboxylate) intercalation into C3A hydrate phases and the role of sulfate present in cement. Cem. Concr. Res. 2010, 40, 45–57. [Google Scholar] [CrossRef]
- Yamada, K.; Takahashi, T.; Hanehara, S.; Matsuhisa, M. Effects of the chemical structure on the properties of poly(carboxylate)-type superplasticizer. Cem. Concr. Res. 2020, 30, 197–207. [Google Scholar] [CrossRef]
- Yoshioka, K.; Tazawa, E.I.; Kawai, K.; Enohata, T. Adsorption characteristics of superplasticizers on cement component minerals. Cem. Concr. Res. 2002, 32, 1507–1513. [Google Scholar] [CrossRef]
- Soukal, F.; Bocian, L.; Novotny, R.; Dlabajova, L.; Sulekova, N.; Hajzler, J.; Koutny, O.; Drdlova, M. The Effects of Silica Fume and Superplasticizer Type on the Properties and Microstructure of Reactive Powder Concrete. Materials 2023, 16, 6670. [Google Scholar] [CrossRef]
- Hamada, H.M.; Abed, F.; Katman, H.Y.; Hamada, A.M.; Al Jawahery, M.S.; Majdi, A.; Yousif, S.T.; Thomas, B.S. Effect of silica fume on the properties of sustainable cement concrete. J. Mater. Res. Technol. 2023, 24, 8887–8908. [Google Scholar] [CrossRef]
- Anish, C.; Krishnaiah, R.V.; Raju, K.V.B. Strength behavior of green concrete by using fly ash and silica fume. Mater. Today Proc. 2023. [Google Scholar] [CrossRef]
- Smarzewski, P. Mechanical and microstructural studies of high performance concrete with condensed silica fume. Appl. Sci. 2023, 13, 2510. [Google Scholar] [CrossRef]
- Khan, M.I.; Abbas, Y.M.; Fares, G. Enhancing Cementitious Concrete Durability and Mechanical Properties through Silica Fume and Micro-Quartz. Sustainability 2023, 15, 15913. [Google Scholar] [CrossRef]
- Moolchandani, K.; Sharma, A.; Kishan, D. Enhancing Concrete Performance with Crumb Rubber and Waste Materials: A Study on Mechanical and Durability Properties. Buildings 2024, 14, 161. [Google Scholar] [CrossRef]
- Pandey, S.P.; Yu, H.; Lau, C.; Ng, K. New Coal Char-Based Building Products: Manufacturing, Engineering Performance, and Techno-Economic Analysis for the USA Market. Sustainability 2024, 16, 1854. [Google Scholar] [CrossRef]
- Mohamed, A.K.; Weekwerth, S.A.; Mishra, R.K.; Heinz, H.; Flatt, R.J. Molecular modeling of chemical admixtures; opportunities and challenges. Cem. Concr. Res. 2022, 156, 106783. [Google Scholar] [CrossRef]
- Koziara, K.B.; Stroet, M.; Malde, A.K.; Mark, A.E. Testing and validation of the Automated Topology Builder (ATB) version 2.0: Prediction of hydration free enthalpies. J. Comput. Aided Mol. Des. 2014, 28, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field version 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Podio-Guidugli, P. On (Andersen-)Parrinello-Rahman molecar dynamics, the related metadynamics, and the use of the Cauchy-Born rule. J. Elast. 2010, 100, 145–153. [Google Scholar] [CrossRef]
- Shah, D.; Karibayev, M.; Adotey, E.K.; Torkmahalleh, A.M. Impact of volatile organic compounds on chromium containing atmospheric particulate: Insights from molecular dynamics simulations. Sci. Rep. 2020, 10, 17387. [Google Scholar] [CrossRef] [PubMed]
- Mudi, A.; Chakravarty, C. Effect of the Berendsen thermostat on the dynamical properties of water. Mol. Phys. 2004, 102, 681–685. [Google Scholar] [CrossRef]
- Berendsen, H.J.C. Transport properties computed by linear response through weak coupling to a bath. Comput. Simul. Mater. Sci. 1991, 205, 139–155. [Google Scholar]
- Harrison, J.A.; Schall, J.D.; Maskey, S.; Milkulski, P.T.; Knippenberg, M.T.; Morrow, B.H. Review of force fields and intermolecular potentials used in atomistic computational materials research. Appl. Phys. Rev. 2018, 5, 1–10. [Google Scholar] [CrossRef]
- Chen, P.; Ogawa, Y.; Nishiyama, Y.; Ismail, A.E.; Mazeau, K. Linear, non-linear and plastic bending deformation of cellulose nanocrystals. Phys. Chem. Chem. Phys. 2016, 18, 19880–19887. [Google Scholar] [CrossRef] [PubMed]
- Rukmani, S.J.; Kupgan, G.; Anstine, D.M.; Colina, C.M. A molecular dynamics study of water-soluble polymers: Analysis of force fields from atomistic simulations. Mol. Simul. 2019, 45, 310–321. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Gokce, H.S.; Hatungimana, D.; Ramyar, K. Effect of fly ash and silica fume on hardened properties of foam concrete. Constr. Build. Mater. 2019, 194, 1–11. [Google Scholar] [CrossRef]
- Guneyisi, E.; Gesoglu, M.; Booya, E. Fresh properties of self-compacting cold bonded fly ash lightweight aggregate concrete with different mineral admixtures. Mater. Struct. 2012, 45, 1849–1859. [Google Scholar] [CrossRef]
- Djelal, C.; Vanhove, Y.; Magnin, A. Tribological behavior of self compacting concrete. Cem. Concr. Res. 2004, 34, 821–828. [Google Scholar] [CrossRef]
- Hassan, A.A.; Lachemi, M.; Hossain, K.M. Effect of metakaolin and silica fume on the durability of self-consolidating concrete. Cem, Concr. Compos. 2012, 34, 801-8-7. [Google Scholar] [CrossRef]
- Memon, F.A.; Nuruddin, M.F.; Shafiq, N. Effect of silica fume on the fresh and hardened properties of fly ash-based self-compacting geopolymer concrete. Int. J. Miner. Metall. Mater. 2013, 20, 205–213. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System Name | PCE Segment and Water | PCE Segment and SiO2 | SiO2 and Water | |
|---|---|---|---|---|
| Ester-based PCE segment | Absence of SiO2 | 19 | - | - |
| Presence of SiO2 | 20 | 0 | 7 | |
| Ether-based PCE segment | Absence of SiO2 | 19 | - | - |
| Presence of SiO2 | 19 | 0 | 6 | |
| Compound | Bond Length in Å | |||
|---|---|---|---|---|
| Ester-based PCE + SiO2 + Ca2+ | Ca-O1 | Ca-O3 | Ca-O17 | Ca-O58 |
| 2.33 | 2.37 | 2.34 | 2.42 | |
| Ether-based PCE+ SiO2 + Ca2+ | Ca-O1 | Ca-O3 | Ca-O52 | - |
| 2.21 | 2.21 | 2.30 | - | |
| Compound | Ester PCE + SiO2 + Ca2+ | Ether PCE+ SiO2 + Ca2+ |
|---|---|---|
| E(interaction), kJ/mol | −71.75 | −67.16 |
| (PCE/SiO2), kJ/mol | 5.91 | 9.09 |
| (PCE/Ca2+), kJ/mol | 3.34 | 4.65 |
| (SiO2/Ca2+), kJ/mol | 2.46 | 2.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakhimbayev, B.; Mukashev, B.; Kusherova, P.; Serikkanov, A.; Kemelbekova, A.; Agybayev, K.; Aldongarov, A.; Almas, N. Atomistic Insight on Effect of Silica Fume on Intermolecular Interactions between Poly(carboxylate) Superplasticizer and Calcium Ions in Concrete. Nanomaterials 2024, 14, 1084. https://doi.org/10.3390/nano14131084
Rakhimbayev B, Mukashev B, Kusherova P, Serikkanov A, Kemelbekova A, Agybayev K, Aldongarov A, Almas N. Atomistic Insight on Effect of Silica Fume on Intermolecular Interactions between Poly(carboxylate) Superplasticizer and Calcium Ions in Concrete. Nanomaterials. 2024; 14(13):1084. https://doi.org/10.3390/nano14131084
Chicago/Turabian StyleRakhimbayev, Berik, Bulat Mukashev, Parasat Kusherova, Abay Serikkanov, Ainagul Kemelbekova, Kamil Agybayev, Anuar Aldongarov, and Nurlan Almas. 2024. "Atomistic Insight on Effect of Silica Fume on Intermolecular Interactions between Poly(carboxylate) Superplasticizer and Calcium Ions in Concrete" Nanomaterials 14, no. 13: 1084. https://doi.org/10.3390/nano14131084