
A First Principles Study of Lithium Adsorption in Nanoporous Graphene


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
3. Results
3.1. Lithium Ion Transport through Graphene Nanopores
3.2. Adsorption Mechanism of Lithium in a Graphene Nanopore
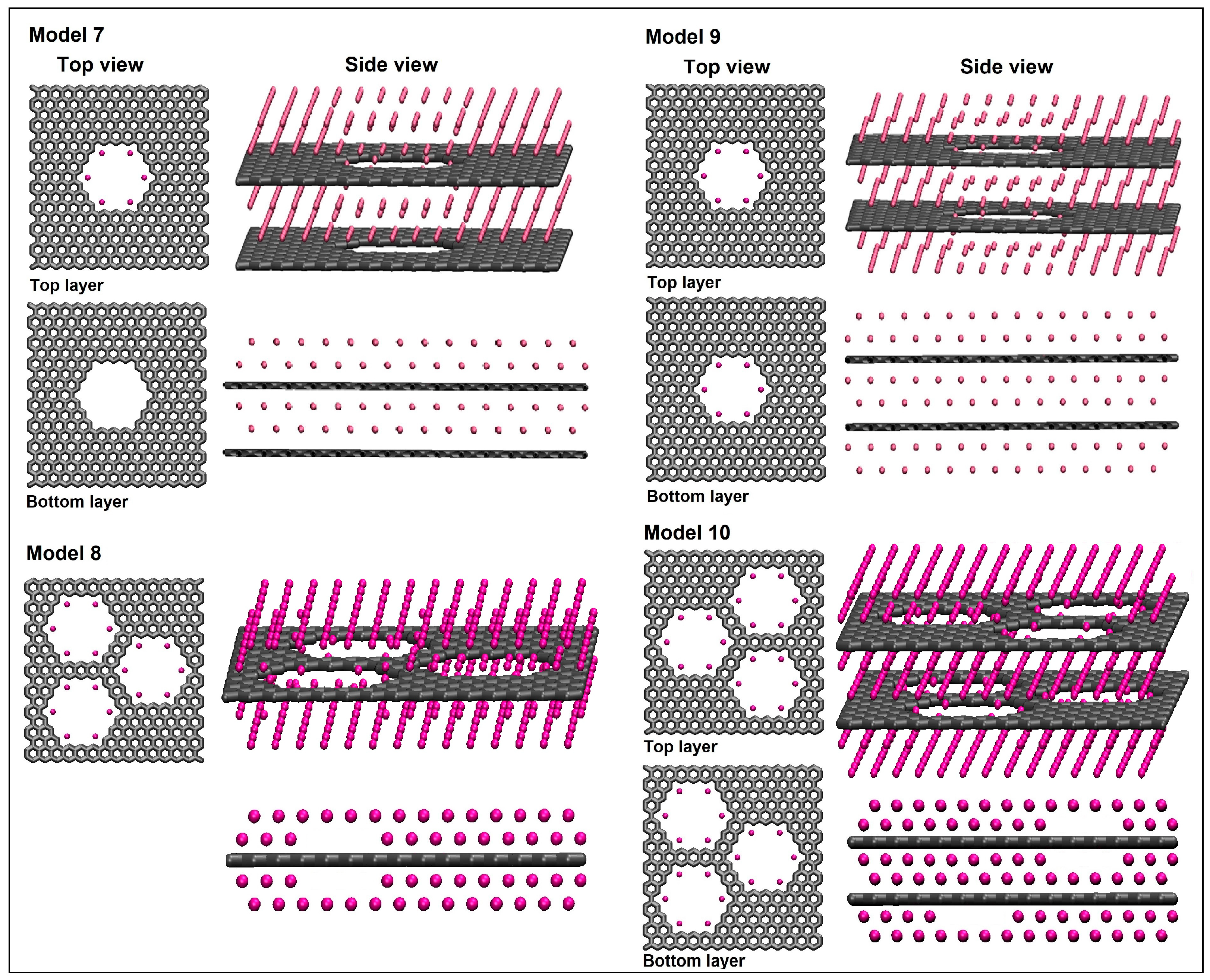
3.3. Multilayer Lithium–Nanoporous Graphene Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhu, P.; Gastol, D.; Marshall, J.; Sommerville, R.; Goodship, V.; Kendrick, E. A review of current collectors for lithium-ion batteries. J. Power Sources 2021, 36, 229321. [Google Scholar] [CrossRef]
- Hou, J.; Qu, S.; Yang, M.; Zhang, J. Materials and electrode engineering of high capacity anodes in lithium ion batteries. J. Power Sources 2020, 450, 227697. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, X.; Zhao, S.; Wang, A.; Luo, J.; Wang, Z.L.; Kang, F.; Lin, Z.; Li, B. Advanced Matrixes for Binder-Free Nanostructured Electrodes in Lithium-Ion Batteries. Adv. Mater. 2020, 32, 1908445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Ren, D.; Wang, L.; He, X. Graphite as anode materials: Fundamental mechanism, recent progress and advances. Energy Storage Mater. 2021, 36, 147–170. [Google Scholar] [CrossRef]
- Choi, C.; Ashby, D.S.; Butts, D.M.; DeBlock, R.H.; Wei, Q.; Lau, J.; Dunn, B. Achieving high energy density and high power density with pseudocapacitive materials. Nat. Rev. Mater. 2020, 5, 5–19. [Google Scholar] [CrossRef]
- Peng, L.; Fang, Z.; Zhu, Y.; Yan, C.; Yu, G. Nanoporous 2D nanomaterials for electrochemical energy storage. Adv. Energy Mater. 2018, 8, 1702179. [Google Scholar] [CrossRef]
- Jia, S.; Zang, J.; Tian, P.; Zhou, S.; Cai, H.; Tian, X.; Wang, Y. A 3-D covalently crosslinked N-doped porous carbon/nanoporous graphene composite for quasi-solid-state supercapacitors. Microporous Mesoporous Mater. 2020, 293, 109796. [Google Scholar] [CrossRef]
- Minakshi, M.; Mujeeb, A.; Whale, J.; Evans, R.; Aughterson, R.; Shinde, P.A.; Ariga, K.; Shrestha, L.K. Synthesis of Porous Carbon Honeycomb Structures Derived from Hemp for Hybrid Supercapacitors with Improved Electrochemistry. Chempluschem 2024, e202400408. [Google Scholar] [CrossRef]
- Minakshi, M.; Samayamanthry, A.; Whale, J.; Aughterson, R.; Shinde, P.A.; Ariga, K.; Shrestha, L.K. Phosphorous—Containing Activated Carbon Derived from Natural Honeydew Peel Powers Aqueous Supercapacitors. Chem. Asian J. 2024, e202400622. [Google Scholar] [CrossRef]
- Allen, M.J.; Tung, V.C.; Kaner, R.B. Honeycomb Carbon: A Review of Graphene. Chem. Rev. 2010, 110, 132–145. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and Graphene Oxide: Synthesis, Properties and Applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, H.; Das, P.; Qin, J.; Li, Y.; Wang, X.; Su, F.; Wen, P.; Li, S.; Lu, P.; et al. The Chemistry and Promising Applications of Graphene and Porous Graphene Materials. Adv. Funct. Mater. 2020, 30, 1909. [Google Scholar] [CrossRef]
- Olabi, A.; Abdelkareem, M.A.; Wilberforce, T.; Sayed, E.T. Application of graphene in energy storage—A review. Renew. Sustain. Energy Rev. 2021, 135, 110026. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, G.; Zhang, F. Multiple roles of graphene in heterogeneous catalysis. Chem. Soc. Rev. 2015, 44, 3023. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Wang, M.; Roy, A. Transparent Graphene/BN-Graphene Stacked Nanofilms for Electrocatalytic Oxygen Evolution. ACS Appl. Nano Mater. 2020, 3, 10. [Google Scholar] [CrossRef]
- Huang, H.; Ying, Y.; Peng, X. Graphene oxide nanosheet: An emerging star material for novel separation membranes. J. Mater. Chem. A 2014, 2, 13772–13782. [Google Scholar] [CrossRef]
- Chung, C.; Kim, Y.-K.; Shin, D.; Ryoo, S.-R.; Hong, B.H.; Min, D.-H. Biomedical applications of graphene and graphene oxide. Acc. Chem. Res. 2013, 46, 2211–2224. [Google Scholar] [CrossRef]
- Perreault, F.; de Faria, A.F.; Elimelech, M. Environmental applications of graphene-based nanomaterials. Chem. Soc. Rev. 2015, 44, 5861–5896. [Google Scholar] [CrossRef]
- Bi, J.; Du, Z.; Sun, J.; Liu, Y.; Wang, K.; Du, H.; Ai, W.; Huang, W. On the Road to the Frontiers of Lithium-Ion Batteries: A Review and Outlook of Graphene Anodes. Adv. Mater. 2023, 35, 2210734. [Google Scholar] [CrossRef]
- Dahn, J.R.; Zheng, T.; Liu, Y.; Xue, J.S. Mechanisms for Lithium Insertion in Carbonaceous Materials. Science 1995, 270, 590–593. [Google Scholar] [CrossRef]
- Pollak, E.; Geng, B.; Jeon, K.-J.; Lucas, I.T.; Richardson, T.J.; Wang, F.; Kostecki, R. The Interaction of Li+ with Single-Layer and Few-Layer Graphene. Nano Lett. 2010, 10, 3386–3388. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Persson, K.A. Li Absorption and Intercalation in Single Layer Graphene and Few Layer Graphene by First Principles. Nano Lett. 2012, 12, 4624–4628. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W. Nobel Lecture: Electronic structure of matterwave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253–1266. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional theory: Its origins, rose to prominence, and future. Rev. Mod. Phys. 2015, 87, 897. [Google Scholar] [CrossRef]
- Botello-Méndez, A.R.; Dubois, S.M.-M.; Lherbier, A.; Charlier, J.-C. Achievements of DFT for the Investigation of Graphene-Related Nanostructures. Acc. Chem. Res. 2014, 47, 3292–3300. [Google Scholar] [CrossRef]
- Zhang, T.; Xue, Q.; Zhang, S.; Dong, M. Theoretical approaches to graphene and graphene based materials. Nano Today 2012, 7, 180–200. [Google Scholar] [CrossRef]
- Nakada, K.; Ishii, A. Migration of adatom on graphene using DFT calculation. Solid State Commun. 2011, 151, 13–16. [Google Scholar] [CrossRef]
- Tau, O.; Lovergine, N.; Prete, P. Adsorption and decomposition steps on Cu(111) of liquid aromatic hydrocarbon precursors for low-temperature CVD of graphene: A DFT study. Carbon 2023, 206, 142–149. [Google Scholar] [CrossRef]
- Buldum, A.; Tetiker, G. First-principles study of graphene-lithium structures for battery applications. J. Appl. Phys. 2013, 113, 154312. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.M.; Yakobson, B.I.; Wood, B.C. Assessing Carbon-Based Anodes for Lithium-Ion Batteries: A Universal Description of Charge-Transfer Binding. Phys. Rev. Lett. 2014, 113, 028305. [Google Scholar] [CrossRef]
- Datta, D.; Li, J.; Koratkar, N.; Shenoy, V.B. Enchanced Lithiation in Defective Graphene. Carbon 2014, 80, 305–310. [Google Scholar] [CrossRef]
- Tsai, Y.-J.; Kuo, C.-L. Effect of Structural Disorders on the Li Storage Capacity of Graphene Nanomaterials: A First-Principles Stidy. ACS Appl. Mater. Interfaces 2020, 12, 22917–22929. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Comm. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Liu, D.C.; Nocedal, J. On the Limited Memory BFGS Method for Large Scale Optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef]
- Ching, W.Y.; Callaway, J. Band structure, cohesive energy, optical conductivity, and Compton profile of lithium. Phys. Rev. B. 1974, 9, 5115. [Google Scholar] [CrossRef]
- Fan, Z.; Yan, J.; Ning, G.; Wei, T.; Zhi, L.; Wei, F. Porous graphene networks as high performance anode materials for lithium ion batteries. Carbon 2013, 60, 558–561. [Google Scholar] [CrossRef]


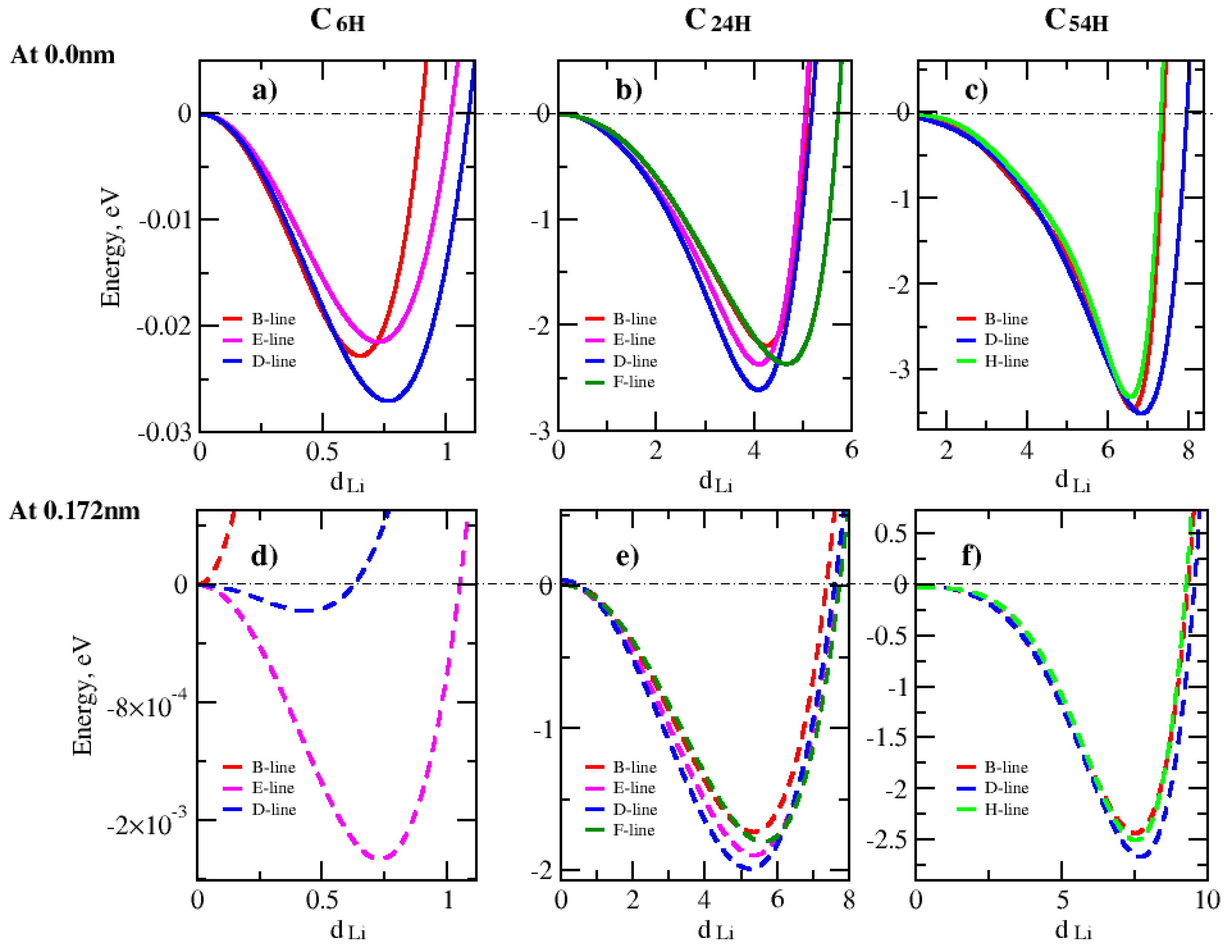
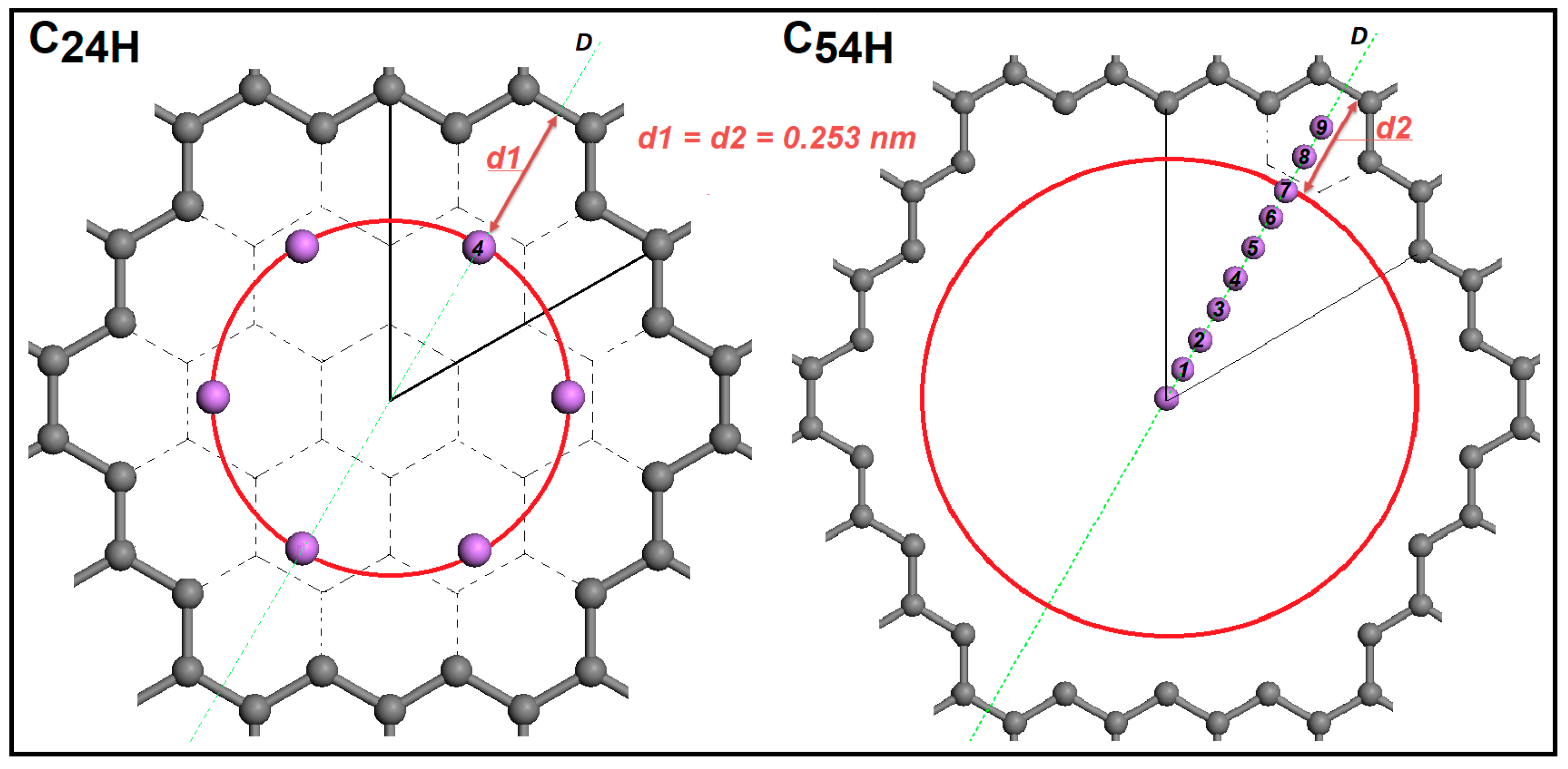
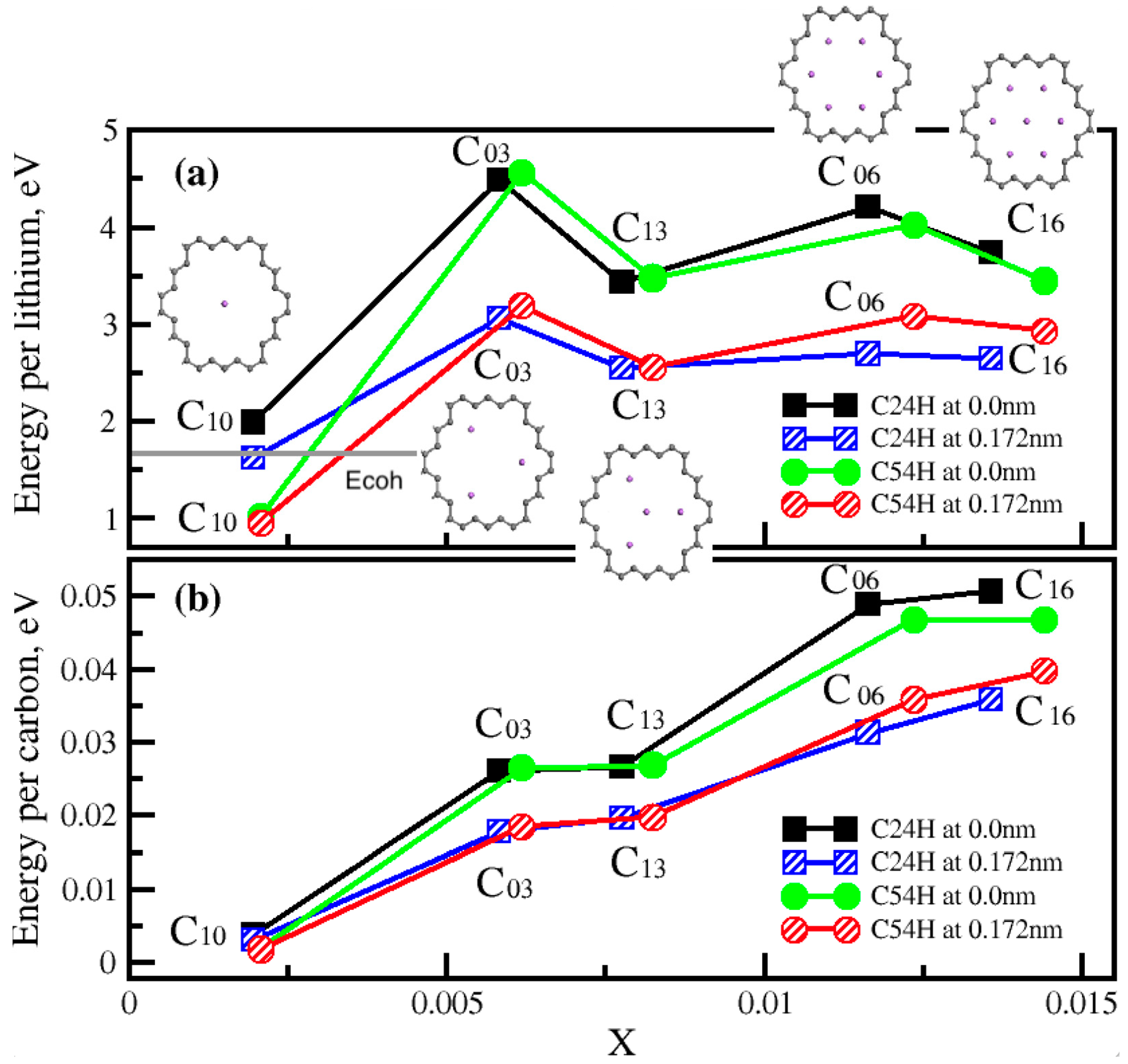

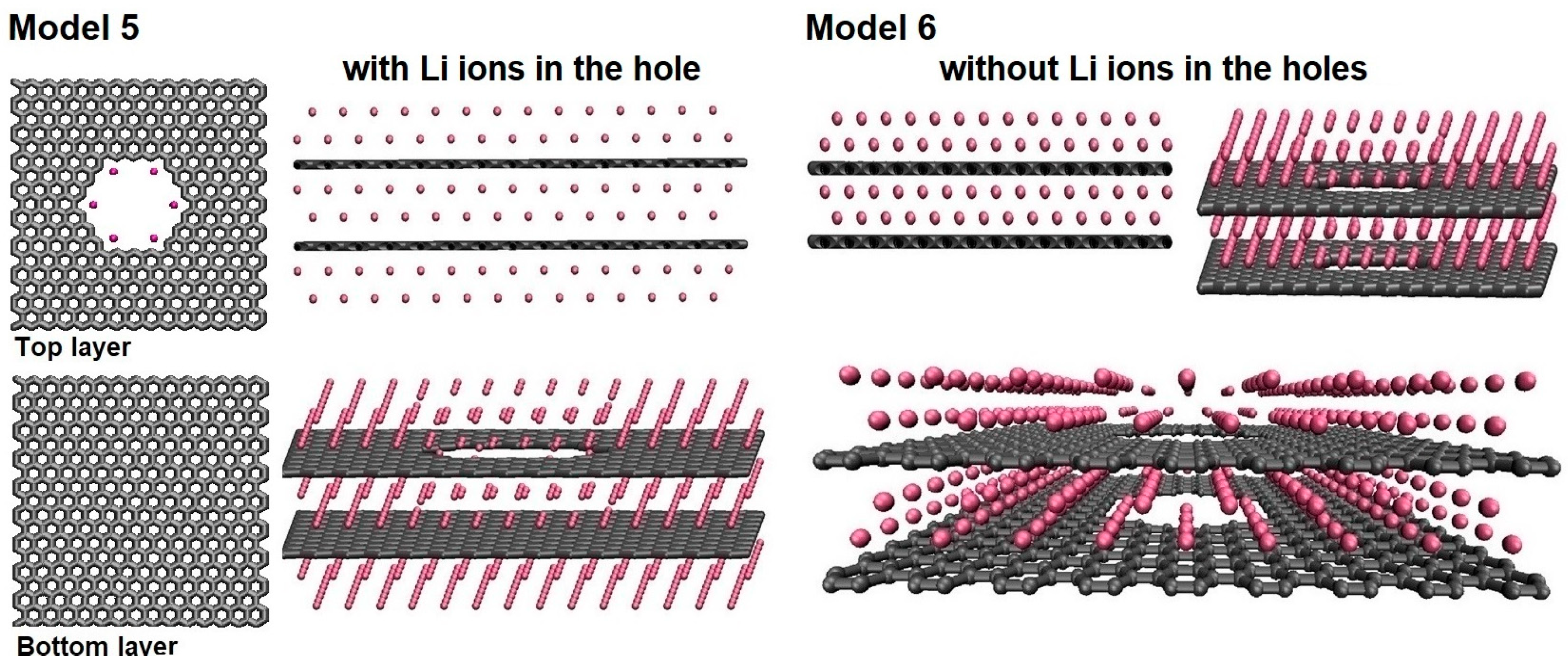

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barabanova, L.; Buldum, A. A First Principles Study of Lithium Adsorption in Nanoporous Graphene. Nanomaterials 2024, 14, 1528. https://doi.org/10.3390/nano14181528
Barabanova L, Buldum A. A First Principles Study of Lithium Adsorption in Nanoporous Graphene. Nanomaterials. 2024; 14(18):1528. https://doi.org/10.3390/nano14181528
Chicago/Turabian StyleBarabanova, Liudmyla, and Alper Buldum. 2024. "A First Principles Study of Lithium Adsorption in Nanoporous Graphene" Nanomaterials 14, no. 18: 1528. https://doi.org/10.3390/nano14181528
APA StyleBarabanova, L., & Buldum, A. (2024). A First Principles Study of Lithium Adsorption in Nanoporous Graphene. Nanomaterials, 14(18), 1528. https://doi.org/10.3390/nano14181528