Microfluidics Facilitates the Development of Single-Cell RNA Sequencing



Abstract
:1. Introduction
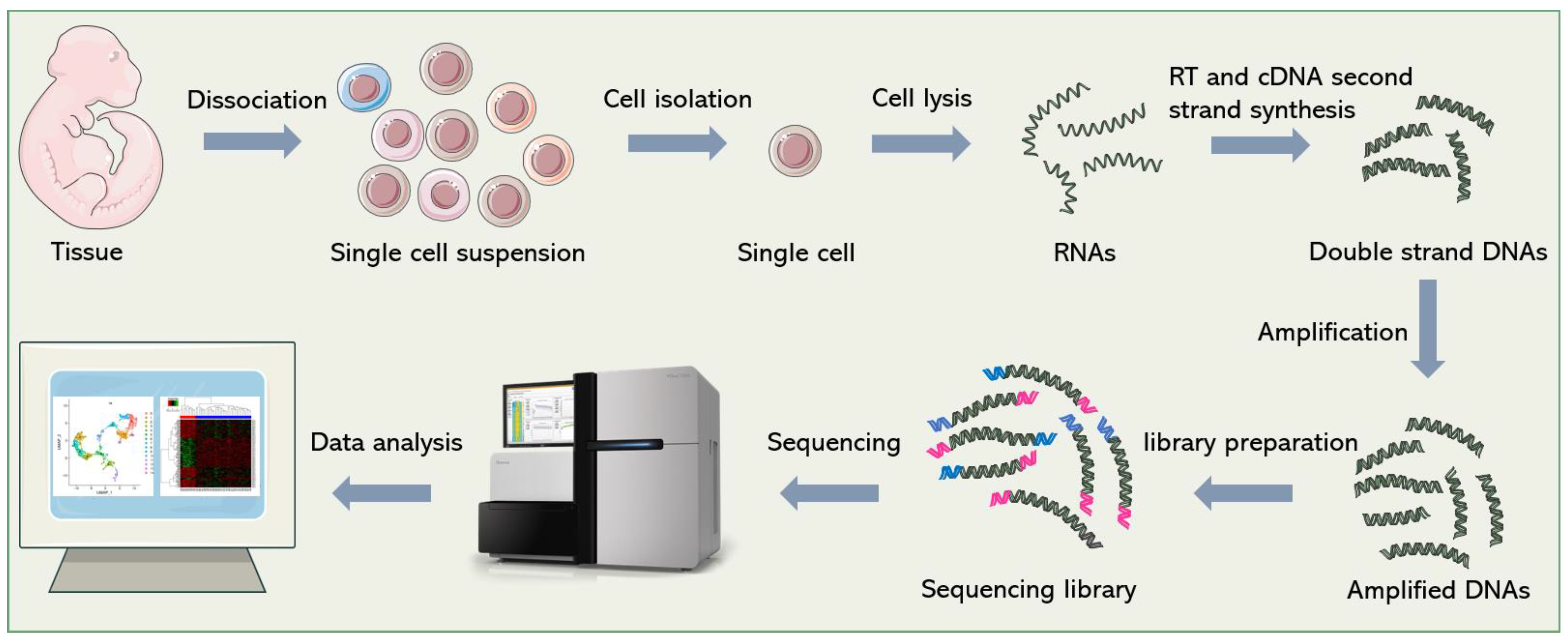
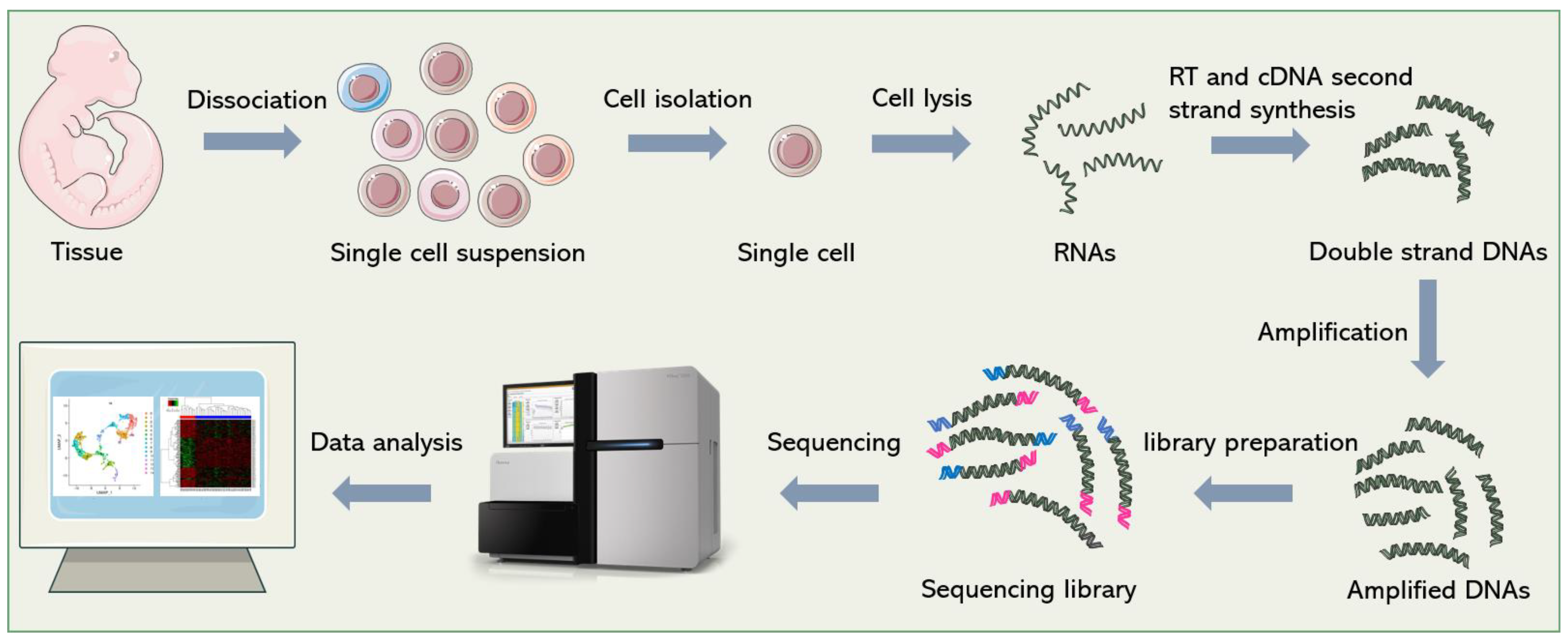
2. General Process of scRNA-seq
3. Developmental Course of scRNA-seq
3.1. Scaling of Sequencing Throughput
3.2. Improvement in Sensitivity
4. Low-Throughput scRNA-seq Methods
4.1. Smart-Seq Chemistry
4.2. CEL-Seq Chemistry
4.3. MATQ-seq Chemistry
5. High-Throughput scRNA-seq Methods
5.1. Application of Microfluidic Technology in scRNA-seq
5.2. Droplet-Based scRNA-seq Methods
5.2.1. inDrop
5.2.2. Drop-Seq
5.2.3. 10x Genomics
5.2.4. BAG-seq
5.3. Microwell-Based scRNA-seq Methods
5.3.1. CytoSeq
5.3.2. RNA Printing
5.3.3. Seq-Well
5.3.4. Microwell-seq
5.4. Comparison of Droplet- and Microwell-Based Methods
6. Future Perspectives
6.1. Improvement in Sensitivity of High-Throughput Sequencing Methods
6.2. Development of Multiomics Joint Sequencing Methods
6.3. Acquisition of Spatial Information of Transcripts
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lönnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.E.J.; Chan, S.Y. Current approaches and developments in transcript profiling of the human placenta. Hum. Reprod. Update 2020, 26, 799–840. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.D.; Wu, Z.; Irizarry, R.A.; Leek, J.T. Sequencing technology does not eliminate biological variability. Nat. Biotechnol. 2011, 29, 572–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Costa, V.; Angelini, C.; de Feis, I.; Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. J. Biomed, Biotechnol. 2010, 2010, 853916. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- Wang, Y.; Mashock, M.; Tong, Z.; Mu, X.; Chen, H.; Zhou, X.; Zhang, H.; Zhao, G.; Liu, B.; Li, X. Changing Technologies of RNA Sequencing and Their Applications in Clinical Oncology. Front. Oncol. 2020, 10, 447. [Google Scholar] [CrossRef] [Green Version]
- Picelli, S.; Faridani, O.R.; Björklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Chuang, H.Y.; Hofree, M.; Ideker, T. A decade of systems biology. Annu. Rev. Cell Dev. Biol. 2010, 26, 721–744. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zernicka-Goetz, M. Principles of Self-Organization of the Mammalian Embryo. Cell 2020, 183, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Choi, K.; Chen, Y.; Skelly, D.A.; Churchill, G.A. Bayesian model selection reveals biological origins of zero inflation in single-cell transcriptomics. Genome Biol. 2020, 21, 183. [Google Scholar] [CrossRef]
- He, Y.; Yuan, H.; Wu, C.; Xie, Z. DISC: A highly scalable and accurate inference of gene expression and structure for single-cell transcriptomes using semi-supervised deep learning. Genome Biol. 2020, 21, 170. [Google Scholar] [CrossRef]
- Yu, Q.; Kilik, U.; Holloway, E.M.; Tsai, Y.H.; Harmel, C.; Wu, A.; Wu, J.H.; Czerwinski, M.; Childs, C.J.; He, Z.; et al. Charting human development using a multi-endodermal organ atlas and organoid models. Cell 2021, 184, 3281–3298.e3222. [Google Scholar] [CrossRef]
- Plass, M.; Solana, J.; Wolf, F.A.; Ayoub, S.; Misios, A.; Glažar, P.; Obermayer, B.; Theis, F.J.; Kocks, C.; Rajewsky, N. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 2018, 360, eaaq1723. [Google Scholar] [CrossRef] [Green Version]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Simmons, S.K.; Guo, A.; Shetty, A.S.; Ko, M.; Nguyen, L.; Jokhi, V.; Robinson, E.; Oyler, P.; Curry, N.; et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 2020, 370, eaaz6063. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Li, Z.; Rong, D.; Zhang, H.; Shi, X.; Yang, W.; Zheng, W.; Sun, G.; Wu, F.; Cao, H.; et al. Single-cell RNA sequencing in cancer: Applications, advances, and emerging challenges. Mol. Ther. Oncolytics 2021, 21, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef]
- Ma, A.; McDermaid, A.; Xu, J.; Chang, Y.; Ma, Q. Integrative Methods and Practical Challenges for Single-Cell Multi-omics. Trends Biotechnol. 2020, 38, 1007–1022. [Google Scholar] [CrossRef]
- Moor, A.E.; Itzkovitz, S. Spatial transcriptomics: Paving the way for tissue-level systems biology. Curr. Opin. Biotechnol. 2017, 46, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 2021, 22, 3–18. [Google Scholar] [CrossRef]
- Rao, A.; Barkley, D.; Franca, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Matuła, K.; Rivello, F.; Huck, W.T.S. Single-Cell Analysis Using Droplet Microfluidics. Adv. Biosyst. 2020, 4, e1900188. [Google Scholar] [CrossRef] [Green Version]
- Curran, S.; McKay, J.A.; McLeod, H.L.; Murray, G.I. Laser capture microscopy. Mol. Pathol. 2000, 53, 64–68. [Google Scholar] [CrossRef]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser capture microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef]
- Gautam, V.; Chatterjee, S.; Sarkar, A.K. Single Cell Type Specific RNA Isolation and Gene Expression Analysis in Rice Using Laser Capture Microdissection (LCM)-Based Method. Methods Mol. Biol. 2021, 2238, 275–283. [Google Scholar] [CrossRef]
- Keays, K.M.; Owens, G.P.; Ritchie, A.M.; Gilden, D.H.; Burgoon, M.P. Laser capture microdissection and single-cell RT-PCR without RNA purification. J. Immunol. Methods 2005, 302, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Nichterwitz, S.; Chen, G.; Aguila, B.J.; Yilmaz, M.; Storvall, H.; Cao, M.; Sandberg, R.; Deng, Q.; Hedlund, E. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat. Commun. 2016, 7, 12139. [Google Scholar] [CrossRef] [Green Version]
- Pontén, F.; Williams, C.; Ling, G.; Ahmadian, A.; Nistér, M.; Lundeberg, J.; Pontén, J.; Uhlén, M. Genomic analysis of single cells from human basal cell cancer using laser-assisted capture microscopy. Mutat. Res. 1997, 382, 45–55. [Google Scholar] [CrossRef]
- Chen, J.; Suo, S.; Tam, P.P.; Han, J.J.; Peng, G.; Jing, N. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat. Protoc. 2017, 12, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Liew, L.C.; Wang, Y.; Peirats-Llobet, M.; Berkowitz, O.; Whelan, J.; Lewsey, M.G. Laser-Capture Microdissection RNA-Sequencing for Spatial and Temporal Tissue-Specific Gene Expression Analysis in Plants. J. Vis. Exp. 2020, 162, e61517. [Google Scholar] [CrossRef]
- Zechel, S.; Zajac, P.; Lönnerberg, P.; Ibáñez, C.F.; Linnarsson, S. Topographical transcriptome mapping of the mouse medial ganglionic eminence by spatially resolved RNA-seq. Genome Biol. 2014, 15, 486. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Zhong, G.; Zhang, Q.; Zhang, L.; Sun, Y.; Zhang, Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 2020, 30, 763–778. [Google Scholar] [CrossRef]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Habib, N.; Li, Y.; Heidenreich, M.; Swiech, L.; Avraham-Davidi, I.; Trombetta, J.J.; Hession, C.; Zhang, F.; Regev, A. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 2016, 353, 925–928. [Google Scholar] [CrossRef] [Green Version]
- Lacar, B.; Linker, S.B.; Jaeger, B.N.; Krishnaswami, S.R.; Barron, J.J.; Kelder, M.J.E.; Parylak, S.L.; Paquola, A.C.M.; Venepally, P.; Novotny, M.; et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 2016, 7, 11022. [Google Scholar] [CrossRef]
- See, K.; Tan, W.L.W.; Lim, E.H.; Tiang, Z.; Lee, L.T.; Li, P.Y.Q.; Luu, T.D.A.; Ackers-Johnson, M.; Foo, R.S. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat. Commun. 2017, 8, 225. [Google Scholar] [CrossRef] [Green Version]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, E.; Zuber, V.; Fernandez-Jimenez, N.; Bilbao, J.R.; Clark, G.R.; Maher, E.R.; Bottolo, L. MethylCal: Bayesian calibration of methylation levels. Nucleic Acids Res. 2019, 47, e81. [Google Scholar] [CrossRef] [Green Version]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xing, D.; Tan, L.; Li, H.; Zhou, G.; Huang, L.; Xie, X.S. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.; Singh, J.M.; Eberwine, J.H. Transcriptome analysis of single cells. J. Vis. Exp. 2011, 50, e2634. [Google Scholar] [CrossRef] [Green Version]
- Balzer, M.S.; Ma, Z.; Zhou, J.; Abedini, A.; Susztak, K. How to Get Started with Single Cell RNA Sequencing Data Analysis. J. Am. Soc. Nephrol. 2021, 32, 1279–1292. [Google Scholar] [CrossRef]
- Grun, D.; van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, K. Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nat. Rev. Nephrol. 2020, 16, 408–421. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Perez, R.K.; Gordon, M.G.; Subramaniam, M.; Kim, M.C.; Hartoularos, G.C.; Targ, S.; Sun, Y.; Ogorodnikov, A.; Bueno, R.; Lu, A.; et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 2022, 376, eabf1970. [Google Scholar] [CrossRef] [PubMed]
- Stoler, D.L.; Stewart, C.C.; Stomper, P.C. Breast epithelium procurement from stereotactic core biopsy washings: Flow cytometry-sorted cell count analysis. Clin. Cancer Res. 2002, 8, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Hochgerner, H.; Lönnerberg, P.; Hodge, R.; Mikes, J.; Heskol, A.; Hubschle, H.; Lin, P.; Picelli, S.; La Manno, G.; Ratz, M.; et al. STRT-seq-2i: Dual-index 5′ single cell and nucleus RNA-seq on an addressable microwell array. Sci. Rep. 2017, 7, 16327. [Google Scholar] [CrossRef] [Green Version]
- DeLaughter, D.M. The Use of the Fluidigm C1 for RNA Expression Analyses of Single Cells. Curr. Protoc. Mol. Biol. 2018, 122, e55. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef] [Green Version]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Holland, C.H.; Tanevski, J.; Perales-Paton, J.; Gleixner, J.; Kumar, M.P.; Mereu, E.; Joughin, B.A.; Stegle, O.; Lauffenburger, D.A.; Heyn, H.; et al. Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 2020, 21, 36. [Google Scholar] [CrossRef] [Green Version]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 2017, 14, 267–270. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Wu, X.; Guo, H.; Hu, Y.; Tang, F.; Huang, Y. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 2015, 16, 148. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.K.; Wadsworth, M.H.N.; Gierahn, T.M.; Do, T.; Weiss, D.; Andrade, P.R.; Ma, F.; de Andrade, S.B.J.; Shao, S.; Tsoi, L.C.; et al. Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immunity 2020, 53, 878–894.e877. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramsköld, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lonnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef] [Green Version]
- Drese, K.S. [Lab on a Chip]. Internist 2019, 60, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Medina-Sanchez, M.; Miserere, S.; Merkoci, A. Nanomaterials and lab-on-a-chip technologies. Lab Chip 2012, 12, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Kim, J.; Ni, M.; Wei, Y.; Okamoto, H.; Lee, J.; Adler, C.; Cavino, K.; Murphy, A.J.; Yancopoulos, G.D.; et al. Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc. Natl. Acad. Sci. USA 2016, 113, 3293–3298. [Google Scholar] [CrossRef] [Green Version]
- Abate, A.R.; Thiele, J.; Weitz, D.A. One-step formation of multiple emulsions in microfluidics. Lab Chip 2011, 11, 253–258. [Google Scholar] [CrossRef]
- Li, S.; Kendall, J.; Park, S.; Wang, Z.; Alexander, J.; Moffitt, A.; Ranade, N.; Danyko, C.; Gegenhuber, B.; Fischer, S.; et al. Copolymerization of single-cell nucleic acids into balls of acrylamide gel. Genome Res. 2020, 30, 49–61. [Google Scholar] [CrossRef]
- Fan, H.C.; Fu, G.K.; Fodor, S.P. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 2015, 347, 1258367. [Google Scholar] [CrossRef]
- Bose, S.; Wan, Z.; Carr, A.; Rizvi, A.H.; Vieira, G.; Pe’Er, D.; Sims, P.A. Scalable microfluidics for single-cell RNA printing and sequencing. Genome Biol. 2015, 16, 120. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e1017. [Google Scholar] [CrossRef] [Green Version]
- Gierahn, T.M.; Wadsworth, M.H.N.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- Mereu, E.; Lafzi, A.; Moutinho, C.; Ziegenhain, C.; McCarthy, D.J.; Álvarez-Varela, A.; Batlle, E.; Sagar; Grün, D.; Lau, J.K.; et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 2020, 38, 747–755. [Google Scholar] [CrossRef]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenblatt-Rosen, O.; Regev, A.; Oberdoerffer, P.; Nawy, T.; Hupalowska, A.; Rood, J.E.; Ashenberg, O.; Cerami, E.; Coffey, R.J.; Demir, E.; et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 2020, 181, 236–249. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar] [CrossRef]
- Zhu, C.; Yu, M.; Huang, H.; Juric, I.; Abnousi, A.; Hu, R.; Lucero, J.; Behrens, M.M.; Hu, M.; Ren, B. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol. 2019, 26, 1063–1070. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, Y.; Li, Y.E.; Lucero, J.; Behrens, M.M.; Ren, B. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods 2021, 18, 283–292. [Google Scholar] [CrossRef]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.; Ponting, C.P.; Voet, T.; et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Lu, P.; Wang, H.; Bian, S.; Wu, X.; Zhang, Y.; Liu, Y.; Fu, D.; Wen, L.; Hao, J.; et al. Integrated single-cell multiomics analysis reveals novel candidate markers for prognosis in human pancreatic ductal adenocarcinoma. Cell Discov. 2022, 8, 13. [Google Scholar] [CrossRef]
- Tirosh, I.; Suva, M.L. Dissecting human gliomas by single-cell RNA sequencing. Neuro Oncol. 2018, 20, 37–43. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e1618. [Google Scholar] [CrossRef] [PubMed]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Guan, D.; Shao, J.; Zhao, Z.; Wang, P.; Qin, J.; Deng, Y.; Boheler, K.R.; Wang, J.; Yan, B. PTHGRN: Unraveling post-translational hierarchical gene regulatory networks using PPI, ChIP-seq and gene expression data. Nucleic Acids Res. 2014, 42, W130–W136. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Field | Publication Year | Name | Barcode | UMI | Amplification Method | Sequencing Method | Throughput * | Conclusion | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Low-throughput methods | 2009 | Tang’s method | no | no | PCR | Nearly full length | + | The first scRNA-seq method | Tang F, et al. Nat Methods. 2009 [35] |
| 2011 | STRT-seq | yes | no | PCR | 5′ sequencing | + | 1. Being able to analyze transcription start sites 2. Cell-specific barcode | Islam S, et al. Genome Res. 2011 [1] | |
| 2012 | Smart-seq | no | no | PCR | full length | + | 1. High sensitivity 2. High coverage 3. Template-switching strategy | Ramsköld D, et al. Nat Biotechnol. 2012 [73] | |
| 2012 | CEL-seq | yes | no | IVT | 3′ sequencing | + | Linear in vitro transcription | Hashimshony T, et al. Cell Rep. 2012 [32] | |
| 2014 | Smart-seq2 | no | no | PCR | full length | + | Optimized conditions | Picelli S, et al. Nat Protoc. 2014 [10] | |
| 2016 | CEL-seq2 | yes | yes | IVT | 3′ sequencing | + | Optimized conditions | Hashimshony T, et al. Genome Biol. 2016 [79] | |
| 2017 | MATQ-seq | no | yes | Multiple annealing | full length | + | The most sensitive scRNA-seq method | Sheng K, et al. Nat Methods. 2017 [74] | |
| 2020 | Smart-seq3 | no | yes | PCR | full length | + | Highly sensitive and isoform-specific | Hagemann-Jensen M, et al. Nat Biotechnol. 2020 [77] | |
| Automatic liquid handling high-throughput method | 2014 | MARS-Seq | yes | yes | IVT | 3′ sequencing | ++ | Combination of FACS and automatic liquid handling | Jaitin DA, et al. Science. 2014 [63] |
| Droplet-based high-throughput methods | 2015 | inDrop | yes | yes | IVT | 3′ sequencing | ++ | 1. High hydrogel packaging efficiency 2. UV-initiated primer release 3. High-throughput CEL-seq method | Klein AM, et al. Cell. 2015 [66] |
| 2015 | Drop-seq | yes | yes | PCR | 3′ sequencing | ++ | High-throughput Smart-seq method | Macosko EZ, et al. Cell. 2015 [33] | |
| 2017 | 10x Chromium | yes | yes | PCR | 3′ sequencing | +++ | The most sensitive high-throughput scRNA-seq method. | Zheng GX, et al. Nat Commun. 2017 [67] | |
| 2020 | BAG-seq | yes | yes | PCR | 3′ sequencing | ++ | Capturing nucleic acid directly in hydrogel | Li S, et al. Genome Res. 2020 [85] | |
| Microwell-based high-throughput methods | 2015 | CytoSeq | yes | yes | PCR | 3′ sequencing | ++ | Using microwell to isolate and label cells | Fan HC, et al. Science. 2015 [86] |
| 2015 | Single-cell RNA printing | yes | no | IVT | 3′ sequencing | ++ | Solid-phase capture of RNA | Bose S, et al. Genome Biol. 2015 [87] | |
| 2017 | Seq-Well | yes | yes | PCR | 3′ sequencing | ++ | Semi-permeable polycarbonate membrane and surface-functionalized PDMS array | Gierahn TM, et al. Nat Methods. 2017 [89] | |
| 2018 | microwell-seq | yes | yes | PCR | 3′ sequencing | ++ | Cheap agarose microarray | Han X, et al. Cell. 2018 [88] | |
| Combinatorial indexing-based high-throughput methods | 2017 | sci-RNA-seq | yes | yes | PCR | 3′ sequencing | ++ | High-throughput and low cost | Cao J, et al. Science. 2017 [68] |
| 2018 | Split-seq | yes | yes | PCR | 3′ sequencing | +++ | High-throughput and low cost | Rosenberg AB, et al. Science. 2018 [69] | |
| Spatial transcriptomics | 2016 | LCM-seq | no | no | PCR | full length | + | Providing spatial information | Nichterwitz S, et al. Nat Commun. 2016 [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Cao, W.; Mu, Y.; Zhu, Q. Microfluidics Facilitates the Development of Single-Cell RNA Sequencing. Biosensors 2022, 12, 450. https://doi.org/10.3390/bios12070450
Pan Y, Cao W, Mu Y, Zhu Q. Microfluidics Facilitates the Development of Single-Cell RNA Sequencing. Biosensors. 2022; 12(7):450. https://doi.org/10.3390/bios12070450
Chicago/Turabian StylePan, Yating, Wenjian Cao, Ying Mu, and Qiangyuan Zhu. 2022. "Microfluidics Facilitates the Development of Single-Cell RNA Sequencing" Biosensors 12, no. 7: 450. https://doi.org/10.3390/bios12070450
APA StylePan, Y., Cao, W., Mu, Y., & Zhu, Q. (2022). Microfluidics Facilitates the Development of Single-Cell RNA Sequencing. Biosensors, 12(7), 450. https://doi.org/10.3390/bios12070450