Plasmonic Biosensing for Label-Free Detection of Two Hallmarks of Cancer Cells: Cell-Matrix Interaction and Cell Division

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
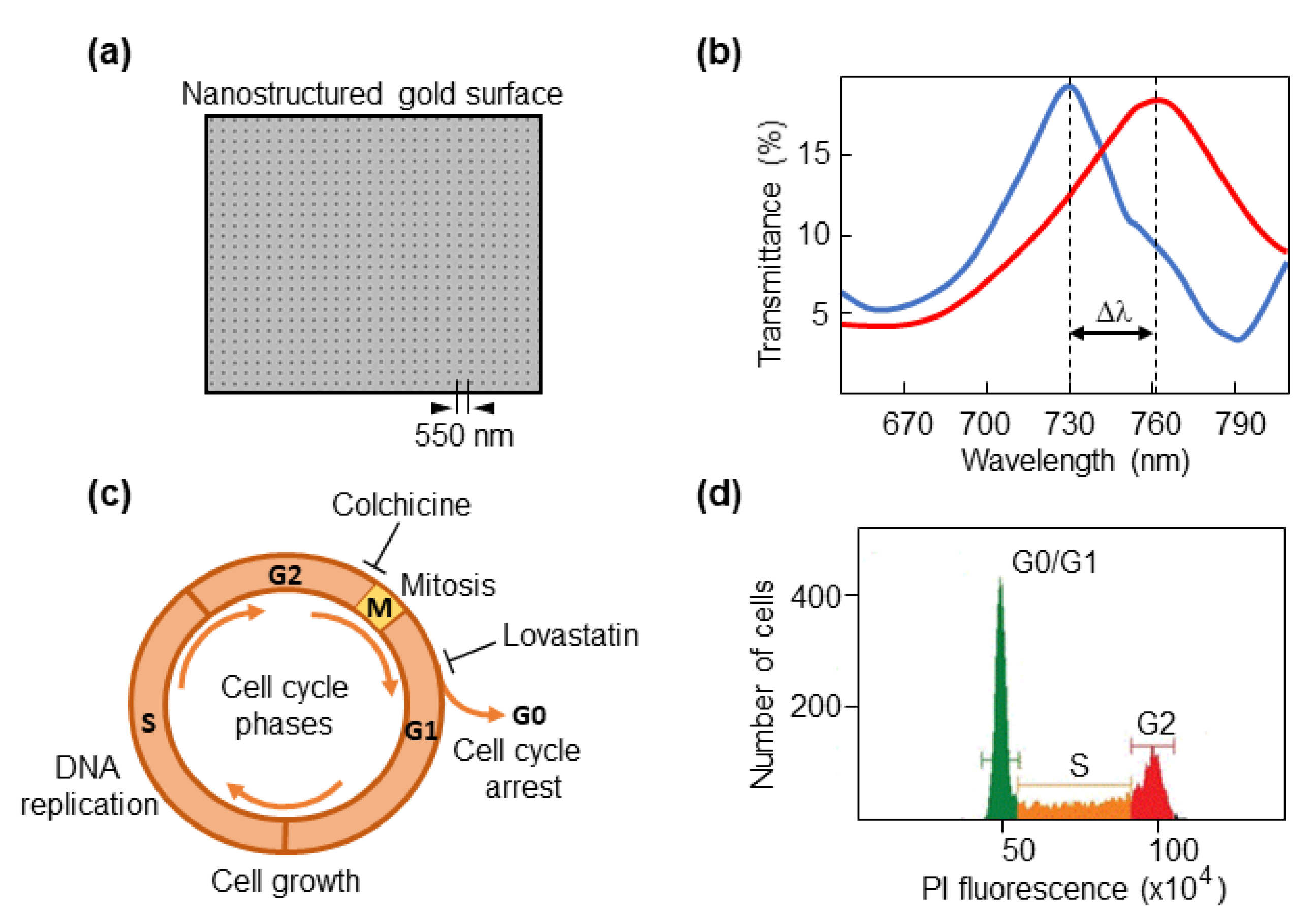
2.1. Optical Biosensor
2.2. Cell Lines
2.3. Cell Cycle Synchronization
2.4. Fluorescence Activated Cell Sorting (FACS)
2.5. Plasma Membrane Staining
2.6. Optical Measurements with the Biosensor
2.7. Statistical Analyses
3. Results and Discussion
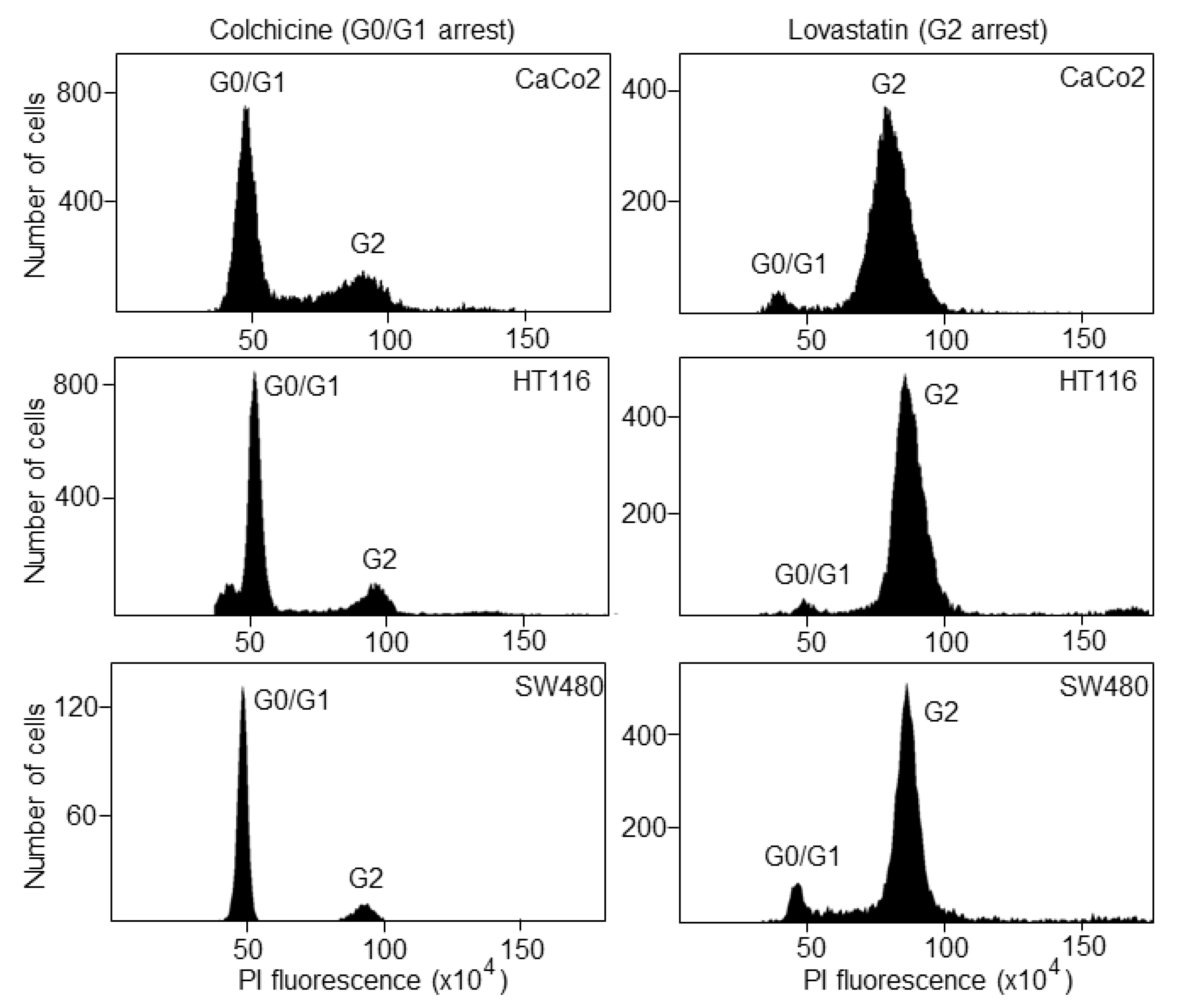
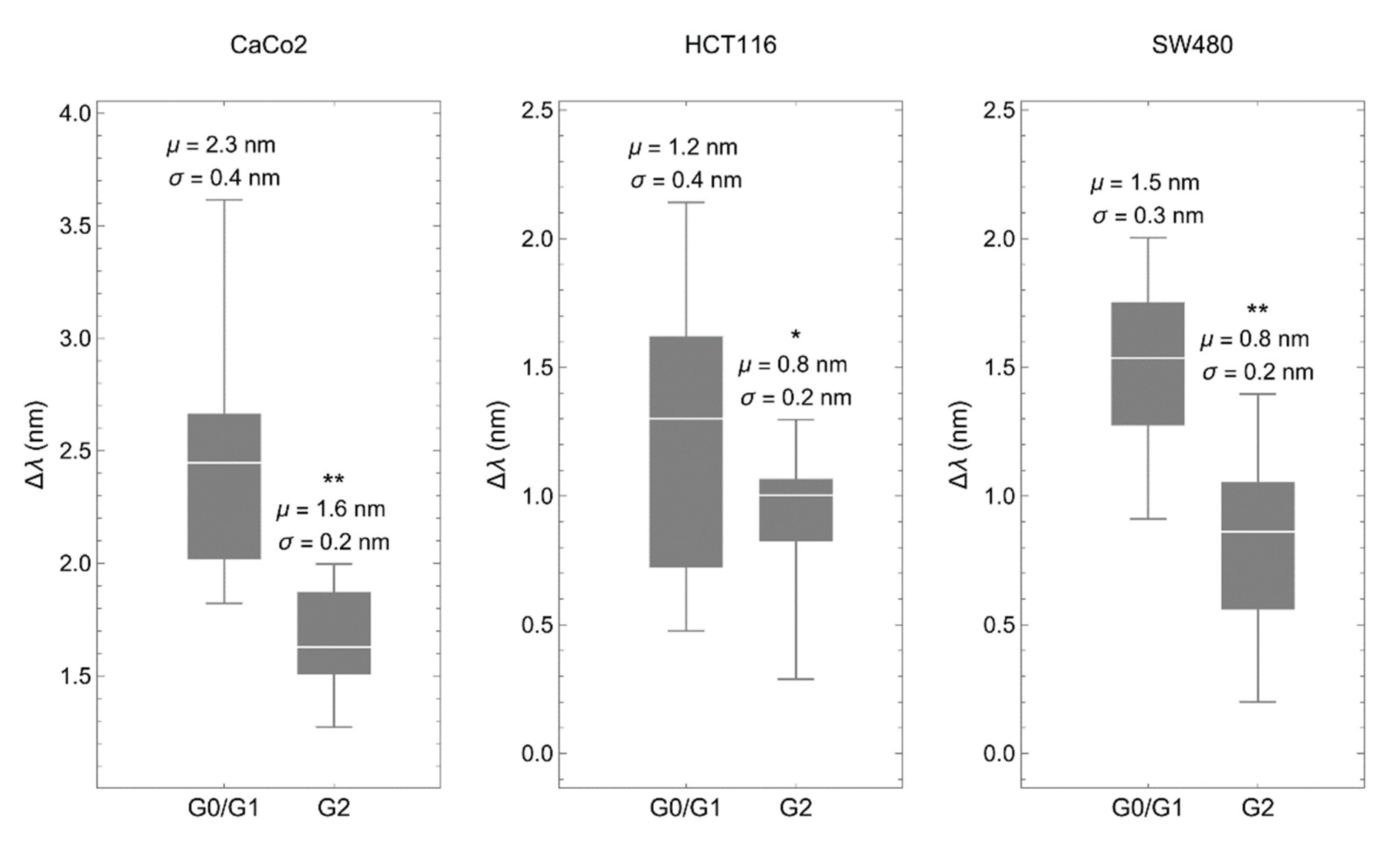
3.1. The Biosensor Is Able to Detect Different Phases of the Cell Cycle
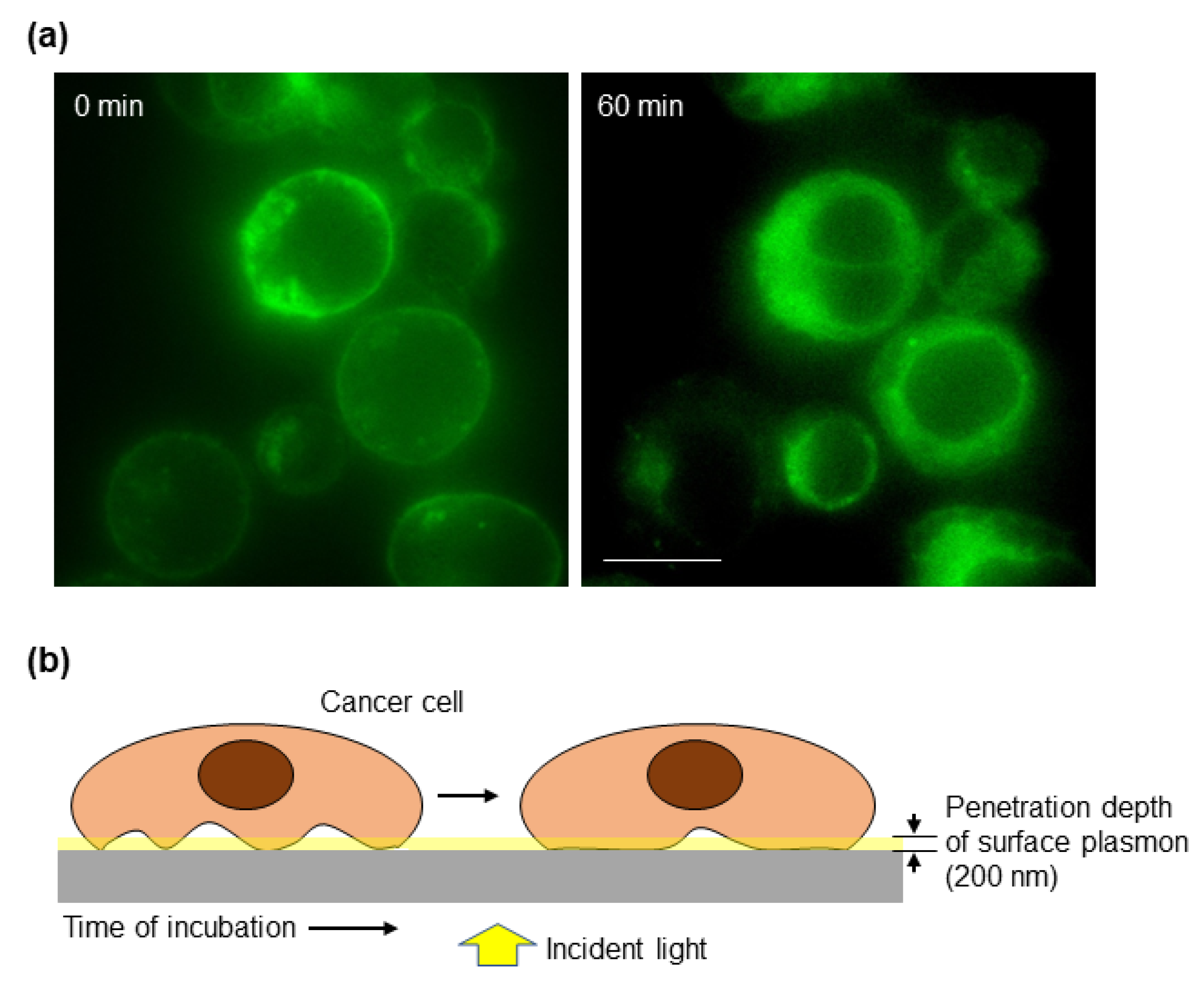
3.2. The Cancer Cell–Matrix Interaction Can Be Monitored by the Optical Biosensor
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, P.R.; Maity, A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007, 254, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, U.; Christofori, G. Cell adhesion in tumor invasion and metastasis: Loss of the glue is not enough. Biochim. Biophys. Acta 2001, 1552, 39–45. [Google Scholar] [CrossRef]
- Tu, L.; Li, X.; Bian, S.; Yu, Y.; Li, J.; Huang, L.; Liu, P.; Wu, Q.; Wang, W. Label-free and real-time monitoring of single cell attachment on template-stripped plasmonic nano-holes. Sci. Rep. 2017, 7, 11020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaks, M.; Giannone, G.; Rottner, K. Actin dynamics in cell migration. Essays Biochem. 2019, 63, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurmeister, S.; Baumann, M.; Balwierz, A.; Keklikoglou, I.; Ward, A.; Uhlmann, S.; Zhang, J.D.; Wiemann, S.; Sahin, Ö. MicroRNA-200c Represses Migration and Invasion of Breast Cancer Cells by Targeting Actin-Regulatory Proteins FHOD1 and PPM1F. Mol. Cell. Biol. 2012, 32, 633–651. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Guo, J.; Zheng, L.; Li, C.; Zheng, T.M.; Tanyi, J.L.; Liang, S.; Benedetto, C.; Mitidieri, M.; Katsaros, D.; et al. The Heterochronic microRNA let-7 Inhibits Cell Motility by Regulating the Genes in the Actin Cytoskeleton Pathway in Breast Cancer. Mol. Cancer Res. 2013, 11, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Priest, A.V.; Maker, A.; Gumbiner, B.; Sivasankar, S. Molecular mechanisms for strengthening E-cadherin adhesion using a monoclonal antibody. Biophys. J. 2022, 121, 526a. [Google Scholar] [CrossRef]
- Li, G.; Suzuki, H.; Asano, T.; Tanaka, T.; Suzuki, H.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-EpCAM Monoclonal Antibody for Various Applications. Antibodies 2022, 11, 41. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chin, L.K.; Ser, W.; Chen, H.F.; Hsieh, C.-M.; Lee, C.-H.; Sung, K.-B.; Ayi, T.C.; Yap, P.H.; Liedberg, B.; et al. Cell refractive index for cell biology and disease diagnosis: Past, present and future. Lab Chip 2016, 16, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Gul, B.; Ashraf, S.; Khan, S.; Nisar, H.; Ahmad, I. Cell refractive index: Models, insights, applications and future perspectives. Photodiagnosis Photodyn. Ther. 2021, 33, 102096. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Sinton, D.; Kavanagh, K.L.; Brolo, A.G. A New Generation of Sensors Based on Extraordinary Optical Transmission. Accounts Chem. Res. 2008, 41, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, C. On-chip nanohole array based sensing: A review. Lab Chip 2013, 13, 2445–2463. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Soler, M.; Özdemir, C.I.; Belushkin, A.; Yesilköy, F.; Altug, H. Plasmonic nanohole array biosensor for label-free and real-time analysis of live cell secretion. Lab Chip 2017, 17, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; Lozano, O.C.; Estevez, M.-C.; Lechuga, L.M. Nanophotonic Biosensors: Driving Personalized Medicine. Opt. Photon News 2020, 31, 24–31. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, Y.; Wang, S.; Liu, X. Surface Plasmon Resonance Microscopy: From Single-Molecule Sensing to Single-Cell Imaging. Angew. Chem. Int. Ed. 2019, 59, 1776–1785. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Liu, Q.; Wu, J.; Tao, N. Mapping Single-Cell–Substrate Interactions by Surface Plasmon Resonance Microscopy. Langmuir 2012, 28, 13373–13379. [Google Scholar] [CrossRef] [Green Version]
- Barreda, .I.; Otaduy, D.; Martín-Rodríguez, R.; Merino, S.; Fernández-Luna, J.L.; González, F.; Moreno, F. Electromagnetic behavior of dielectric objects on metallic periodically nanostructured substrates. Opt. Express 2018, 26, 11222–11237. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Romet-Lemonne, G.; Jégou, A. Mechanotransduction down to individual actin filaments. Eur. J. Cell Biol. 2013, 92, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Couture, M.; Live, L.S.; Dhawan, A.; Masson, J.-F. EOT or Kretschmann configuration? Comparative study of the plasmonic modes in gold nanohole arrays. Analyst 2012, 137, 4162–4170. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Vidal, V.; Gómez, M.; Gutiérrez, O.; Martino, M.; González, F.; Moreno, F.; Fernández-Luna, J.L. A label-free optical system with a nanohole array biosensor for discriminating live single cancer cells from normal cells. Nanophotonics 2022, 11, 315–328. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, J.; Zhang, G.; Fan, S.; Ge, W.; Hu, W.; Huang, P.; Hou, D.; Zheng, S. Characterization and discrimination of human colorectal cancer cells using terahertz spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 256, 119713. [Google Scholar] [CrossRef]
- Zadka, Ł.; Buzalewicz, I.; Ulatowska-Jarża, A.; Rusak, A.; Kochel, M.; Ceremuga, I.; Dzięgiel, P. Label-Free Quantitative Phase Imaging Reveals Spatial Heterogeneity of Extracellular Vesicles in Select Colon Disorders. Am. J. Pathol. 2021, 191, 2147–2171. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Zhang, H.; Min, C.; Zhang, Y.; Zhang, C.; Xin, Z.; Zhu, S.; Yang, Y.; Burge, R.E.; et al. Graphene-based confocal refractive index microscopy for label-free differentiation of living epithelial and mesenchymal cells. ACS Sensors 2020, 5, 510–518. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Moreno-Cencerrado, A.; Iturri, J.; Pecorari, I.; Vivanco, M.D.; Sbaizero, O.; Toca-Herrera, J.L. Investigating cell-substrate and cell-cell interactions by means of single-cell-probe force spectroscopy. Microsc. Res. Tech. 2017, 80, 124–130. [Google Scholar] [CrossRef]
- Helenius, J.; Heisenberg, C.-P.; Gaub, H.E.; Muller, D.J. Single-cell force spectroscopy. J. Cell Sci. 2008, 121, 1785–1791. [Google Scholar] [CrossRef] [Green Version]
- Huebsch, N.D.; Mooney, D. Fluorescent resonance energy transfer: A tool for probing molecular cell–biomaterial interactions in three dimensions. Biomaterials 2007, 28, 2424–2437. [Google Scholar] [CrossRef] [Green Version]
- Kemp-O’Brien, K.; Parsons, M. Using FRET to analyse signals controlling cell adhesion and migration. J. Microsc. 2013, 251, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Sederstrom, J.M. Assaying Cell Cycle Status Using Flow Cytometry. Curr. Protoc. Mol. Biol. 2015, 111, 28.6.1–28.6.11. [Google Scholar] [CrossRef] [PubMed]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.; Otaduy, D.; Barreda, A.; Fernández-Luna, J.; Merino, S.; González, F.; Moreno, F. Optical inspection of manufactured nanohole arrays to bridge the lab-industry gap. Opt. Laser Technol. 2019, 116, 48–57. [Google Scholar] [CrossRef] [Green Version]
- PMartínez-Camblor, P.; Pardo-Fernandez, J.C. The Youden Index in the Generalized Receiver Operating Characteristic Curve Context. Int. J. Biostat. 2019, 15. [Google Scholar] [CrossRef]
- JavanMoghadam-Kamrani, S.; Keyomarsi, K. Synchronization of the cell cycle using Lovastatin. Cell Cycle 2008, 7, 2434–2440. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.S.; Rao, U.S. Statins decrease the expression of c-Myc protein in cancer cell lines. Mol. Cell. Biochem. 2020, 476, 743–755. [Google Scholar] [CrossRef]
- Wójcik, C.; DeMartino, G.N. Intracellular localization of proteasomes. Int. J. Biochem. Cell Biol. 2003, 35, 579–589. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Li, L.; Kraus, V.B. Colchicine: Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Chugh, P.; Clark, A.G.; Smith, M.B.; Cassani, D.A.D.; Dierkes, K.; Ragab, A.; Roux, P.P.; Charras, G.; Salbreux, G.; Paluch, E. Actin cortex architecture regulates cell surface tension. Nat. Cell Biol. 2017, 19, 689–697. [Google Scholar] [CrossRef]
- Charras, G.T.; Hu, C.-K.; Coughlin, M.; Mitchison, T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Golias, C.; Charalabopoulos, A. Cell proliferation and cell cycle control: A mini review. Int. J. Clin. Pract. 2004, 58, 1134–1141. [Google Scholar] [CrossRef]
- Ungai-Salánki, R.; Gerecsei, T.; Fürjes, P.; Orgovan, N.; Sándor, N.; Holczer, E.G.; Horvath, R.; Szabó, B. Automated single cell isolation from suspension with computer vision. Sci. Rep. 2016, 6, 20375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Saha, S.; Sinha, B. Cell spread area and traction forces determine myosin-II-based cortex thickness regulation. Biochim. Biophys. Acta 2019, 1866, 118516. [Google Scholar] [CrossRef] [PubMed]
- Stolarska, M.A.; Rammohan, A.R. Center or periphery? Modeling the effects of focal adhesion placement during cell spreading. PLoS ONE 2017, 12, e0171430. [Google Scholar] [CrossRef]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Grigoriev, I.; Akhmanova, A. Microtubule Dynamics at the Cell Cortex Probed by TIRF Microscopy. Methods Cell Biol. 2010, 97, 91–109. [Google Scholar] [CrossRef]
- Zieber, F.; Aranson, I.S. Computational approaches to substrate-based cell motility. Comput. Mater. 2016, 2, 16019. [Google Scholar] [CrossRef] [Green Version]
- Zamir, E.; Katz, M.; Posen, Y.; Erez, N.; Yamada, K.; Katz, B.-Z.; Lin, S.; Lin, D.C.; Bershadsky, A.; Kam, Z.; et al. Dynamics and segregation of cell–matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2000, 2, 191–196. [Google Scholar] [CrossRef]
- Chalut, K.J.; Paluch, E.K. The Actin Cortex: A Bridge between Cell Shape and Function. Dev. Cell 2016, 38, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Kuwada, S.K.; Li, X. Integrin α5/β1 Mediates Fibronectin-dependent Epithelial Cell Proliferation through Epidermal Growth Factor Receptor Activation. Mol. Biol. Cell 2000, 11, 2485–2496. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-Y.; Xu, K.-S.; Pan, Z.-Q.; Zhang, Z.-Y.; Mi, Y.-T.; Wang, J.-S.; Chen, R.; Niu, J. Integrin alphavbeta6 mediates the potential for colon cancer cells to colonize in and metastasize to the liver. Cancer Sci. 2008, 99, 879–887. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carcelen, M.; Vidal, V.; Franco, A.; Gomez, M.; Moreno, F.; Fernandez-Luna, J.L. Plasmonic Biosensing for Label-Free Detection of Two Hallmarks of Cancer Cells: Cell-Matrix Interaction and Cell Division. Biosensors 2022, 12, 674. https://doi.org/10.3390/bios12090674
Carcelen M, Vidal V, Franco A, Gomez M, Moreno F, Fernandez-Luna JL. Plasmonic Biosensing for Label-Free Detection of Two Hallmarks of Cancer Cells: Cell-Matrix Interaction and Cell Division. Biosensors. 2022; 12(9):674. https://doi.org/10.3390/bios12090674
Chicago/Turabian StyleCarcelen, Maria, Veronica Vidal, Alfredo Franco, Marcos Gomez, Fernando Moreno, and Jose L Fernandez-Luna. 2022. "Plasmonic Biosensing for Label-Free Detection of Two Hallmarks of Cancer Cells: Cell-Matrix Interaction and Cell Division" Biosensors 12, no. 9: 674. https://doi.org/10.3390/bios12090674