Electrospun Nanofibers including Organic/Inorganic Nanohybrids: Polystyrene- and Clay-Based Architectures in Immunosensor Preparation for Serum Amyloid A




Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Apparatus
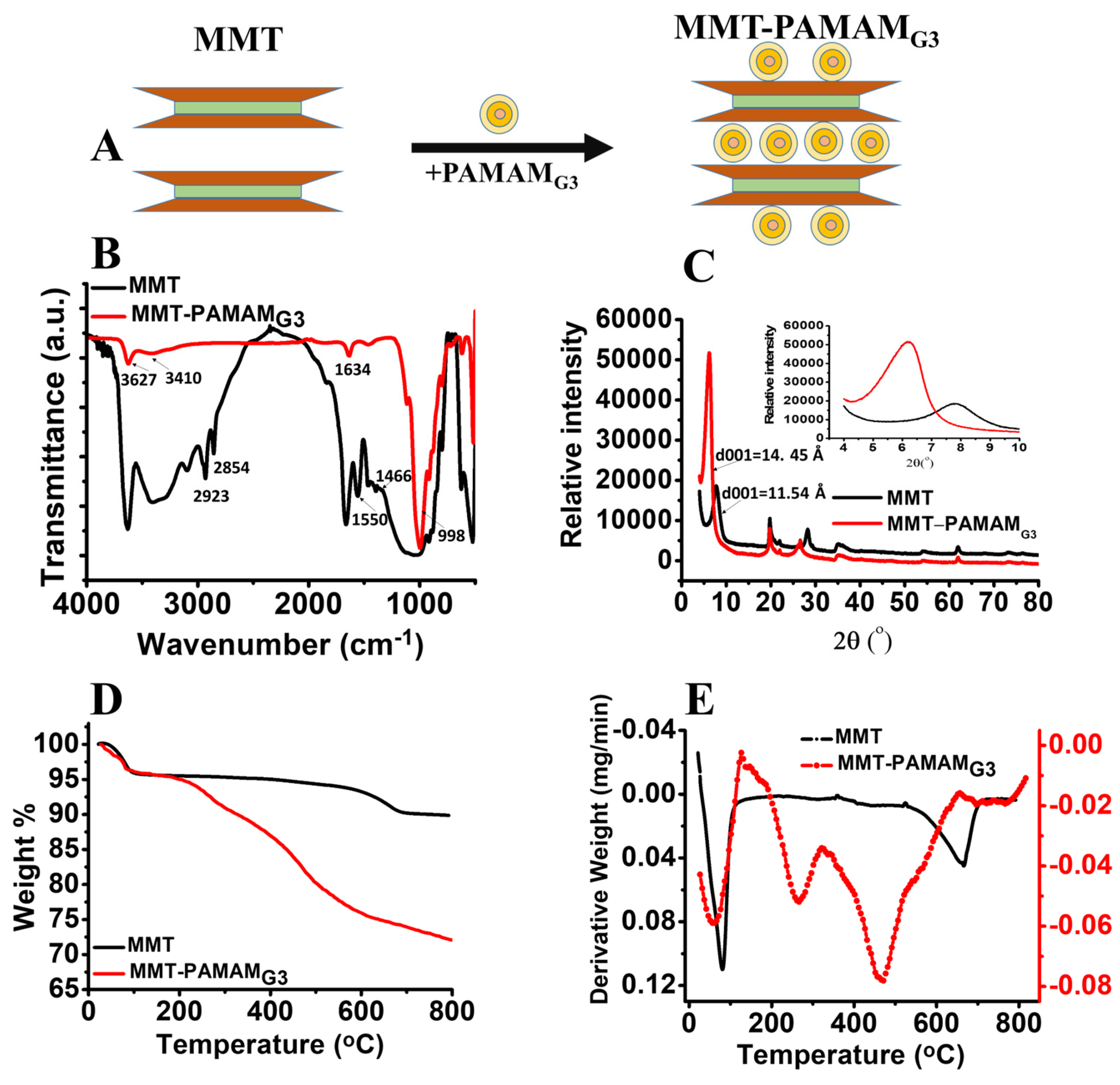
2.3. Preparation of MMT-PAMAMG3
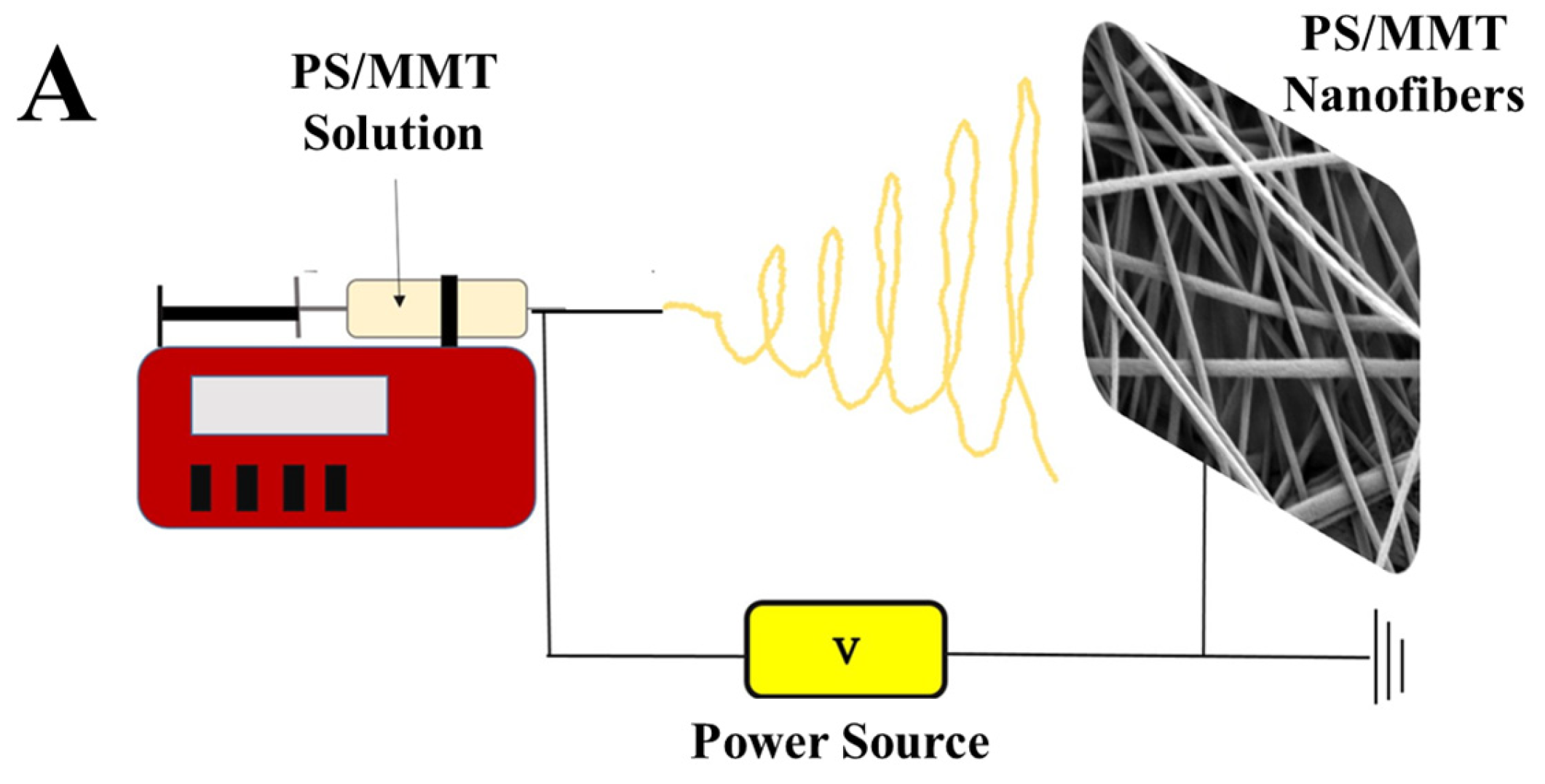
2.4. Fabrication of PS/MMT-PAMAMG3 Electrospun Nanofibers
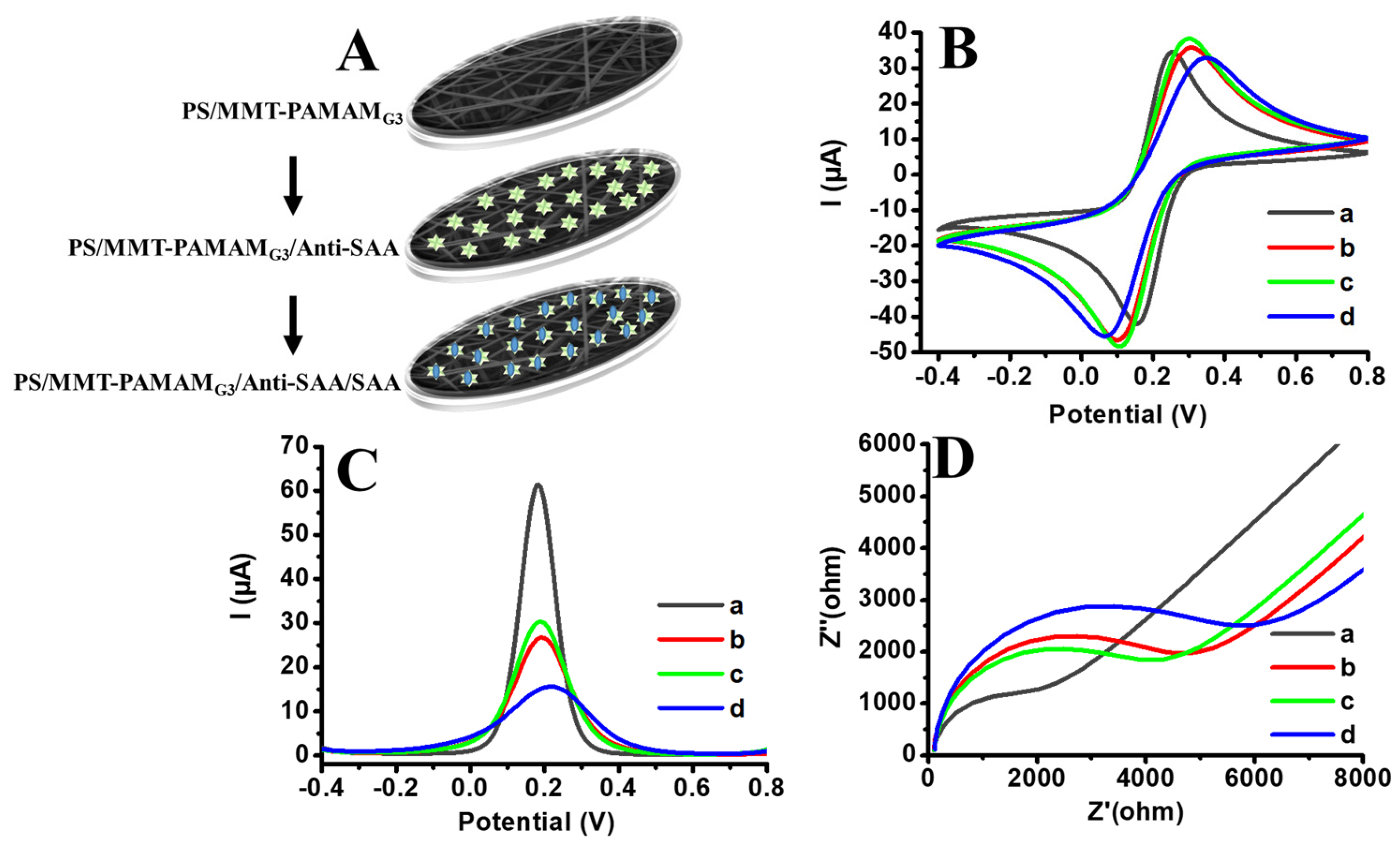
2.5. Preparation of PS/MMT-PAMAMG3/Anti-SAA Immonusensor
2.6. Electrochemical Measurements
3. Results
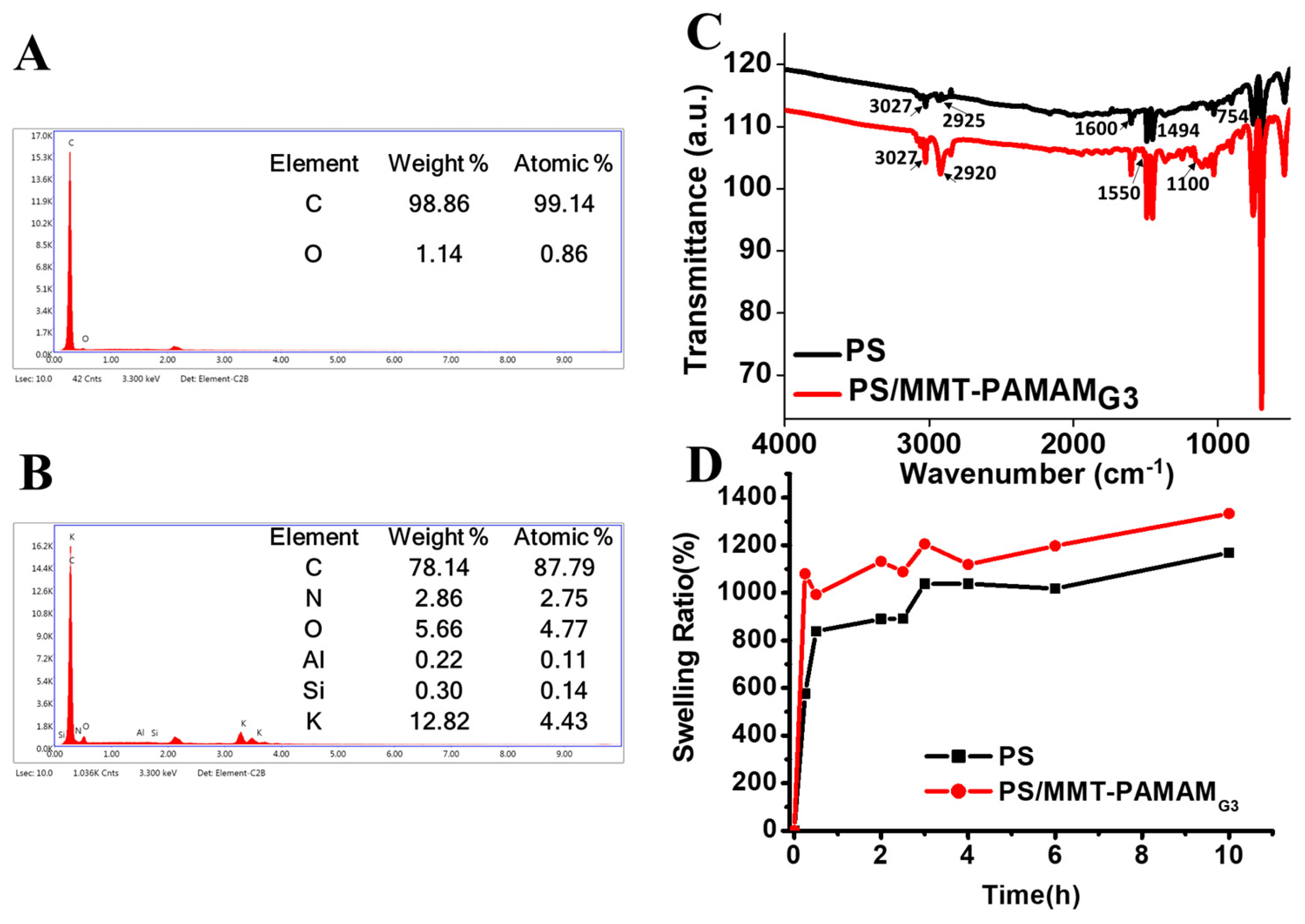
3.1. Characterization of MMT-PAMAMG3
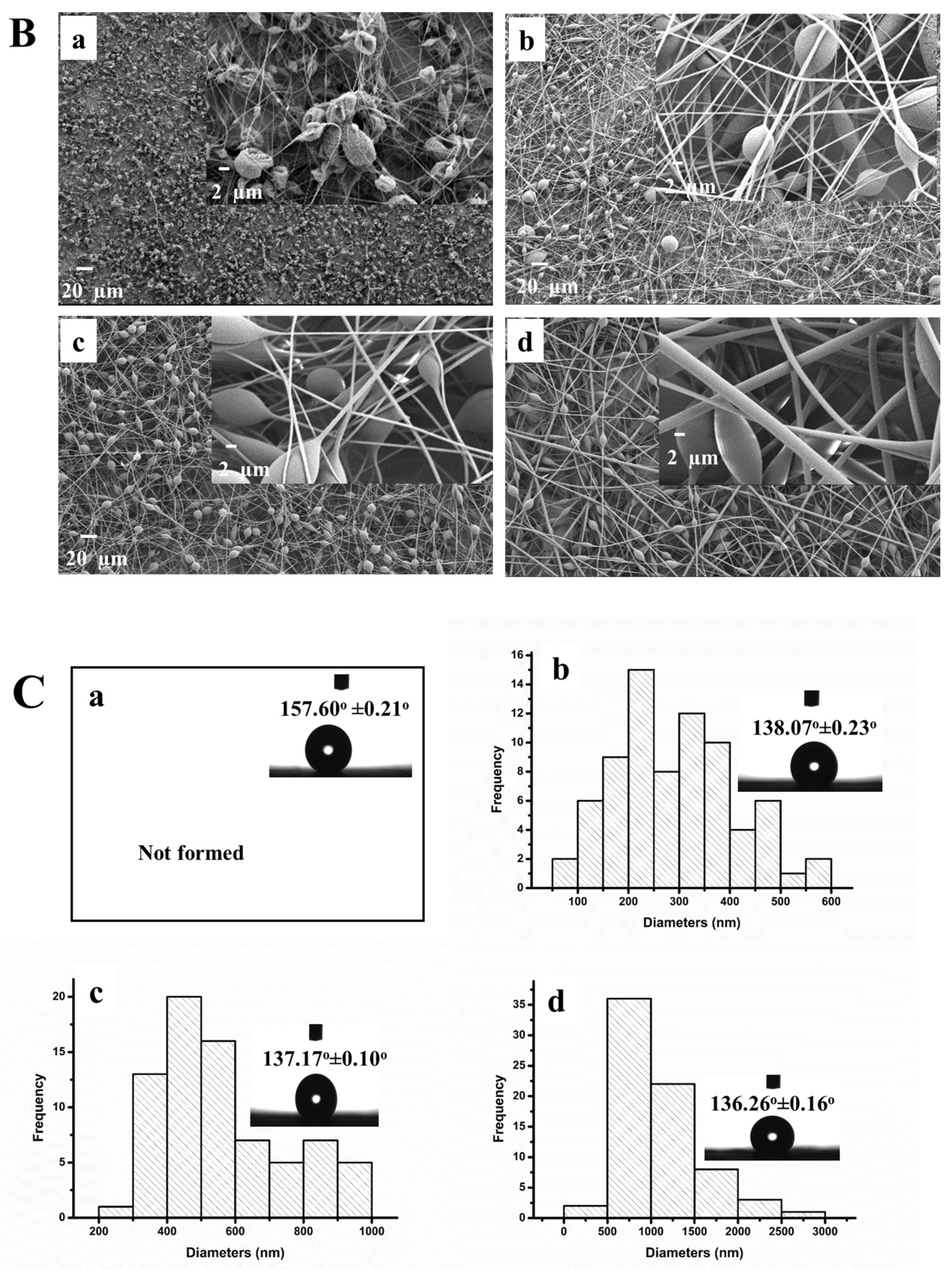
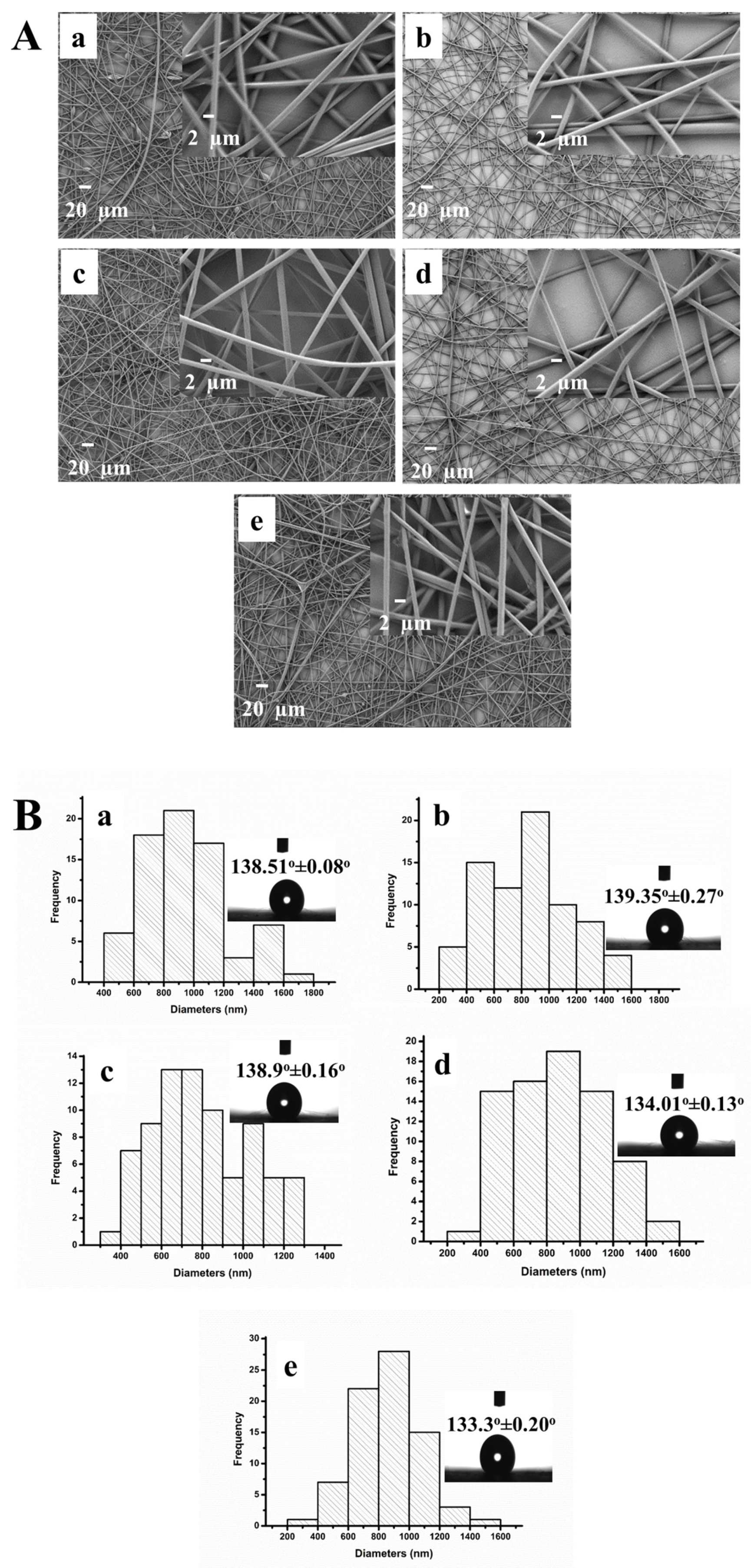
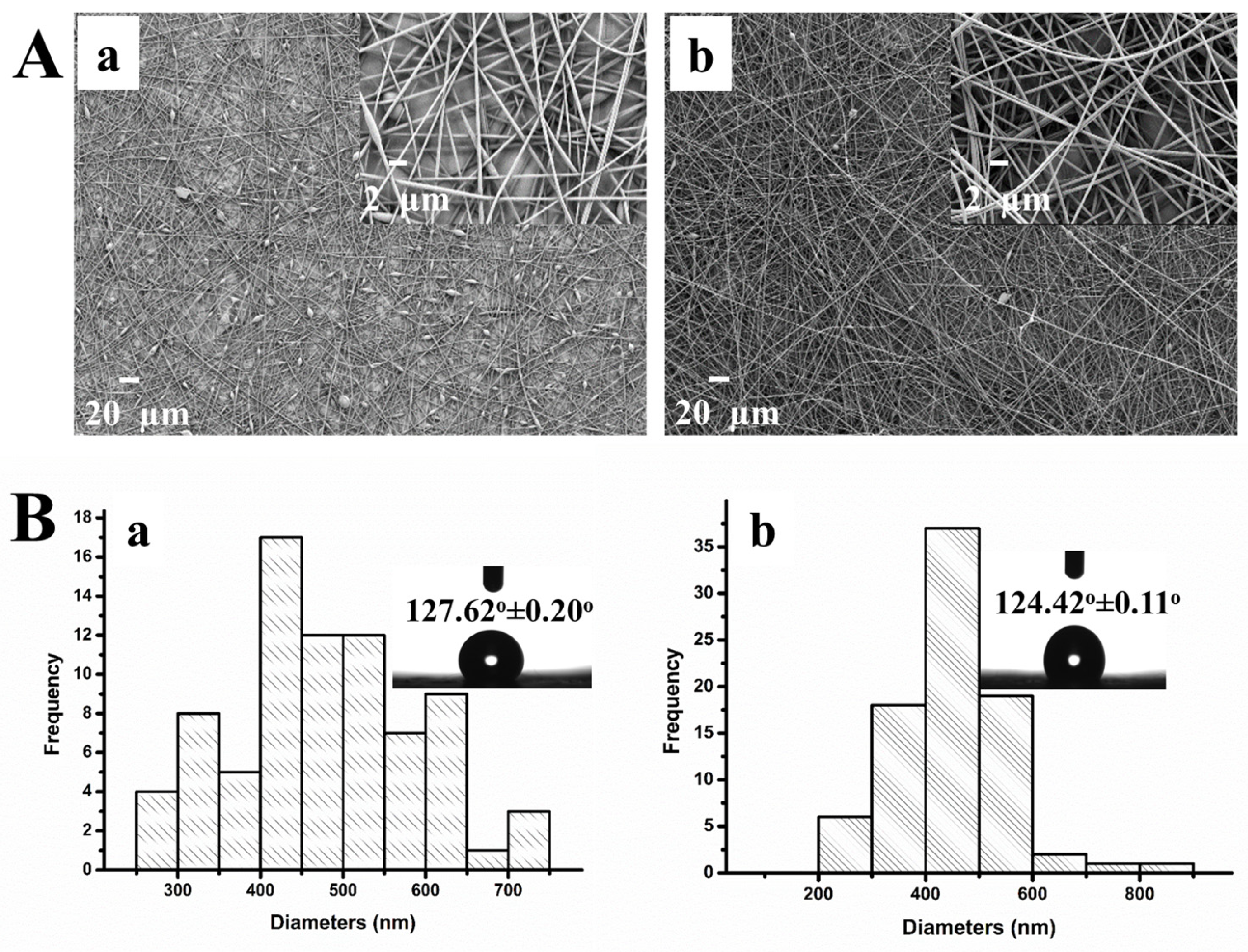
3.2. Characterization of the PS/MMT-PAMAMG3 Electrospun Nanofibers
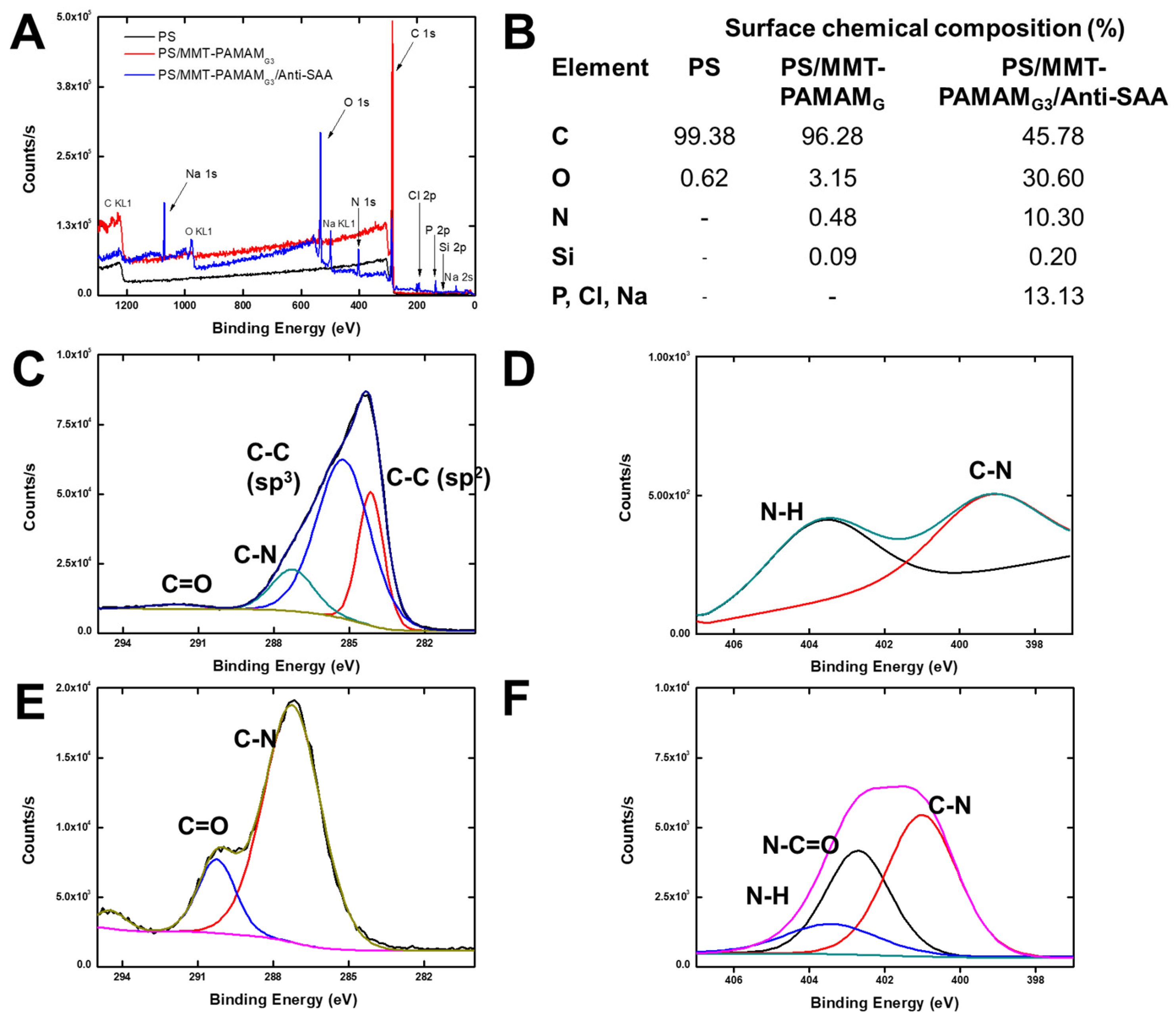
3.3. Characterization of the PS/MMT-PAMAMG3/Anti-SAA Immunosensor
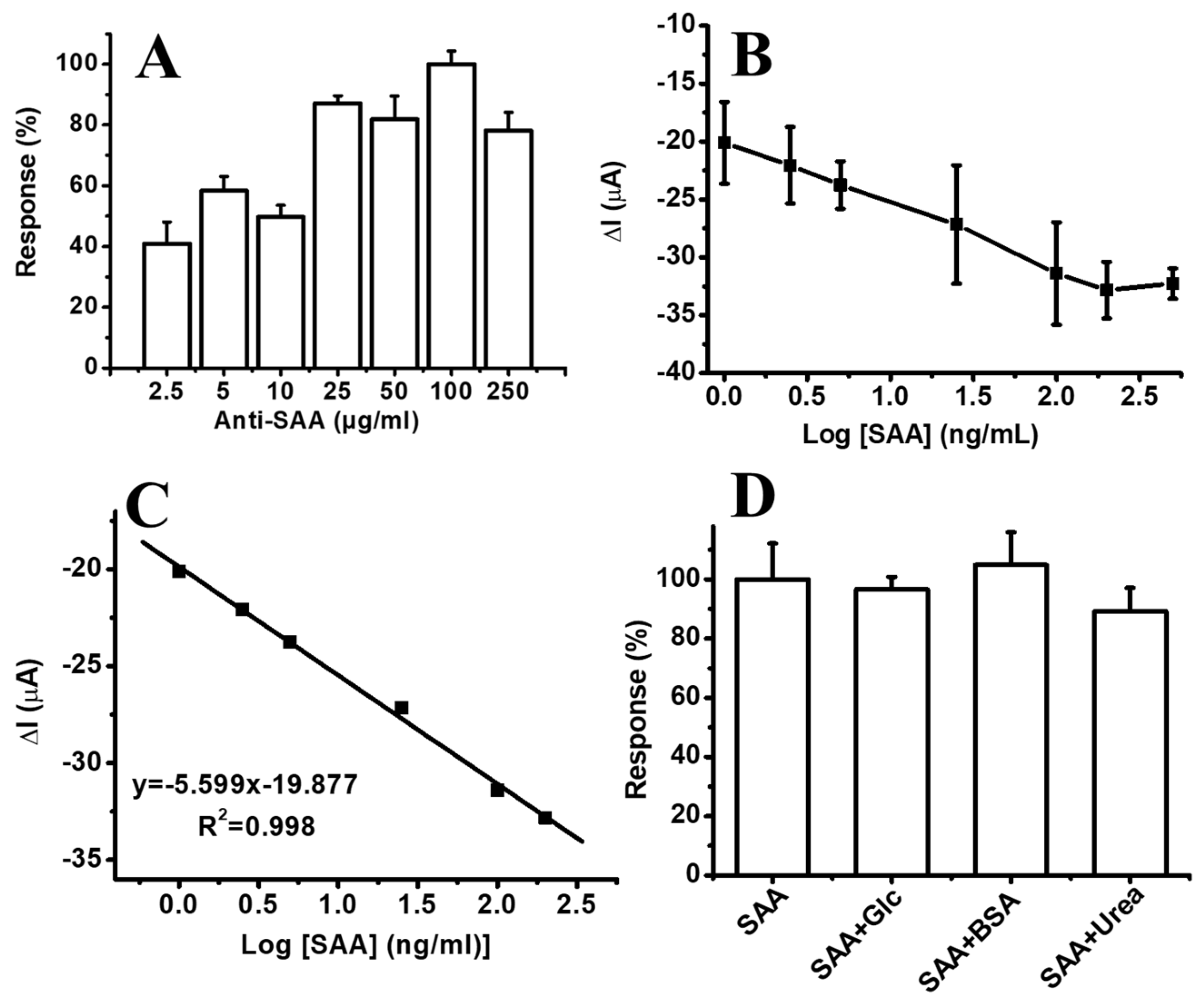
3.4. Electrochemical SAA Detection Based on PS/MMT-PAMAMG3/Anti-SAA
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gruys, E.; Toussaint, M.J.; Niewold, T.A.; Koopmans, S.J. Acute phase reaction and acute phase proteins. J. Zhejiang Univ. Sci. B 2005, 6, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sack, G.H. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, E.; Monteagudo, P.T.; Martins Marrocos, M.S.; Campa, A. Interaction between serum amyloid A and leukocytes—A possible role in the progression of vascular complications in diabetes. Immunol. Lett. 2007, 108, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Vietri, L.; Fui, A.; Bergantini, L.; d’Alessandro, M.; Cameli, P.; Sestini, P.; Rottoli, P.; Bargagli, E. Serum amyloid A: A potential biomarker of lung disorders. Respir. Investig. 2020, 58, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Raynes, J.G.; Cooper, E.H. Comparison of serum amyloid a protein and C-reactive protein concentrations in cancer and non-malignant disease. J. Clin. Pathol. 1983, 36, 798–803. [Google Scholar] [PubMed] [Green Version]
- Cheng, L.; Yang, J.Z.; Bai, W.H.; Li, Z.Y.; Sun, L.F.; Yan, J.J.; Zhou, C.L.; Tang, B.P. Prognostic value of serum amyloid A in patients with COVID-19. Infection 2020, 48, 715–722. [Google Scholar] [CrossRef]
- Pieri, M.; Ciotti, M.; Nuccetelli, M.; Perrone, M.A.; Caliò, M.T.; Lia, M.S.; Minieri, M.; Bernardini, S. Serum Amyloid A Protein as a useful biomarker to predict COVID-19 patients severity and prognosis. Int. Immunopharmacol. 2021, 95, 107512. [Google Scholar] [CrossRef]
- Almusalami, E.M.; Lockett, A.; Ferro, A.; Posner, J. Serum amyloid A—A potential therapeutic target for hyper-inflammatory syndrome associated with COVID-19. Front. Med. 2023, 10, 1135695. [Google Scholar] [CrossRef]
- Wakai, M.; Hayashi, R.; Tanaka, S.; Naito, T.; Kumada, J.; Nomura, M.; Takigawa, H.; Oka, S.; Ueno, Y.; Ito, M.; et al. Serum amyloid A is a better predictive biomarker of mucosal healing than C-reactive protein in ulcerative colitis in clinical remission. BMC Gastroenterol. 2020, 20, 85. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.G.; Ray, S.; Karlsson, R.; Srivastava, S. Calibration-free concentration analysis of protein biomarkers in human serum using surface plasmon resonance. Talanta 2015, 144, 801–808. [Google Scholar] [CrossRef]
- Carbone, T.; Pafundi, V.; Schievano, C.; Assunta, D.; Padula, M.C.; Giordano, M.; Canora, G.; Lazzari, C.; Padula, A.A.; D’Angelo, S. Serum amyloid A in healthy subjects: Assessment of reference value using ELISA method. J. Immunoass. Immunochem. 2021, 42, 129–137. [Google Scholar] [CrossRef]
- Trenchevska, O.; Yassine, H.N.; Borges, C.R.; Nelson, R.W.; Nedelkov, D. Development of quantitative mass spectrometric immunoassay for serum amyloid A. Biomarkers 2016, 21, 743–751. [Google Scholar] [CrossRef]
- Waugh, E.M.; Haining, H.; Harvie, J.; Ridyard, A.E.; Eckersall, P.D. Validation of an automated immunoturbidimetric assay for feline serum amyloid A. BMC Vet. Res. 2022, 18, 359. [Google Scholar] [CrossRef]
- Yamada, T.; Nomatal, Y.; Sugita, O.; Okada, M. A rapid method for measuring serum amyloid A protein by latex agglutination nephelometric immunoassay. Ann. Clin. Biochem. 1993, 30, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Li, Y.; Yuan, G.; Guo, Y.; Yu, C. Immunoassay for serum amyloid A using a glassy carbon electrode modified with carboxy-polypyrrole, multiwalled carbon nanotubes, ionic liquid and chitosan. Microchim. Acta 2015, 182, 1395–1402. [Google Scholar] [CrossRef]
- Karadag, M.; Geyik, C.; Demirkol, D.O.; Ertas, F.N.; Timur, S. Modified gold surfaces by 6-(ferrocenyl) hexanethiol/dendrimer/gold nanoparticles as a platform for the mediated biosensing applications. Mater. Sci. Eng. C 2013, 33, 634–640. [Google Scholar] [CrossRef]
- Çakar, İ.; Özdokur, K.V.; Demir, B.; Yavuz, E.; Demirkol, D.O.; Koçak, S.; Timur, S.; Ertaş, F.N. Molybdenum oxide/platinum modified glassy carbon electrode: A novel electrocatalytic platform for the monitoring of electrochemical reduction of oxygen and its biosensing applications. Sens. Actuators B Chem. 2013, 185, 331–336. [Google Scholar] [CrossRef]
- Tuncagil, S.; Odaci, D.; Varis, S.; Timur, S.; Toppare, L. Electrochemical polymerization of 1-(4-nitrophenyl)-2,5-di(2-thienyl)-1H-pyrrole as a novel immobilization platform for microbial sensing. Bioelectrochemistry 2009, 76, 169–174. [Google Scholar] [CrossRef]
- Kırgöz, Ü.A.; Odaci, D.; Timur, S.; Merkoçi, A.; Pazarlıoğlu, N.; Telefoncu, A.; Alegret, S. Graphite epoxy composite electrodes modified with bacterial cells. Bioelectrochemistry 2006, 69, 128–131. [Google Scholar] [CrossRef]
- Demirkol, D.O.; Yildiz, H.B.; Sayın, S.; Yilmaz, M. Enzyme immobilization in biosensor constructions: Self-assembled monolayers of calixarenes containing thiols. RSC Adv. 2014, 4, 19900–19907. [Google Scholar] [CrossRef]
- Xia, N.; Wang, X.; Zhou, B.; Wu, Y.; Mao, W.; Liu, L. Electrochemical detection of amyloid-β oligomers based on the signal amplification of a network of silver nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 19303–19311. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, B.; Zhou, J.; Tang, J.; Tang, D. Additional Molecular Biological Amplification Strategy for Enhanced Sensitivity of Monitoring Low-Abundance Protein with Dual Nanotags. ACS Appl. Mater. Interfaces 2013, 5, 4479–4485. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.; Brasiunas, B.; Kausaite-Minkstimiene, A.; Ramanaviciene, A. Metal Nanoparticle and Quantum Dot Tags for Signal Amplification in Electrochemical Immunosensors for Biomarker Detection. Chemosensors 2021, 9, 85. [Google Scholar] [CrossRef]
- Lakard, B. Electrochemical Biosensors Based on Conducting Polymers: A Review. Appl. Sci. 2020, 10, 6614. [Google Scholar] [CrossRef]
- Yashini, M.; Auddy, I.; Shanmugasundaram, S.; Vidyalakshmi, R.; Sunil, C.K. Characterization of Antibody Immobilization on Chitosan/Gelatin-Modified Electrode and Its Application to Bacillus cereus Detection in Cereal-Based Food. Food Anal. Methods 2022, 15, 2382–2393. [Google Scholar] [CrossRef]
- Hartati, Y.W.; Irkham, I.; Zulqaidah, S.; Syafira, R.S.; Kurnia, I.; Noviyanti, A.R.; Topkaya, S.N. Recent advances in hydroxyapatite-based electrochemical biosensors: Applications and future perspectives. Sens. Bio-Sens. Res. 2002, 38, 100542. [Google Scholar] [CrossRef]
- Police Patil, A.V.; Chuang, Y.-S.; Li, C.; Wu, C.C. Recent Advances in Electrochemical Immunosensors with Nanomaterial Assistance for Signal Amplification. Biosensors 2023, 13, 125. [Google Scholar] [CrossRef]
- Kirbay, F.O.; Yalcinkaya, E.E.; Atik, G.; Evren, G.; Unal, B.; Demirkol, D.O.; Timur, S. Biofunctionalization of PAMAM-montmorillonite decorated poly(ε-caprolactone)-chitosan electrospun nanofibers for cell adhesion and electrochemical cytosensing. Biosens. Bioelectron. 2018, 109, 286–294. [Google Scholar] [CrossRef]
- Unal, B.; Yalcinkaya, E.E.; Demirkol, D.O.; Timur, S. An electrospun nanofiber matrix based on organo-clay for biosensors: PVA/PAMAM-Montmorillonite. Appl. Surf. Sci. 2018, 444, 542–551. [Google Scholar] [CrossRef]
- Atilgan, H.; Unal, B.; Yalcinkaya, E.E.; Evren, G.; Atik, G.; Ozturk Kirbay, F.; Kilic, N.M.; Odaci, D. Development of an Enzymatic Biosensor Using Glutamate Oxidase on Organic–Inorganic-Structured, Electrospun Nanofiber-Modified Electrodes for Monosodium Glutamate Detection. Biosensors 2023, 13, 430. [Google Scholar] [CrossRef]
- Bongartz, R.; Ag, D.; Seleci, M.; Walter, J.G.; Yalcinkaya, E.E.; Demirkol, D.O.; Stahl, F.; Timur, S.; Scheper, T. Folic acid-modified clay: Targeted surface design for cell culture applications. J. Mater. Chem. B 2013, 1, 522–528. [Google Scholar] [CrossRef]
- Mushtaq, M.; Saba, H.; Wang, W.; Naeem, M.A.; Wei, Q. Fabrication and characterization of electrospun membranes from Poly(lactic acid) and hexadecyl trimethyl ammonium chloride-modified montmorillonite clay. J. Ind. Text. 2019, 50, 415–424. [Google Scholar] [CrossRef]
- Cai, J.; Lei, M.; Zhang, Q.; He, J.R. Electrospun composite nanofiber mats of Cellulose@Organically modified montmorillonite for heavy metal ion removal: Design, characterization, evaluation of absorption performance. Compos. Part A Appl. Sci. Manuf. 2017, 92, 10–16. [Google Scholar] [CrossRef]
- Yezer, I.; Demirkol, D.O. Cellulose acetate–chitosan based electrospun nanofibers for bio-functionalized surface design in biosensing. Cellulose 2020, 27, 10183–10197. [Google Scholar] [CrossRef]
- Gal, J.Y.; Fovet, Y.; Adib-Yadzi, M. About a synthetic saliva for in vitro studies. Talanta 2001, 53, 1103–1115. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Tripathi, K.; Driskell, J.D. Quantifying Bound and Active Antibodies Conjugated to Gold Nanoparticles: A Comprehensive and Robust Approach to Evaluate Immobilization Chemistry. ACS Omega 2018, 3, 8253–8259. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.; Evers, T.H.; Prins, M.W.J. How Antibody Surface Coverage on Nanoparticles Determines the Activity and Kinetics of Antigen Capturing for Biosensing. Anal. Chem. 2014, 86, 8158–8166. [Google Scholar] [CrossRef] [Green Version]
- Damar, K.; Demirkol, D.O. Modified gold surfaces by poly(amidoamine) dendrimers and fructose dehydrogenase for mediated fructose sensing. Talanta 2011, 87, 67–73. [Google Scholar] [CrossRef]
- Meng, Z.; Lv, F.; Zhang, Y.; Zhang, Q.; Zhang, Z.; Ai, S. Modified Na-Montmorillonite with Quaternary Ammonium Salts: Application for Removal of Salicylic Acid From Aqueous Solutions. Clean Soil Air Water 2015, 43, 1150–1156. [Google Scholar] [CrossRef]
- Vazquez, A.; López, M.; Kortaberria, G.; Martín, L.; Mondragon, I. Modification of montmorillonite with cationic surfactants. Thermal and chemical analysis including CEC determination. Appl. Clay Sci. 2008, 41, 24–36. [Google Scholar] [CrossRef]
- Seleci, M.; Ag, D.; Yalcinkaya, E.E.; Demirkol, D.O.; Guler, C.; Timur, S. Amine-intercalated montmorillonite matrices for enzyme immobilization and biosensing applications. RSC Adv. 2012, 2, 2112–2118. [Google Scholar] [CrossRef]
- Zeybekler, S.E.; Odaci, D. Carbon Nanotube-Incorporated Nanofibers for Immunosensor Preparation against CD36. ACS Omega 2023, 8, 5776–5786. [Google Scholar] [CrossRef]
- Nitanan, T.; Opanasopit, P.; Akkaramongkolporn, P.; Rojanarata, T.; Ngawhirunpat, T.; Supaphol, P. Effects of processing parameters on morphology of electrospun polystyrene nanofibers. Korean J. Chem. Eng. 2012, 29, 173–181. [Google Scholar] [CrossRef]
- Shin, C.; Chase, G. Nanofibers from recycle waste expanded polystyrene using natural solvent. Polym. Bull. 2005, 55, 209–215. [Google Scholar] [CrossRef]
- Mostafa, M.; Kandile, N.G.; Mahmoud, M.K.; Ibrahim, H.M. Synthesis and characterization of polystyrene with embedded silver nanoparticle nanofibers to utilize as antibacterial and wound healing biomaterial. Heliyon 2022, 8, e08772. [Google Scholar] [CrossRef]
- Avossa, J.; Herwig, G.; Toncelli, C.; Itel, F.; Rossi, R.M. Electrospinning based on benign solvents: Current definitions, implications and strategies. Green Chem. 2022, 24, 2347–2375. [Google Scholar] [CrossRef]
- El-Newehy, M.H.; Kim, H.Y.; Khattab, T.A.; El-Naggar, M.E. Development of highly photoluminescent electrospun nanofibers for dual-mode secure authentication. Ceram. Int. 2022, 48, 3495–3503. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, H.Y.; Bang, H.J.; Jung, Y.H.; Lee, S.G. The change of bead morphology formed on electrospun polystyrene fibers. Polymer 2003, 44, 4029–4034. [Google Scholar] [CrossRef]
- Uyar, T.; Besenbacher, F. Electrospinning of uniform polystyrene fibers: The effect of solvent conductivity. Polymer 2008, 49, 5336–5343. [Google Scholar] [CrossRef]
- Demir, M.M. Investigation on glassy skin formation of porous polystyrene fibers electrospun from DMF. Express Polym. Lett. 2010, 4, 2–8. [Google Scholar] [CrossRef]
- An, J.; Wang, X.; Li, Y.; Kang, W.; Lian, K. Polystyrene nanofibers as an effective sorbent for the adsorption of clonazepam: Kinetic and thermodynamic studies. RSC Adv. 2022, 12, 3394–3401. [Google Scholar] [CrossRef]
- Er, S.; Demirkol, D.O. Graphene oxide incorporated polystyrene electrospun nanofibers for immunosensing of CD36 as a marker of diabetic plasma. Bioelectrochemistry 2022, 145, 108083. [Google Scholar] [CrossRef]
- Razmshoar, P.; Bahrami, S.H.; Akbari, S. Functional hydrophilic highly biodegradable PCL nanofibers through direct aminolysis of PAMAM dendrimer. Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 1069–1080. [Google Scholar] [CrossRef]
- Wu, H.-D.; Wu, S.-C.; Wu, I.-D.; Chang, F.C. Novel determination of the crystallinity of syndiotactic polystyrene using FTIR spectrum. Polymer 2001, 42, 4719–4725. [Google Scholar] [CrossRef]
- Armbruster, D.A.; Tillman, M.D.; Hubbs, L.M. Limit of Detection (LQD)/Limit of Quantitation (LOQ): Comparison of the Empirical and the Statistical Methods Exemplified with GC-MS Assays of Abused Drugs. Clin. Chem. 1994, 40 Pt 1, 1233–1238. [Google Scholar] [CrossRef]
- Vashist, S.K.; Luong, J.H. Bioanalytical Requirements and Regulatory Guidelines for Immunoassays. In Handbook of Immunoassay Technologies; Elsevier: Amsterdam, The Netherlands, 2018; pp. 81–95. ISBN 9780128117941. [Google Scholar]
- Hosman, I.S.; Kos, I.; Lamot, L. Serum Amyloid A in Inflammatory Rheumatic Diseases: A Compendious Review of a Renowned Biomarker. Front. Immunol. 2021, 11, 631299. [Google Scholar] [CrossRef]
- Balayan, S.; Chauhan, N.; Chandra, R.; Jain, U. Molecular imprinting based electrochemical biosensor for identification of serum amyloid A (SAA), a neonatal sepsis biomarker. Int. J. Biol. Macromol. 2022, 195, 589–597. [Google Scholar] [CrossRef]
- Liu, X.; Yang, X.; Li, K.; Liu, H.; Xiao, R.; Wang, W.; Wang, C.; Wang, S. Fe3O4@Au SERS tags-based lateral flow assay for simultaneous detection of serum amyloid A and C-reactive protein in unprocessed blood sample. Sens. Actuators B Chem. 2020, 320, 128350. [Google Scholar] [CrossRef]
- Gul, O.; Calay, E.; Sezerman, U.; Basaga, H.; Gurbuz, Y. Sandwich-type, antibody microarrays for the detection and quantification of cardiovascular risk markers. Sens. Actuators B Chem. 2007, 125, 581–588. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, F.; Li, N.; Wu, R.; Li, J.; Shen, H.; Li, L.S.; Guo, F. Development of dual quantum dots-based fluorescence-linked immunosorbent assay for simultaneous detection on inflammation biomarkers. Sens. Actuators B Chem. 2019, 301, 127118. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sensing Mode | Material | Recognition Molecule | Linear Range | LOD | Applied Sample | Ref |
|---|---|---|---|---|---|---|
| ELEC | MWCNTs MnO2NSs Co3O4NPs | MIP | 0.12 pg/mL–12 μg/mL | 0.12 pg/mL | Serum | [59] |
| LFA | Fe3O4@Au SERS tags | Anti-SAA | 0.1–500 ng/mL | 0.10 ng/mL | Blood | [60] |
| FL | Sandwich-type, antibody microarrays | Antibody | 5.9–478 ng/mL | 5.90 ng/mL | Serum | [61] |
| ELEC | MWCNTs/IL/Chit/GCE | Anti-SAA | 0.001–900 ng/mL | 0.30 pg/mL | Serum | [15] |
| FL | dQDs-FLISA | Anti-SAA | 10–1000 ng/mL | 2.39 ng/mL | Serum | [62] |
| ELEC | PS/MMT-PAMAMG3 | Anti-SAA | 1.0–200 ng/mL | 0.57 ng/mL | Synthetic saliva and serum | This study |
| Synthetic Sample | Added SAA (ng/mL) | * Found SAA (ng/mL) | Recovery % |
|---|---|---|---|
| Saliva | 25 | 24.82 | 99.29 |
| Serum | 25 | 24.35 | 97.41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evren, G.; Er, E.; Yalcinkaya, E.E.; Horzum, N.; Odaci, D. Electrospun Nanofibers including Organic/Inorganic Nanohybrids: Polystyrene- and Clay-Based Architectures in Immunosensor Preparation for Serum Amyloid A. Biosensors 2023, 13, 673. https://doi.org/10.3390/bios13070673
Evren G, Er E, Yalcinkaya EE, Horzum N, Odaci D. Electrospun Nanofibers including Organic/Inorganic Nanohybrids: Polystyrene- and Clay-Based Architectures in Immunosensor Preparation for Serum Amyloid A. Biosensors. 2023; 13(7):673. https://doi.org/10.3390/bios13070673
Chicago/Turabian StyleEvren, Gizem, Eray Er, Esra Evrim Yalcinkaya, Nesrin Horzum, and Dilek Odaci. 2023. "Electrospun Nanofibers including Organic/Inorganic Nanohybrids: Polystyrene- and Clay-Based Architectures in Immunosensor Preparation for Serum Amyloid A" Biosensors 13, no. 7: 673. https://doi.org/10.3390/bios13070673
APA StyleEvren, G., Er, E., Yalcinkaya, E. E., Horzum, N., & Odaci, D. (2023). Electrospun Nanofibers including Organic/Inorganic Nanohybrids: Polystyrene- and Clay-Based Architectures in Immunosensor Preparation for Serum Amyloid A. Biosensors, 13(7), 673. https://doi.org/10.3390/bios13070673