Live Cell Monitoring of Separase Activity, a Key Enzymatic Reaction for Chromosome Segregation, with Chimeric FRET-Based Molecular Sensor upon Cell Cycle Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
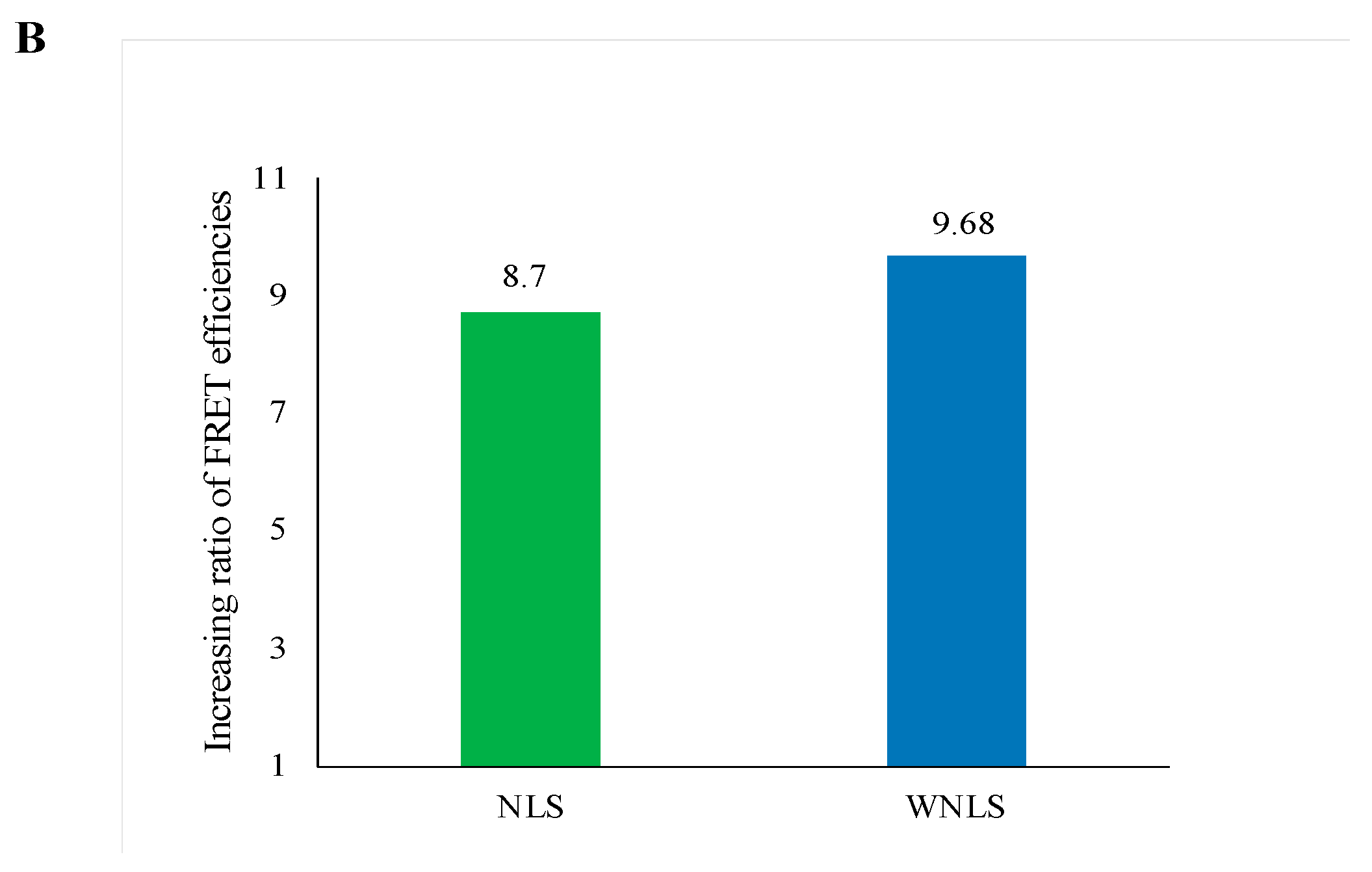{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction for Separase Activity Detection with or without Nuclear Localization Sequence (NLS)
2.2. Chimeric FRET-Based Molecular Sensor Preparation
2.3. Evaluations for Separase Activity with Chimeric FRET-Based Molecular Sensor
2.4. Introduction of Chimeric FRET-Based Molecular Sensor into Cell-Cycle-Controlled HeLa Cells
2.5. Flow Cytometry
2.6. Verification of Cell Cycle Contorol by Serum Starvation through DNA Amount Estimation
2.7. Separase Activations through Cell Cycle Progression
2.8. Fluorescence Microscope Observation of Bioprobe Localization inside Cells
2.9. Imaging Analysis
2.10. General Image Acquisitions for FRET-Based Molecular Sensor Localizations in HeLa Cells
2.11. Western Blot Analysis
2.12. Separase Activity Estimation Included in Cell-Cycle-Progressed Cell Lysate Preparation with FRET-Based Molecular Sensors
3. Results and Discussion
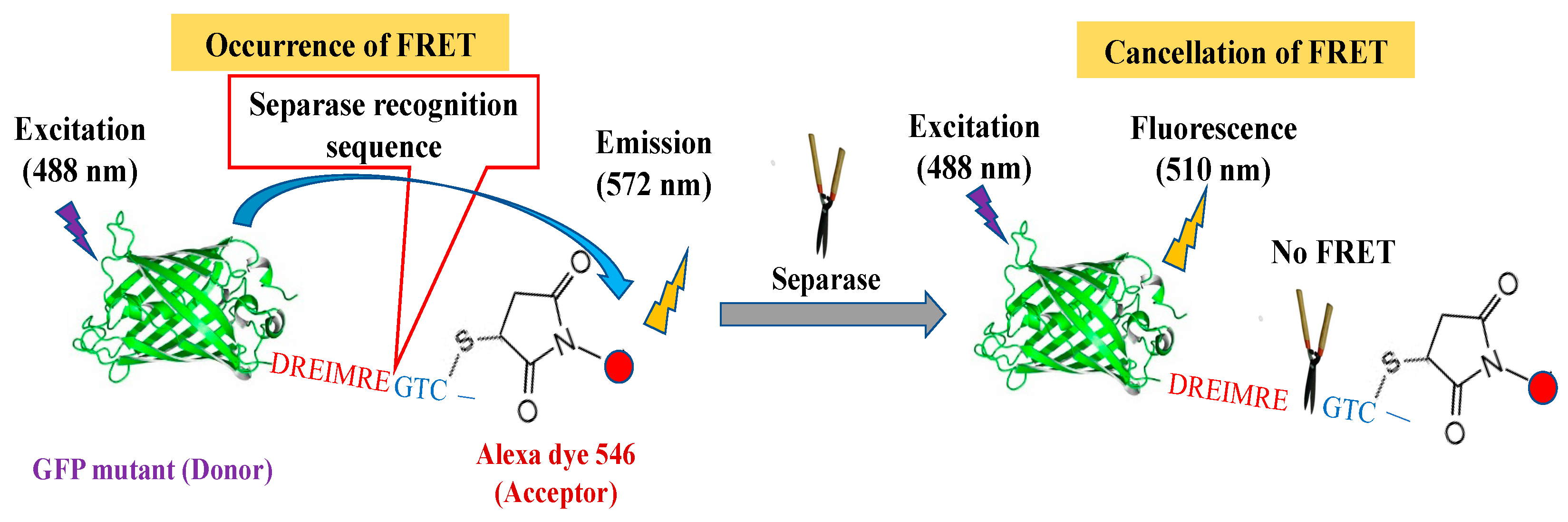
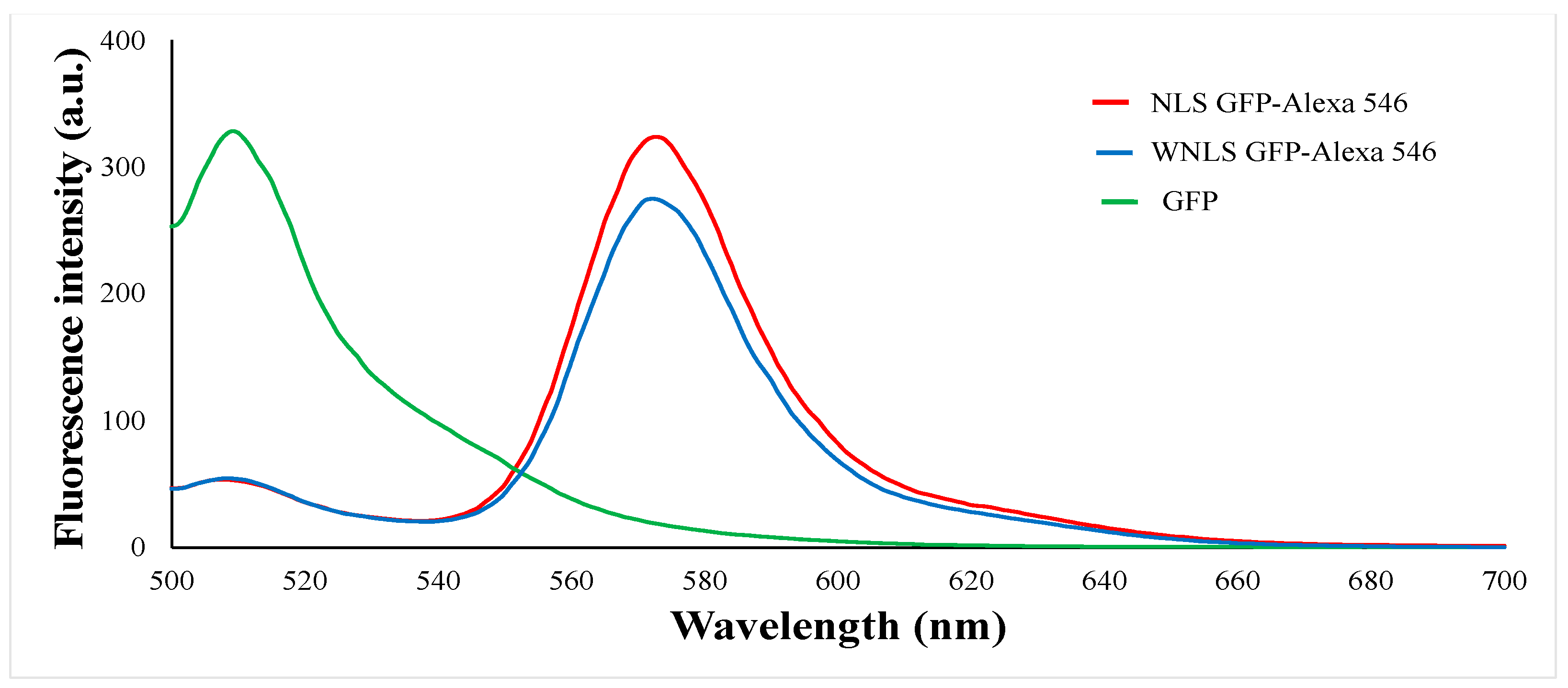
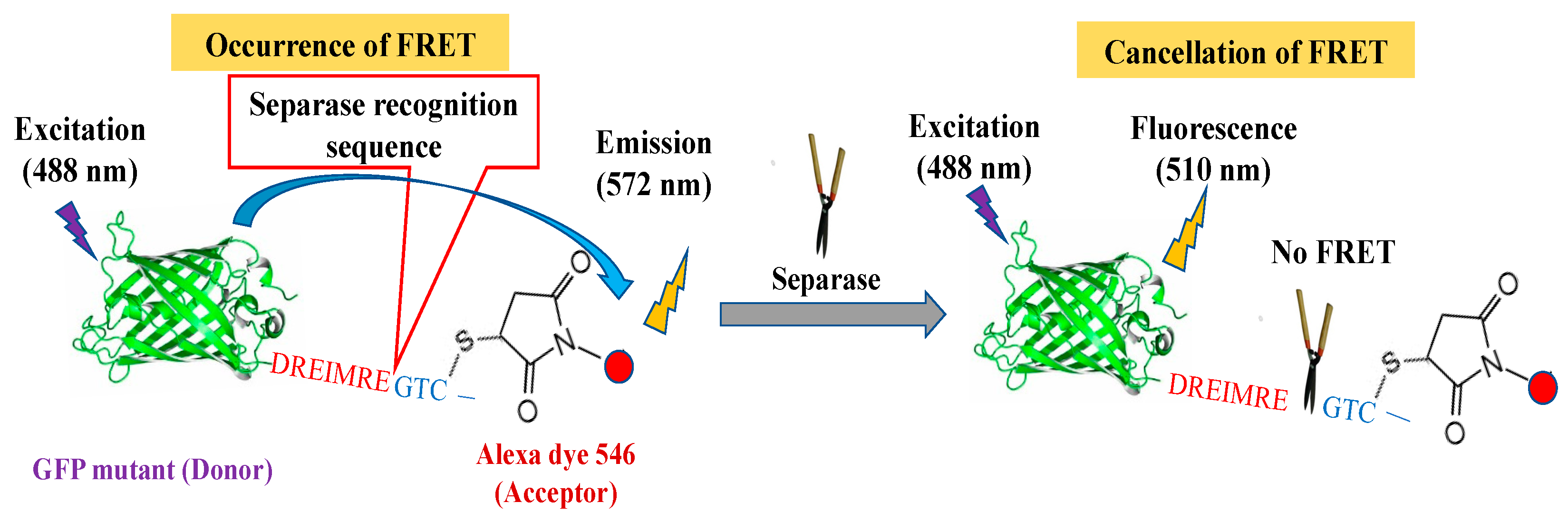
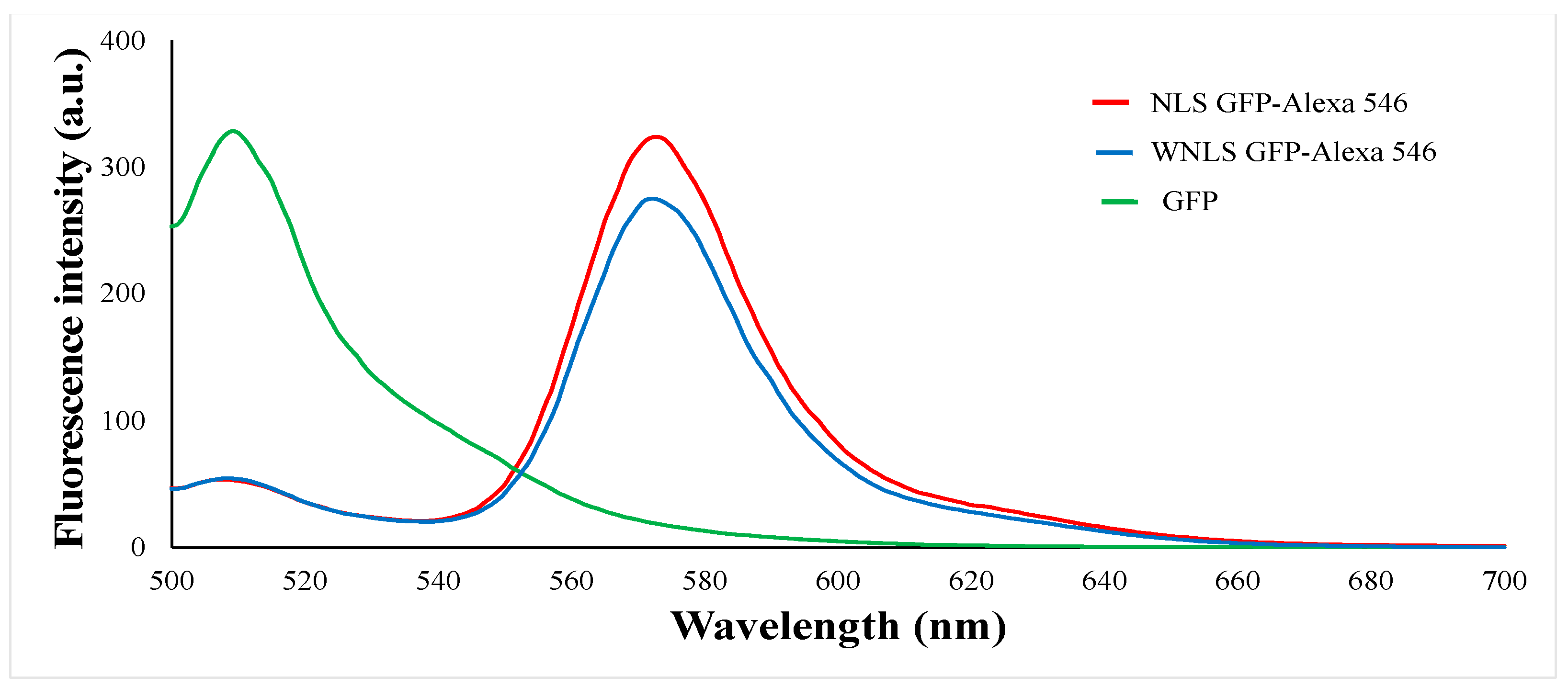
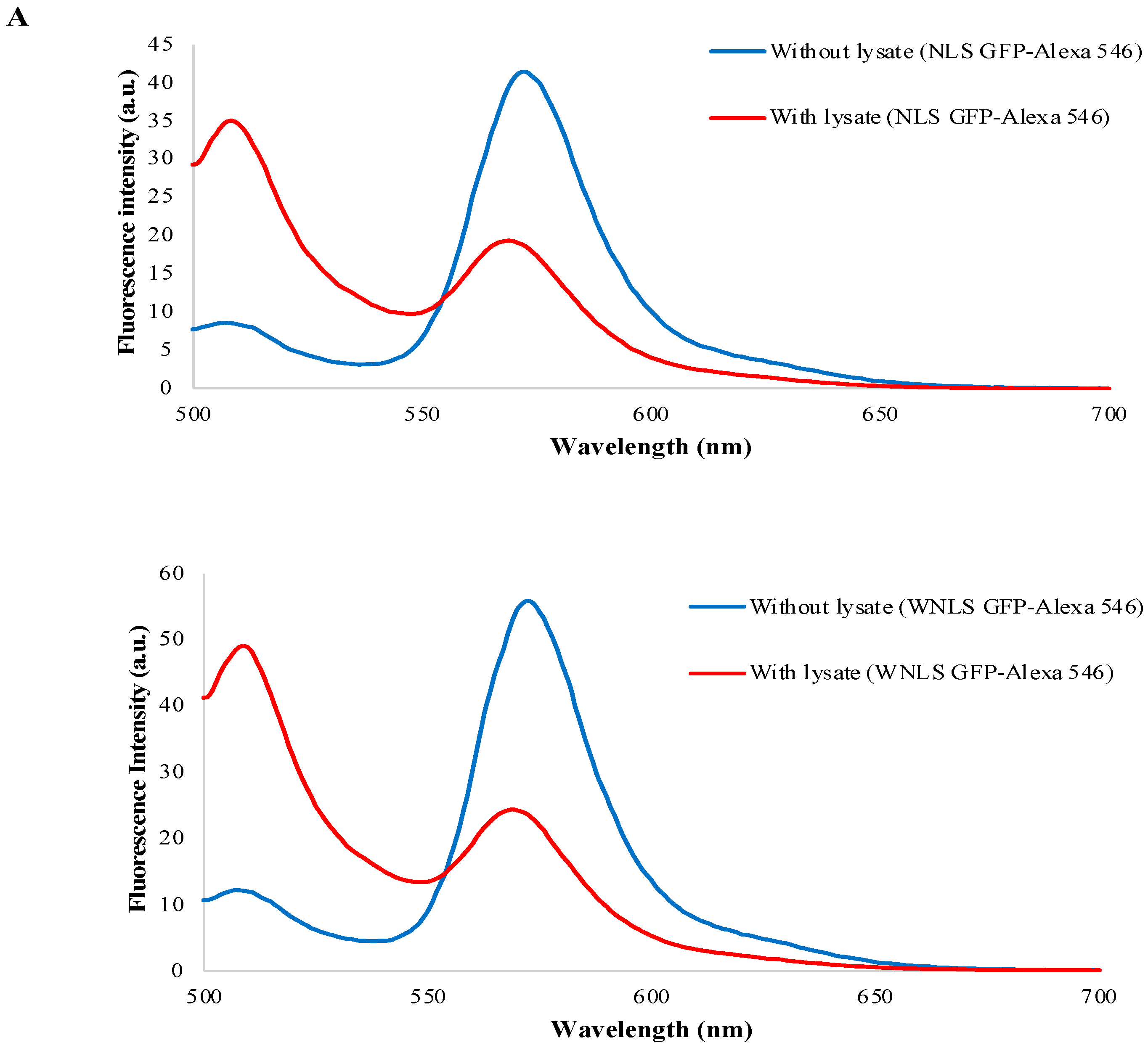
3.1. FRET-Based Molecular Sensor Preparations and Their Fluorescence Properties
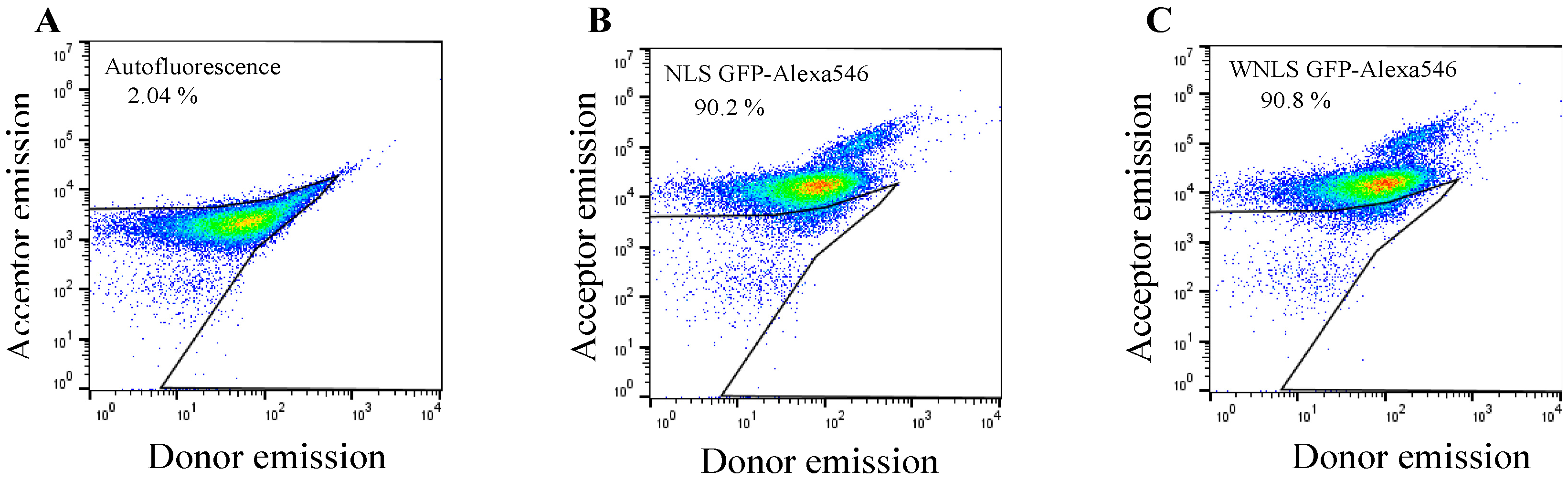
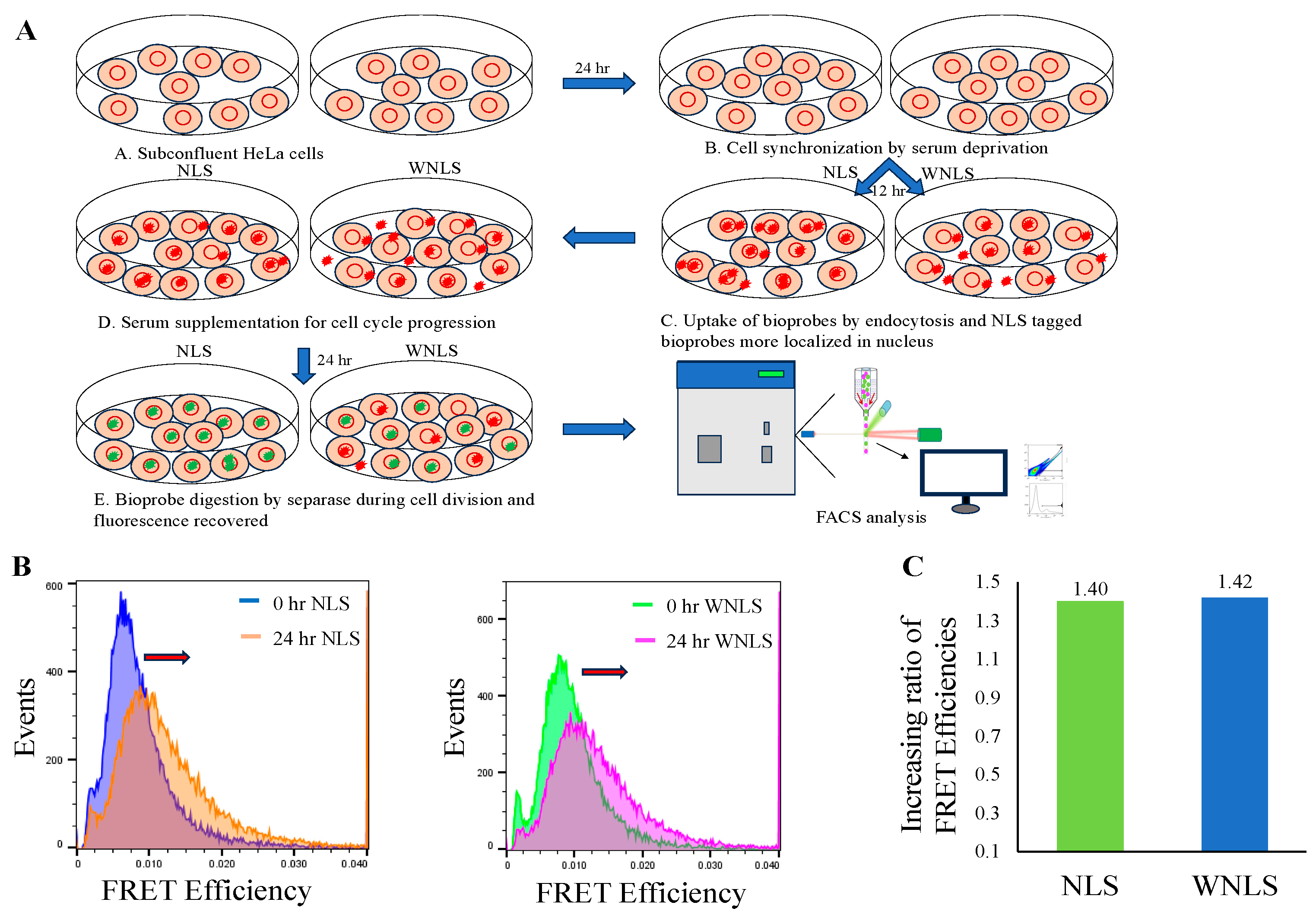
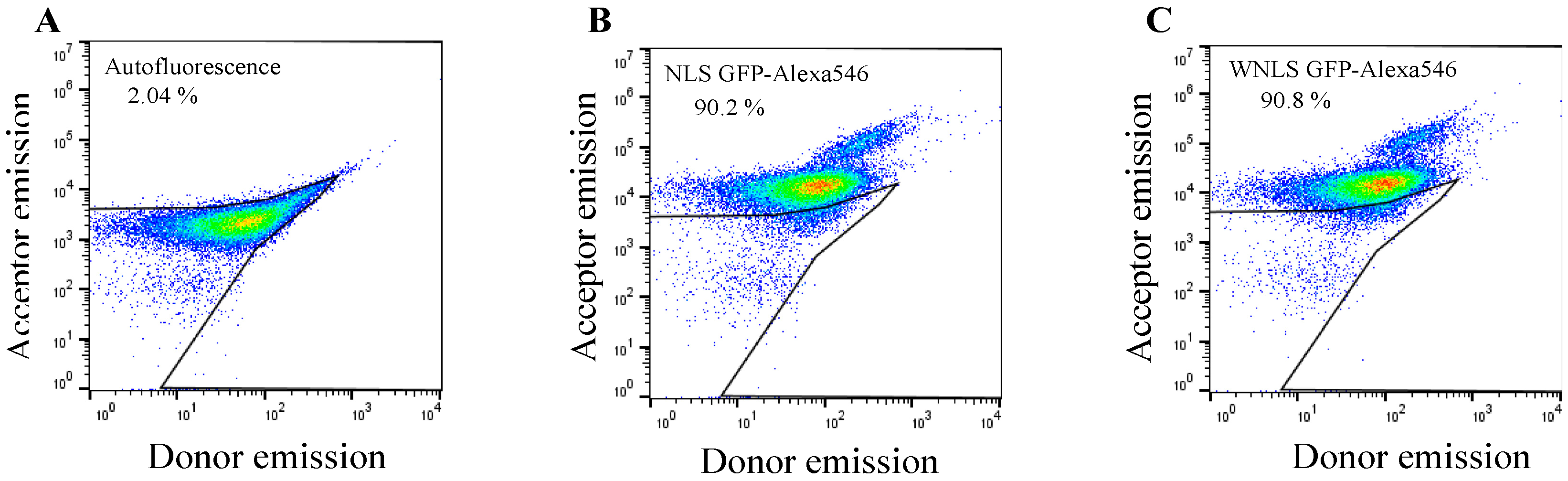
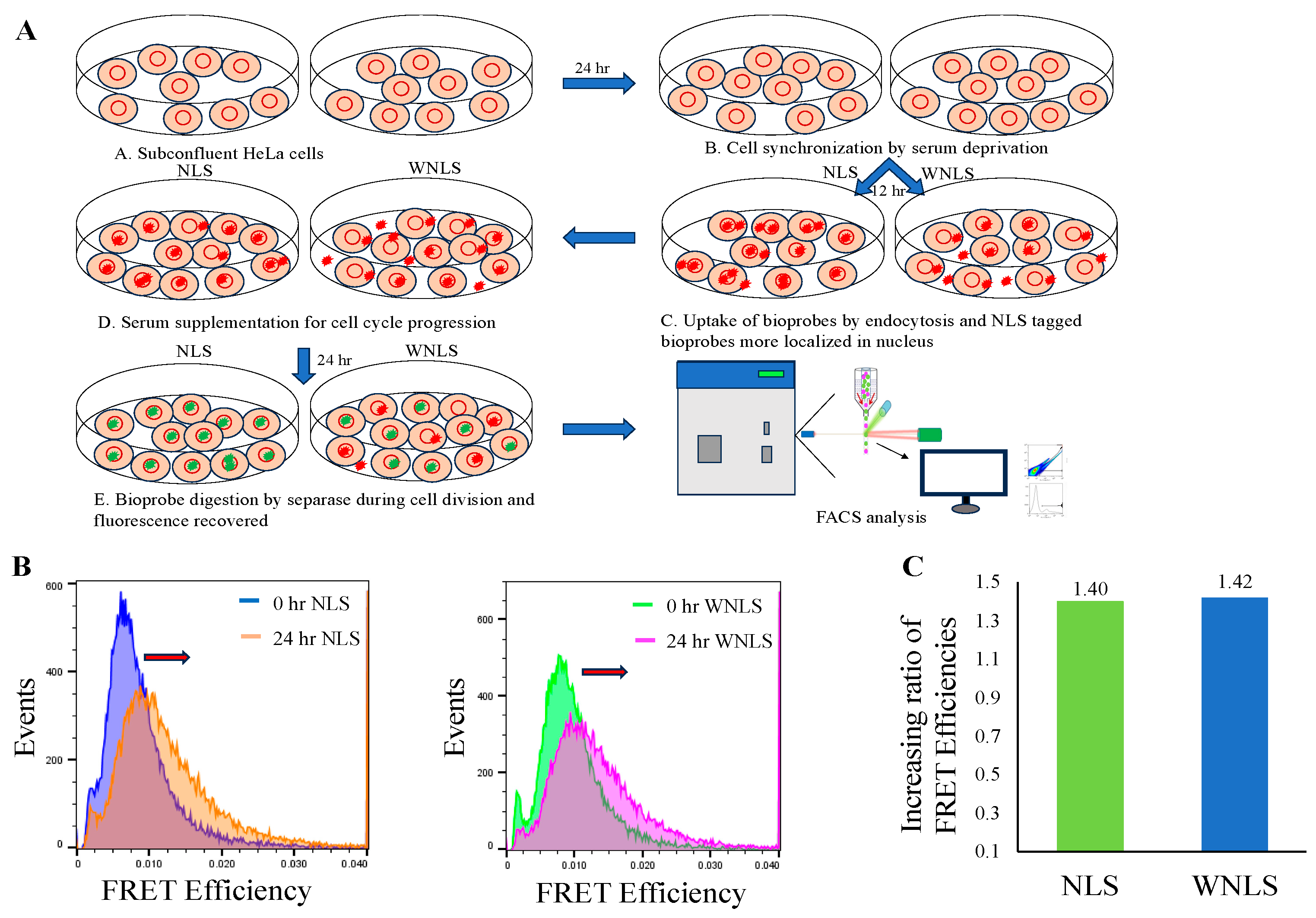
3.2. Introduction of Chimeric FRET-Based Molecular Sensors into Cell-Cycle-Controlled Hela Cells and Flow Cytometry
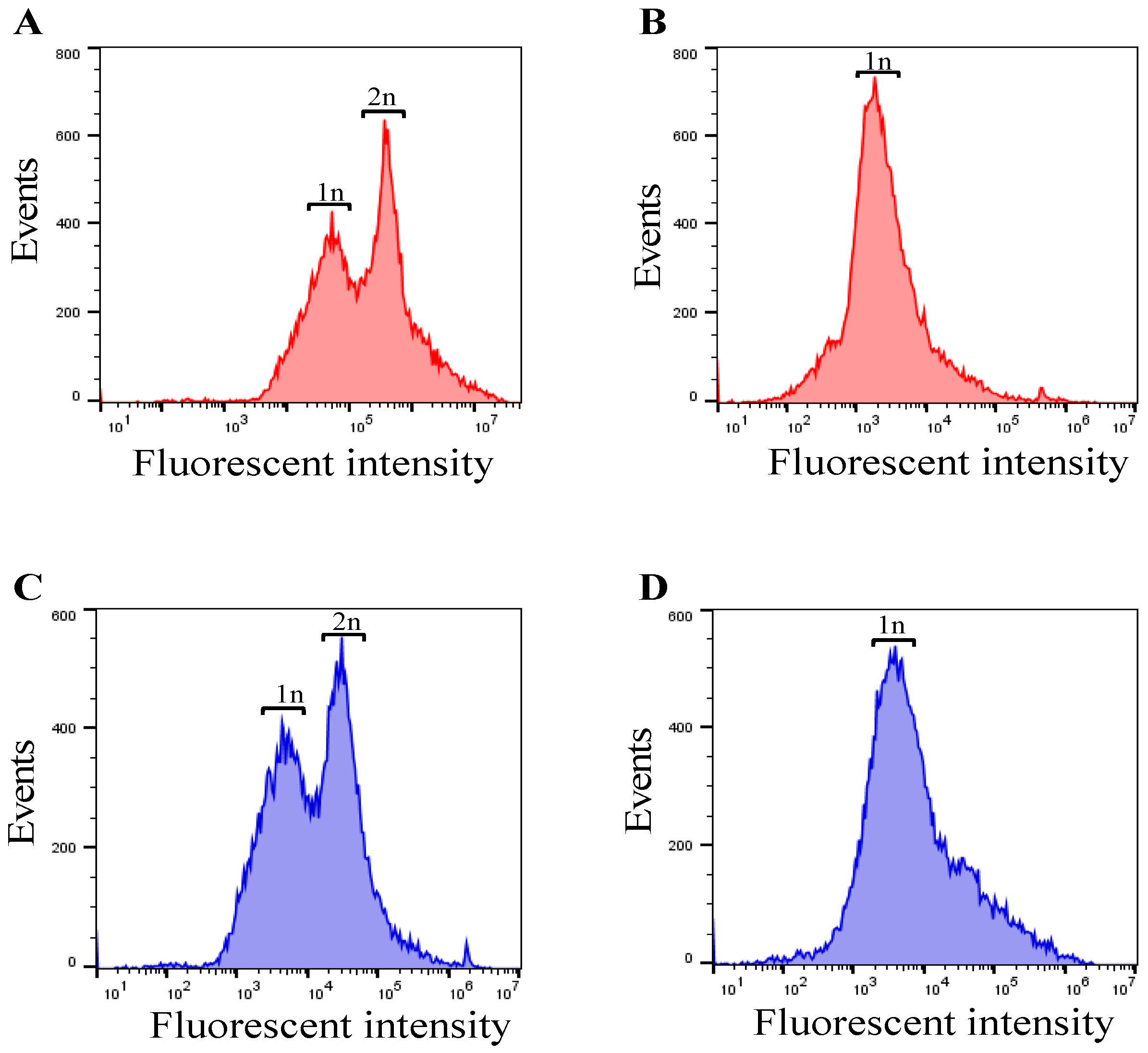
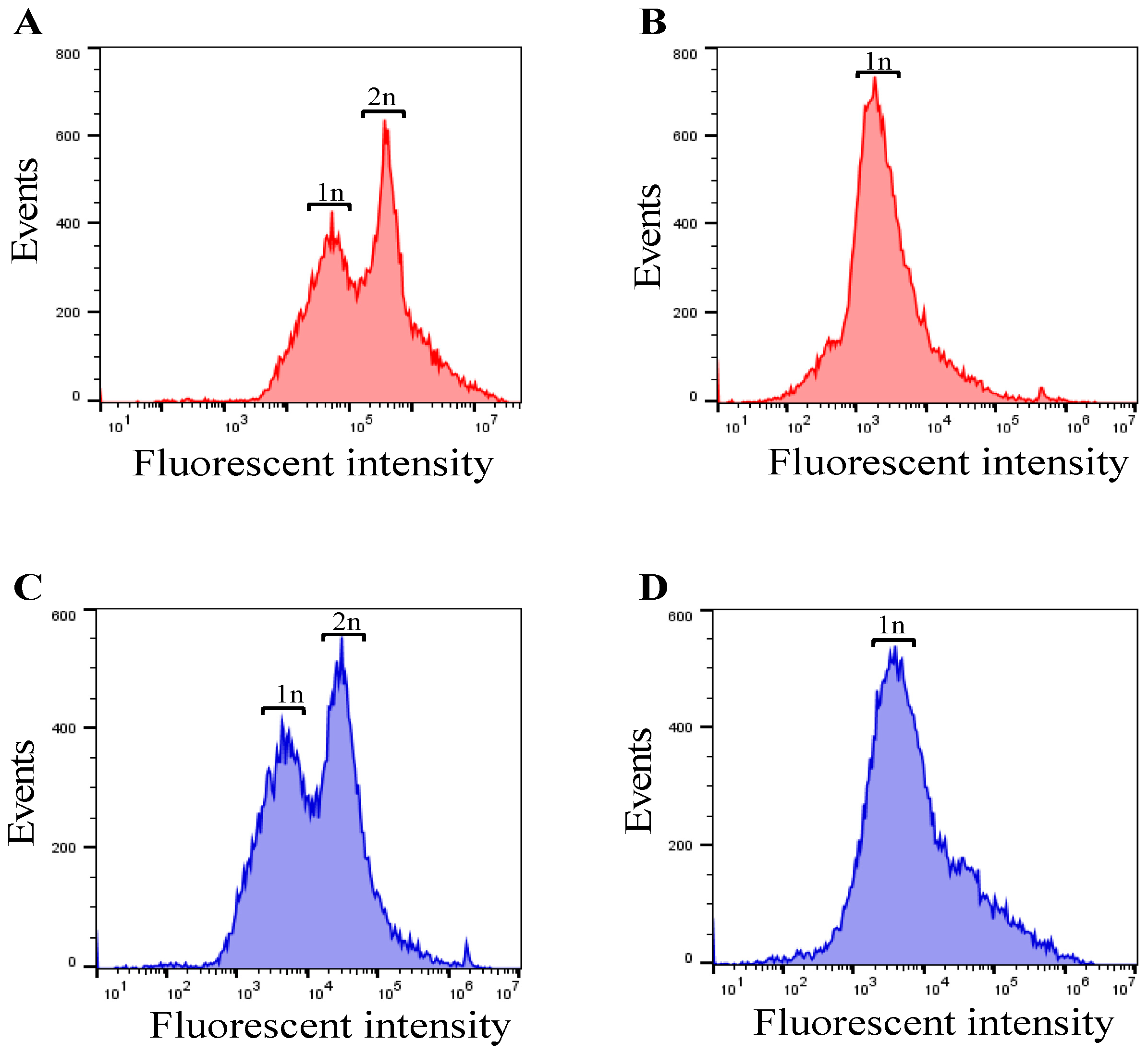
3.3. Checking Cell Cycle Control/Synchronization by DNA Quantification
3.4. Separase Activations through Cell Cycle Progression
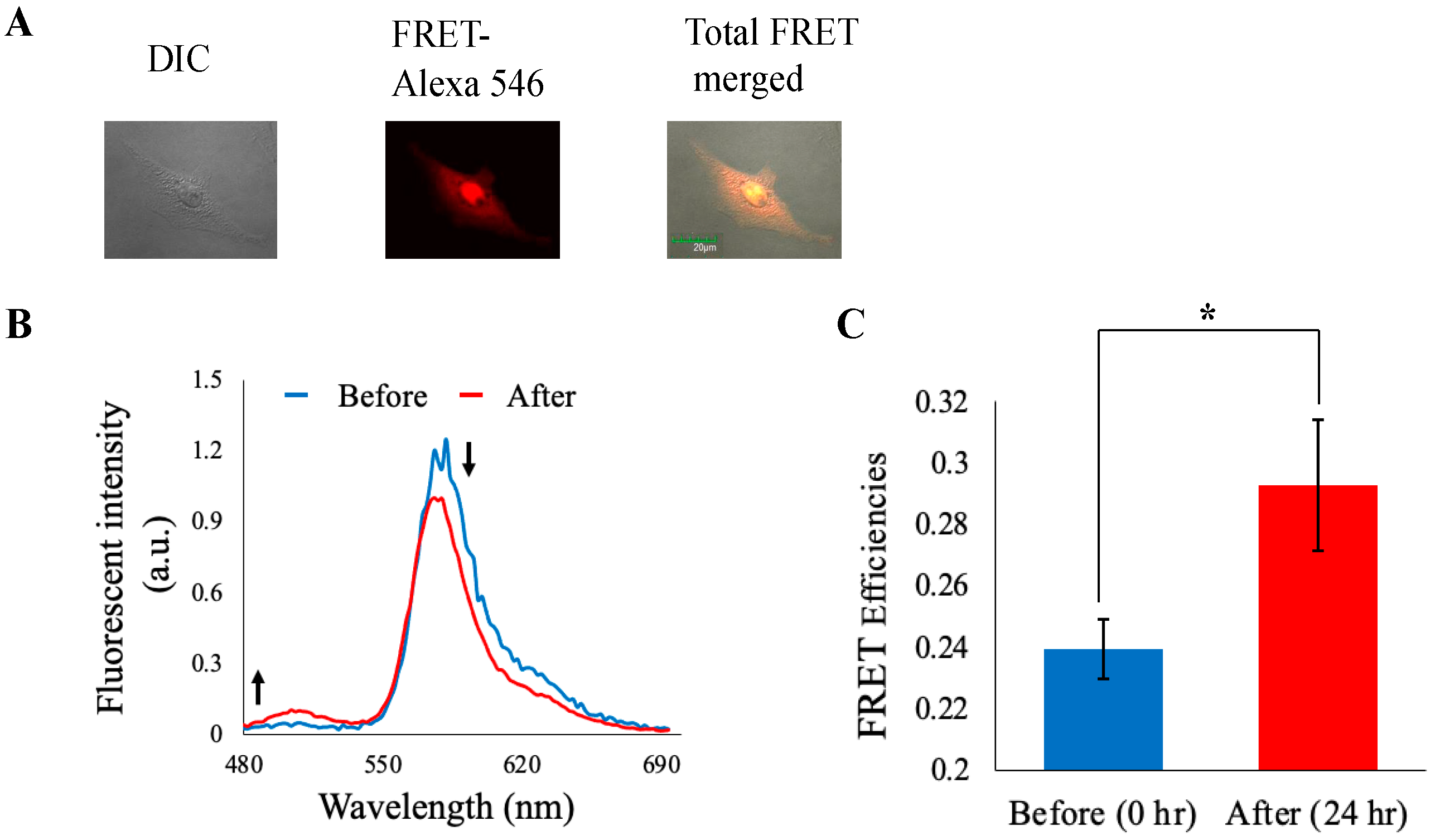
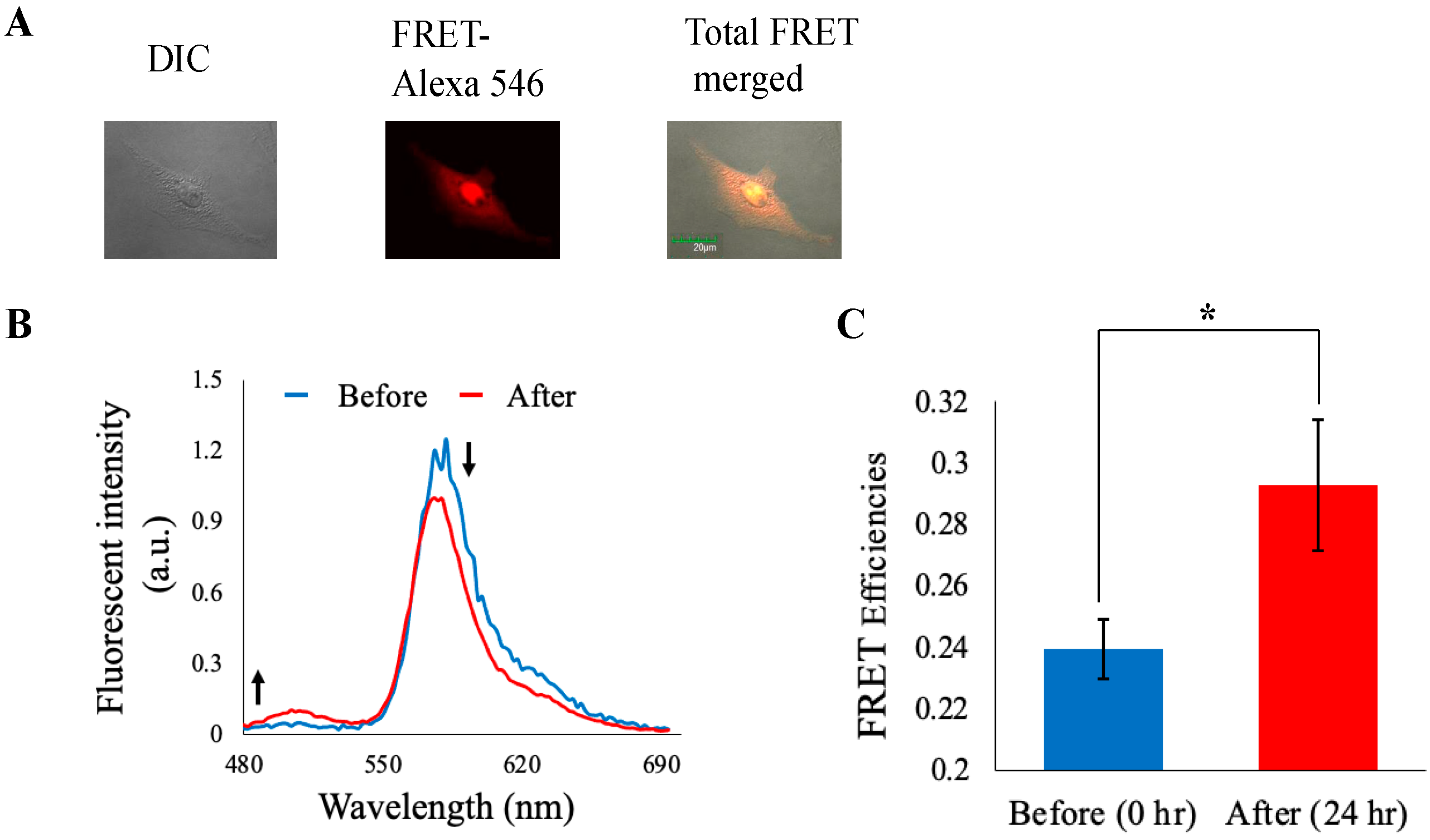
3.5. Imaging Analysis
3.6. Western Blot Analysis for Separase Expression
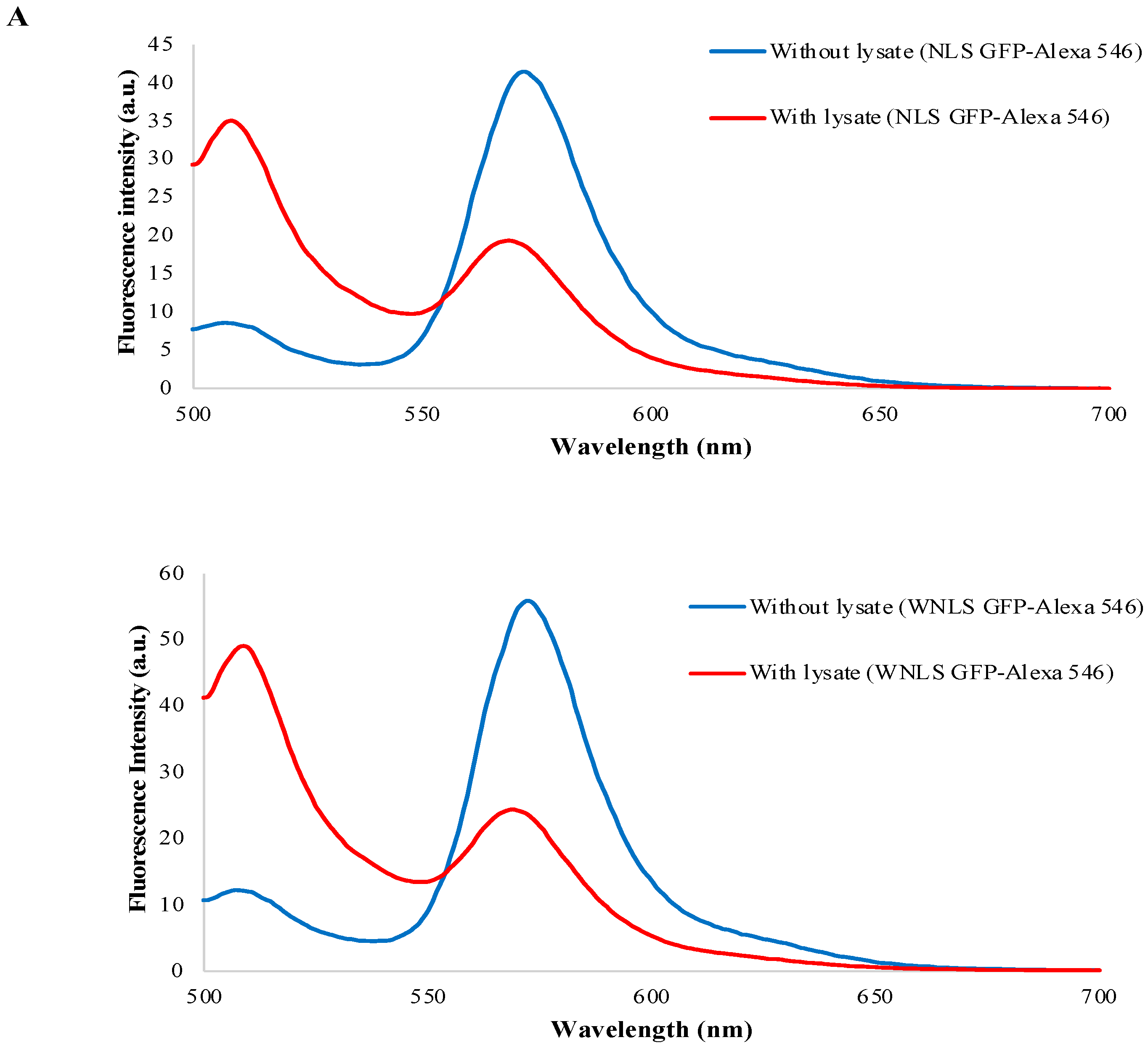
3.7. Separase Activity Estimation Derived from Cell-Cycle-Progressed Cell Lysate
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uhlmann, F.; Wernic, D.; Poupart, M.A.; Koonin, E.V.; Nasmyth, K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 2000, 103, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ciosk, R.; Zachariae, W.; Michaelis, C.; Shevchenko, A.; Mann, M.; Nasmyth, K. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell 1998, 93, 1067–1076. [Google Scholar] [CrossRef]
- Uhlmann, F.; Lottspeich, F.; Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999, 400, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Henschke, L.; Frese, M.; Hellmuth, S.; Marx, A.; Stemmann, O.; Mayer, T.U. Identification of Bioactive Small Molecule Inhibitors of Separase. ACS Chem. Biol. 2019, 14, 2155–2159. [Google Scholar] [CrossRef] [PubMed]
- Jallepalli, P.V.; Lengauer, C. Chromosome segregation and cancer: Cutting through the mystery. Nat. Rev. Cancer 2001, 1, 109–117. [Google Scholar] [CrossRef]
- Hornig, N.C.; Knowles, P.P.; McDonald, N.Q.; Uhlmann, F. The dual mechanism of separase regulation by securin. Curr. Biol. 2002, 12, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Waizenegger, I.; Giménez-Abián, J.F.; Wernic, D.; Peters, J.-M. Regulation of human separase by securin binding and autocleavage. Curr. Biol. 2002, 12, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Gorr, I.H.; Boos, D.; Stemmann, O. Mutual inhibition of separase and Cdk1 by two-step complex formation. Molecular Cell 2005, 19, 135–141. [Google Scholar] [CrossRef]
- Sanchez-Puig, N.; Veprintsev, D.B.; Fersht, A.R. Human full-length Securin is a natively unfolded protein. Protein Sci. 2005, 14, 1410–1418. [Google Scholar] [CrossRef]
- Csizmok, V.; Felli, I.C.; Tompa, P.; Banci, L.; Bertini, I. Structural and dynamic characterization of intrinsically disordered human securin by NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 16873–16879. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, S.B.C.; Clyne, R.K.; Fuchs, J.; Loidl, J.; Uhlmann, F.; Nasmyth, K. Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 2000, 103, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Adachi, Y.; Yanagida, M. Separase-mediated cleavage of cohesin at interphase is required for DNA repair. Nature 2004, 430, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- McAleenan, A.; Clemente-Blanco, A.; Cordon-Preciado, V.; Sen, N.; Esteras, M.; Jarmuz, A.; Aragón, L. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 2013, 493, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, S.; Gutiérrez-Caballero, C.; Llano, E.; Pendás, A.M.; Stemmann, O. Local activation of mammalian separase in interphase promotes double-strand break repair and prevents oncogenic transformation. EMBO J. 2018, 37, e99184. [Google Scholar] [CrossRef] [PubMed]
- Tsou, M.F.; Wang, W.J.; George, K.A.; Uryu, K.; Stearns, T.; Jallepalli, P.V. Polo kinase and separase regulatethe mitotic licensing of centriole duplication in human cells. Dev. Cell 2009, 17, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Rhee, K. Separase-dependent cleavage of pericentrin B is necessary and sufficient for centriole disengagement during mitosis. Cell Cycle 2012, 11, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- Papi, M.; Berdougo, E.; Randall, C.L.; Ganguly, S.; Jallepalli, P.V. Multiple roles for separase auto-cleavage during the G2/M transi- tion. Nat. Cell Biol. 2005, 7, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Baskerville, C.; Segal, M.; Reed, S.I. The protease activity of yeast separase (esp1) is required for anaphase spindle elongation independently of its role in cleavage of cohesin. Genetics 2008, 178, 2361–2372. [Google Scholar] [CrossRef]
- Thein, K.H.; Kleylein-Sohn, J.; Nigg, E.A.; Gruneberg, U. Astrin is required for the maintenance of sister chromatid cohesion and centrosome integrity. J. Cell Biol. 2007, 178, 345–354. [Google Scholar] [CrossRef]
- Nakamura, A.; Arai, H.; Fujita, N. Centrosomal Aki1 and cohesin function in separase-regulated centriole disengagement. J. Cell Biol. 2009, 187, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Hauf, S.; Waizenegger, I.C.; Peters, J.M. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science 2001, 293, 1320–1323. [Google Scholar] [CrossRef]
- Schockel, L.; Mockel, M.; Mayer, B.; Boos, D.; Stemmann, O. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat. Cell Biol. 2011, 13, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Ohsumi, K.; Iwabuchi, M.; Kawamata, T.; Ono, Y.; Takahashi, M. Kendrin is a novel substrate for separase involved in the licensing of centriole duplication. Curr. Biol. 2012, 22, 915–921. [Google Scholar] [CrossRef]
- Funabiki, H.; Kumada, K.; Yanagida, M. Fission yeast Cut1 and Cut2 are essential for sister chromatid separation, concentrate along the metaphase spindle and form large complexes. EMBO J. 1996, 15, 6617–6628. [Google Scholar] [CrossRef]
- Cohen-Fix, O.; Peters, J.M.; Kirschner, M.W.; Koshland, D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996, 10, 3081–3093. [Google Scholar] [CrossRef]
- Rao, H.; Uhlmann, F.; Nasmyth, K.; Varshavsky, A. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature 2001, 410, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, W.; Shin, T.H.; Galova, M.; Obermaier, B.; Nasmyth, K. Identification of subunits of the anaphasepromoting complex of Saccharomyces cerevisiae. Science 1996, 274, 1201–1204. [Google Scholar] [CrossRef]
- Peters, J.-M. The anaphase-promoting complex: Proteolysis in mitosis andbeyond. Mol. Cell 2002, 9, 931–943. [Google Scholar] [CrossRef]
- Zou, H.; Stemman, O.; Anderson, J.S.; Mann, M.; Kirschner, M.W. Anaphase specific auto-cleavage of separase. FEBS Lett. 2002, 528, 246–250. [Google Scholar] [CrossRef]
- Herzig, A.; Lehner, C.F.; Heidmann, S. Proteolytic cleavage of the THR subunit during anaphase limits Drosophila separase function. Genes Dev. 2002, 16, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Chestukhin, A.; Pfeffer, C.; Milligan, S.; DeCaprio, J.A.; Pellman, D. Processing, localization, and requirement of human separase for normal anaphase progression. Proc. Natl. Acad. Sci. USA 2003, 100, 4574–4579. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Böttger, F.; Stemmann, O.; Taylor, S.S. Protein phosphatase 2A and separase form a complex regulated by separase autocleavage. J. Biol. Chem. 2007, 282, 24623–24632. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, S.; Böttger, F.; Pan, C.; Mann, M.; Stemmann, O. PP2A delays APC/C-dependent degradation of separase-associated but not free securin. EMBO J. 2014, 33, 1134–1147. [Google Scholar] [CrossRef]
- Agarwal, R.; Cohen-Fix, O. Phosphorylation of the mitotic regulator Pds1/securin by Cdc28 is required for efficient nuclear localization of Esp1/separase. Genes Dev. 2002, 16, 1371–1382. [Google Scholar] [CrossRef]
- Lin, Z.; Luo, X.; Yu, H. Structural basis of cohesin cleavage by separase. Nature 2016, 532, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Tong, L. Molecular mechanism for the regulation of yeast separase by securin. Nature 2017, 542, 255–259. [Google Scholar] [CrossRef]
- Boland, A.; Martin, T.G.; Zhang, Z.; Yang, J.; Bai, X.C.; Chang, L.; Scheres, S.H.; Barford, D. Cryo-EM structure of a metazoan separase-securin complex at near-atomic resolution. Nat. Struct. Mol. Biol. 2017, 24, 414–418. [Google Scholar] [CrossRef]
- Yu, J.; Raia, P.; Ghent, C.M.; Raisch, T.; Sadian, Y.; Cavadini, S.; Sabale, P.M.; Barford, D.; Raunser, S.; Morgan, D.O.; et al. Structural basis of human separase regulation by securin and CDK1-cyclin B1. Nature 2021, 596, 138–142. [Google Scholar] [CrossRef]
- Hellmuth, S.; Rata, S.; Brown, A.; Heidmann, S.; Novak, B.; Stemmann, O. Human chromosome segregation involves multi-layered regulation of separase by the peptidyl-prolyl-isomerase Pin1. Mol. Cell 2015, 58, 495–506. [Google Scholar] [CrossRef]
- Zhang, W.; Suzuki, M.; Ito, Y.; Douglas, K.T. A Chemically Modified Green-Fluorescent Protein that Responds to Cleavage of an Engineered Disulphide Bond by Fluorescence Resonance Energy Transfer (FRET)-Based Changes. Chem. Lett. 2005, 34, 766–767. [Google Scholar] [CrossRef]
- Suzuki, M.; Shindo, Y.; Yamanaka, R.; Oka, K. Live imaging of apoptotic signaling flow using tunable combinatorial FRET-based bioprobes for cell population analysis of caspase cascades. Sci. Rep. 2022, 12, 21160. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Matsushima, Y.; Sugimura, T.; Terada, M. A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res. 1991, 19, 2785. [Google Scholar] [CrossRef] [PubMed]
- Alnemri, T.F.; Armstrong, R.C.; Krebs, J.; Srinivasula, S.M.; Wang, L.; Bullrich, F.; Fritz, L.C.; Trapani, J.A.; Tomaselli, K.J.; Litwack, G.; et al. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl. Acad. Sci. USA 1996, 93, 7464–7469. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Tanaka, S.; Ito, Y.; Inoue, M.; Sakai, T.; Nishigaki, K. Simple and tunable Förster resonance energy transfer-based bioprobes for high-throughput monitoring of caspase-3 activation in living cells by using flow cytometry. Biochim Biophys Acta. 2012, 1823, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Nakayama, Y.; Kinjo, M. Efficient and dynamic nuclear localization of green fluorescent protein via RNA binding. Biochem. Biophys. Res. Commun. 2015, 463, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sakata, I.; Sakai, T.; Tomioka, H.; Nishigaki, K.; Tramier, M.; Coppey-Moisan, M. A high-throughput direct fluorescence resonance energy transfer-based assay for analyzing apoptotic proteases using flow cytometry and fluorescence lifetime measurements. Anal. Biochem. 2015, 291, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Taguma, K.; Bandaranayake, U.K.; Ikeda, W. Development of Ratiometric Fluorescence Sensing Molecule for Caspase-14, a Key Enzyme of Epidermis Metabolism. J. Biomed. Res. Environ. Sci. 2023, 4, 793–800. [Google Scholar] [CrossRef]
- Chen, M.; Huang, J.; Yang, X.; Liu, B.; Zhang, W.; Huang, L. Serum Starvation Induced Cell Cycle Synchronization Facilitates Human Somatic Cells Reprogramming. PLoS ONE 2012, 7, e28203. [Google Scholar] [CrossRef]
- Konecna, M.; Abbasi Sani, S.; Anger, M. Separase and Roads to Disengage Sister Chromatids during Anaphase. Int. J. Mol. Sci. 2023, 24, 4604. [Google Scholar] [CrossRef] [PubMed]
- Shindo, N.; Kumada, K.; Iemura, K.; Yasuda, J.; Fujimori, H.; Mochizuki, M.; Tamai, K.; Tanaka, K.; Hirota, T. Autocleavage of separase suppresses its premature activation by promoting binding to cyclin B1. Cell Rep. 2022, 41, 111723. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, S.; Stemman, O. Separase-triggered apoptosis enforces minimal length mitosis. Nature 2020, 580, 542–547. [Google Scholar] [CrossRef] [PubMed]

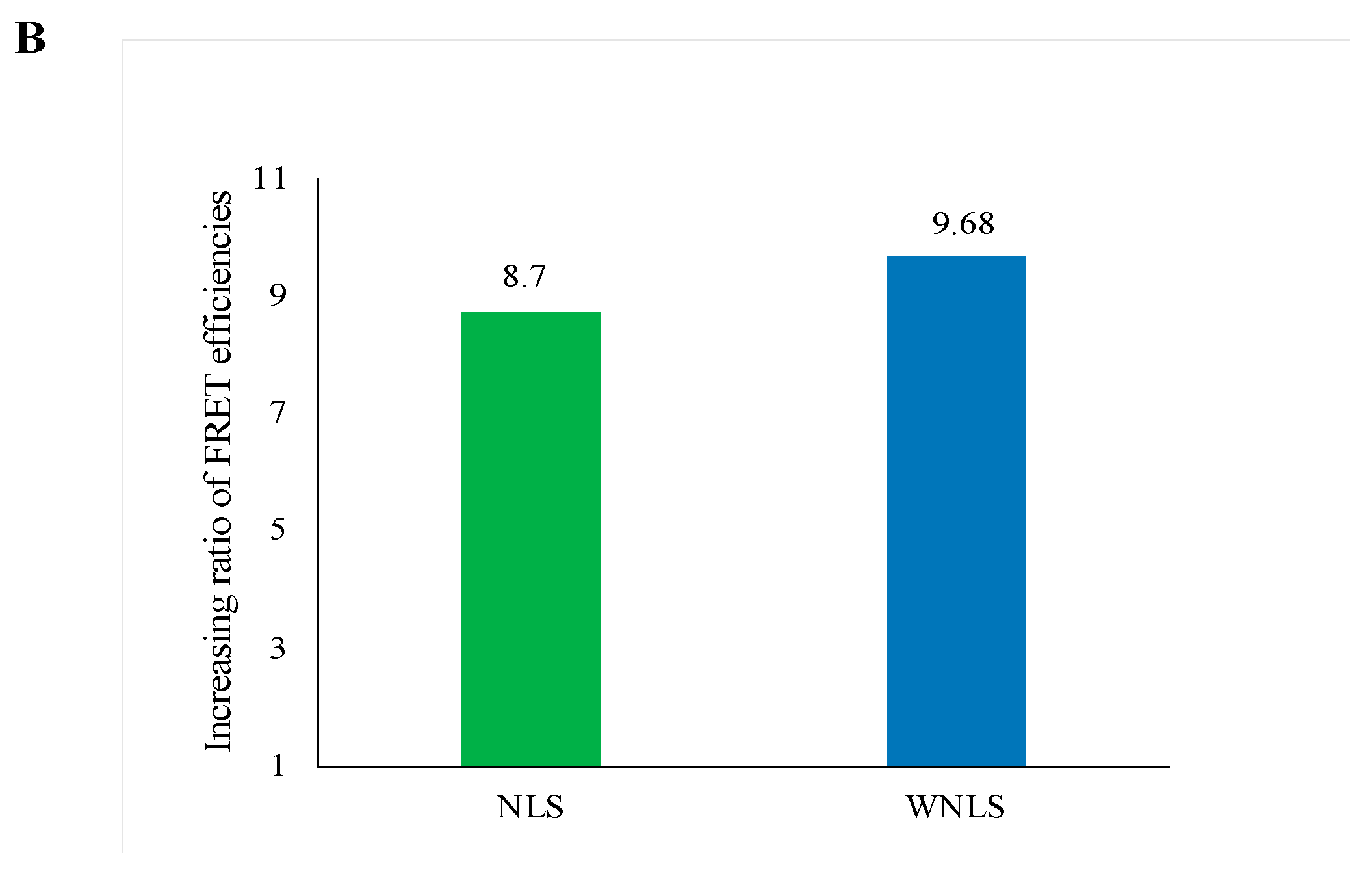
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.S.; Shindo, Y.; Oka, K.; Ikeda, W.; Suzuki, M. Live Cell Monitoring of Separase Activity, a Key Enzymatic Reaction for Chromosome Segregation, with Chimeric FRET-Based Molecular Sensor upon Cell Cycle Progression. Biosensors 2024, 14, 192. https://doi.org/10.3390/bios14040192
Rahman MS, Shindo Y, Oka K, Ikeda W, Suzuki M. Live Cell Monitoring of Separase Activity, a Key Enzymatic Reaction for Chromosome Segregation, with Chimeric FRET-Based Molecular Sensor upon Cell Cycle Progression. Biosensors. 2024; 14(4):192. https://doi.org/10.3390/bios14040192
Chicago/Turabian StyleRahman, Md. Shazadur, Yutaka Shindo, Kotaro Oka, Wataru Ikeda, and Miho Suzuki. 2024. "Live Cell Monitoring of Separase Activity, a Key Enzymatic Reaction for Chromosome Segregation, with Chimeric FRET-Based Molecular Sensor upon Cell Cycle Progression" Biosensors 14, no. 4: 192. https://doi.org/10.3390/bios14040192
APA StyleRahman, M. S., Shindo, Y., Oka, K., Ikeda, W., & Suzuki, M. (2024). Live Cell Monitoring of Separase Activity, a Key Enzymatic Reaction for Chromosome Segregation, with Chimeric FRET-Based Molecular Sensor upon Cell Cycle Progression. Biosensors, 14(4), 192. https://doi.org/10.3390/bios14040192