Advanced Liver-on-a-Chip Model for Evaluating Drug Metabolism and Hepatotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Routine Cell Culture
2.2. Liver-on-Chip Design and Preparation
2.3. Diffusion Evaluation in Hydrogel
2.4. Three-Dimensional (3D) Liver Aggregates Formation
2.5. Three-Dimensional (3D) Liver Aggregates Transfer to the Liver-on-Chip
2.6. Morphological Changes Evaluation
2.7. Live/Dead Cell Imaging
2.8. AlamarBlue® Assay
2.9. CellTracker® Labeling
2.10. Immunostaining
2.11. Urea Production
2.12. Albumin Production
2.13. Cytochrome P450 Activity
2.14. Drug Metabolism Evaluation
2.15. Statistical Analysis
3. Results
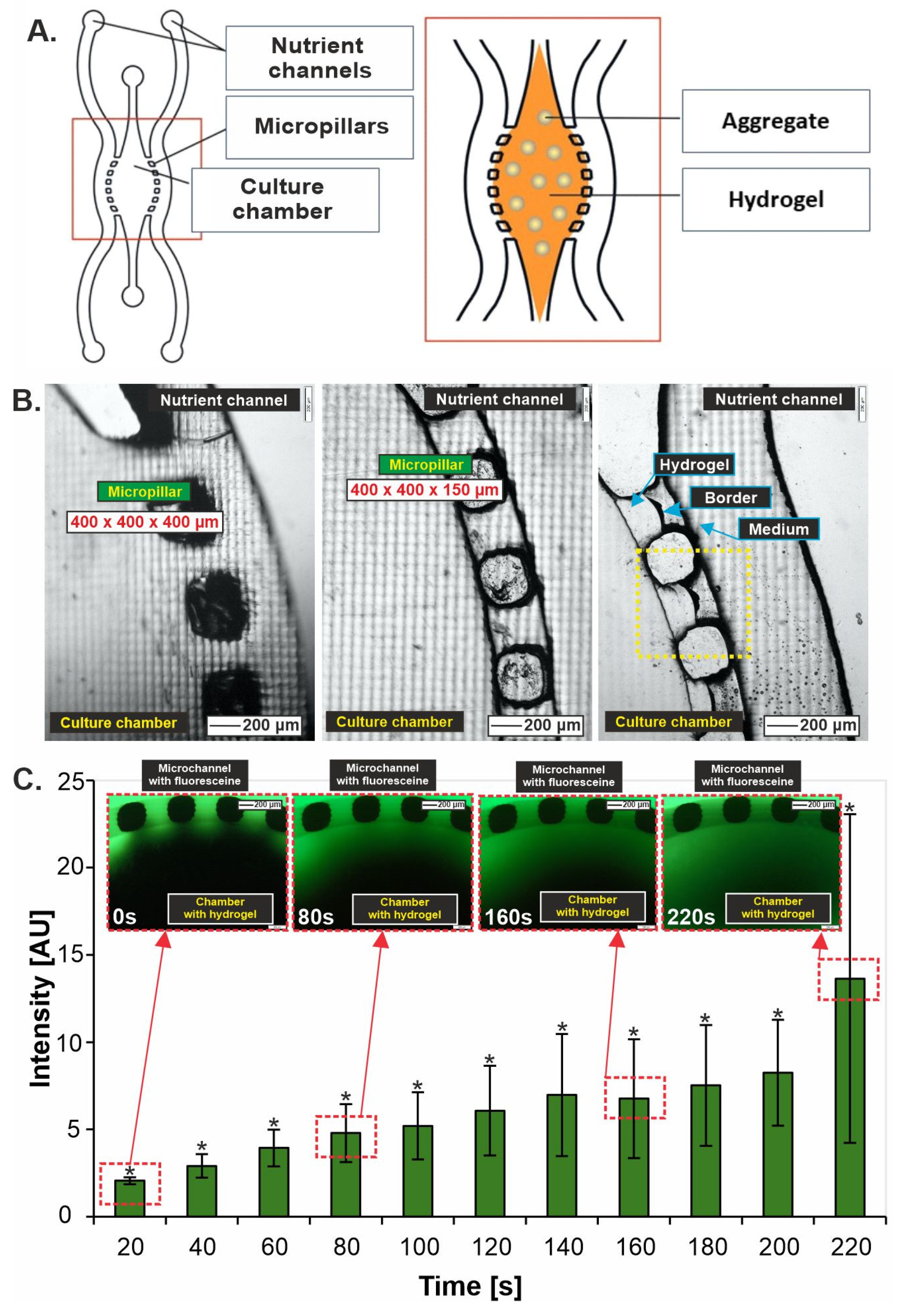
3.1. Liver-on-Chip Design Optimization
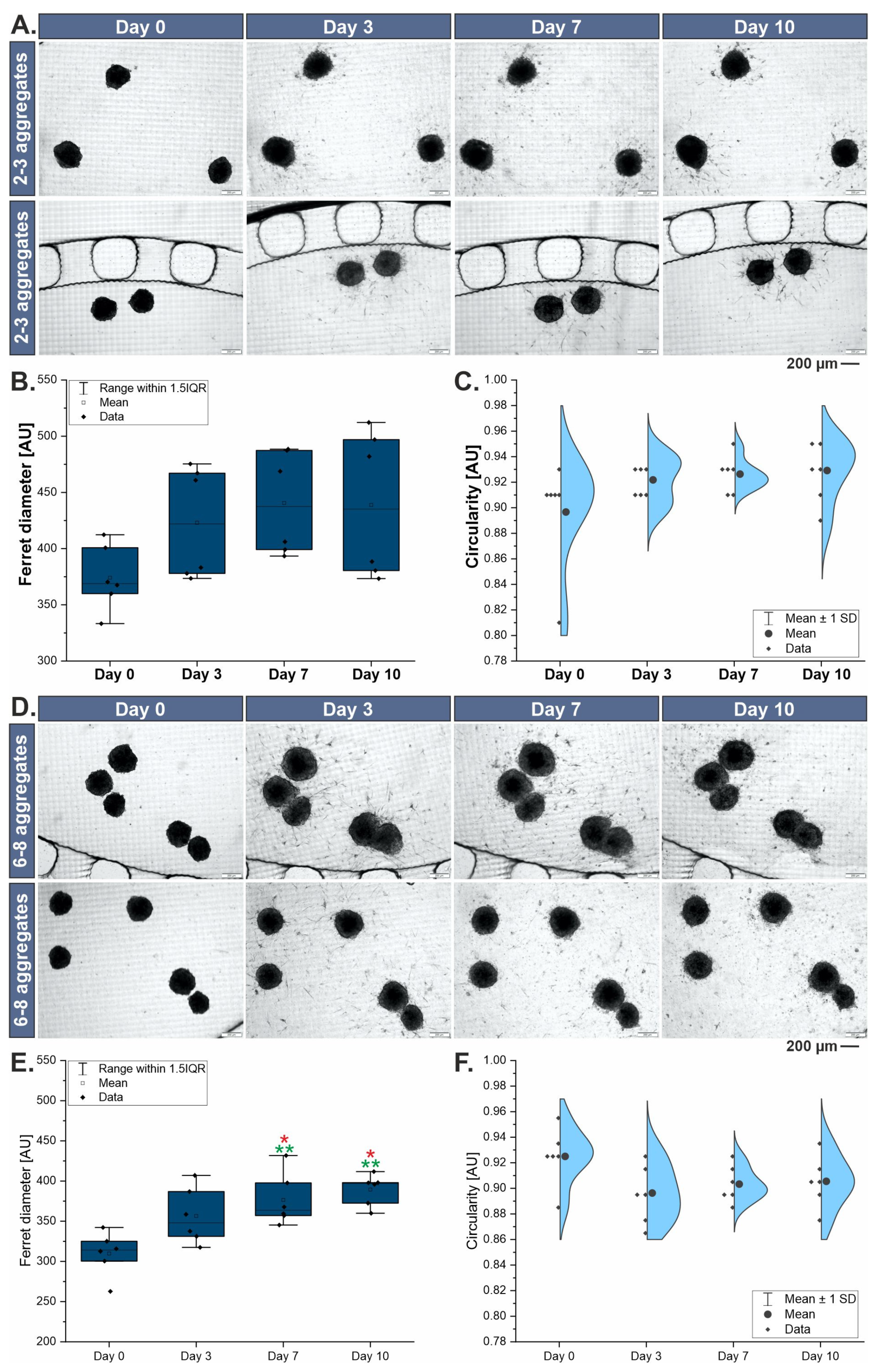
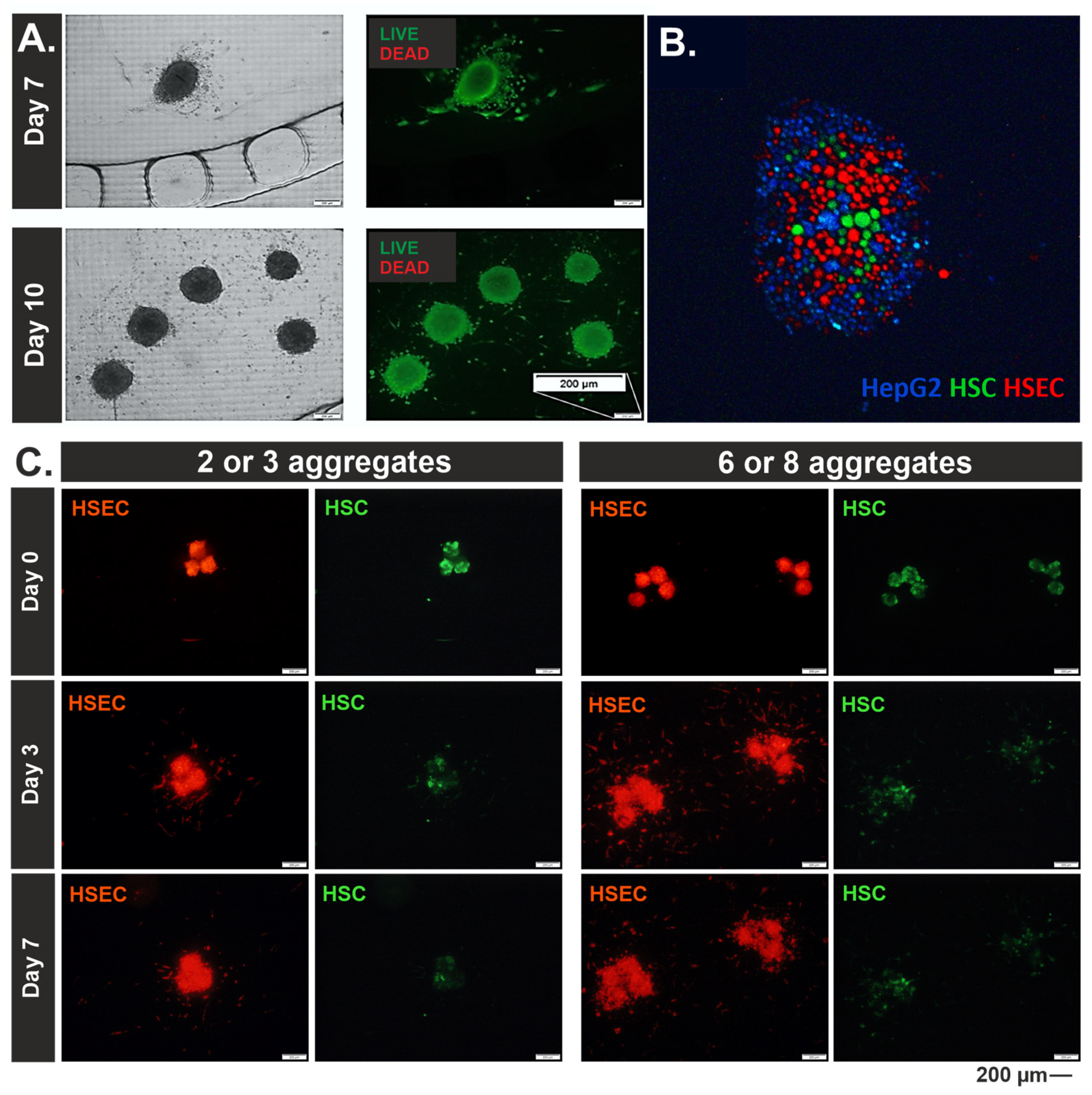
3.2. Morphological Characterization of 3D Liver Models
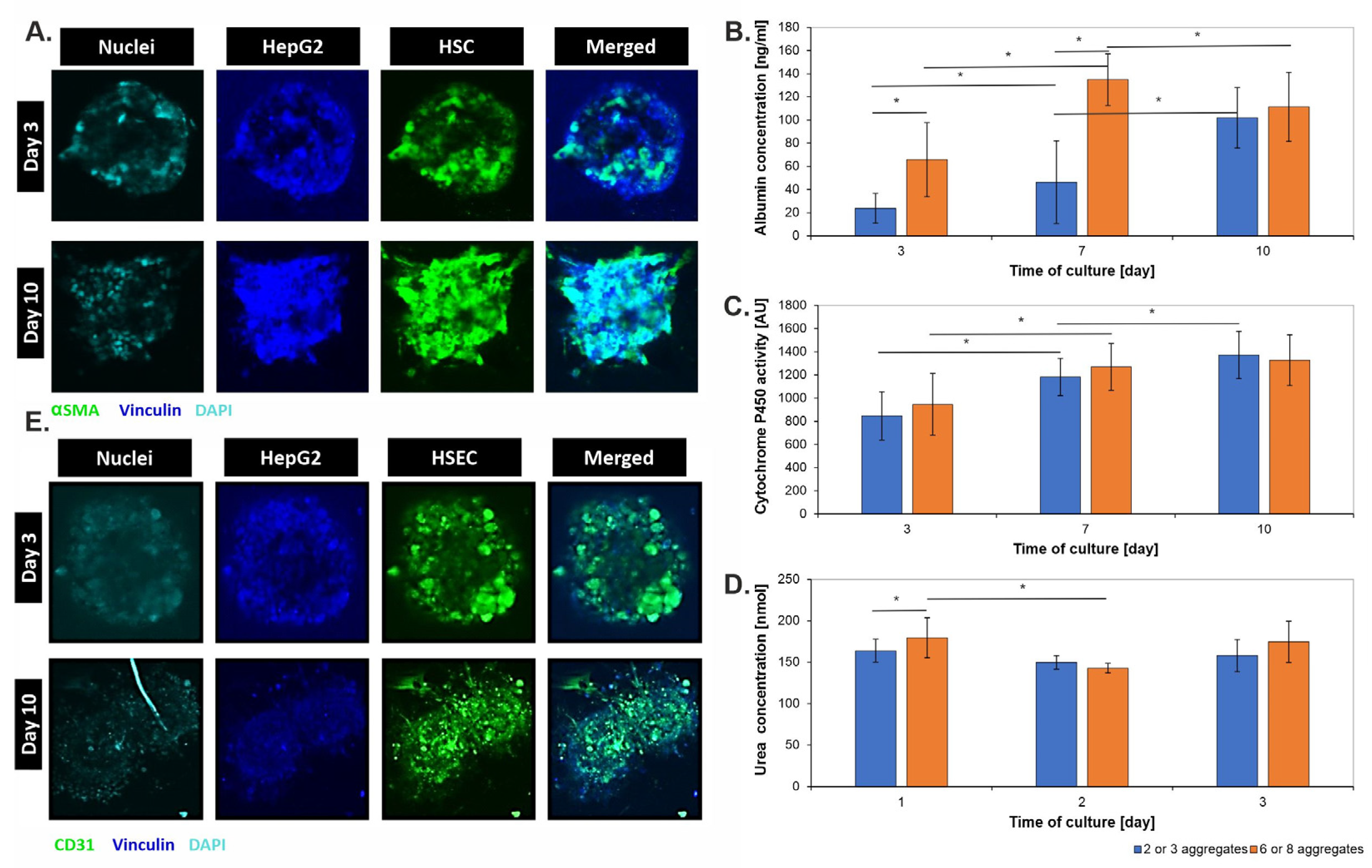
3.3. Metabolic Characterization of 3D Liver Models
3.4. Drug Metabolism Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalra, A.; Yetiskul, E.; Wehrle, C.J.; Tuma, F. Physiology, Liver. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK535438/ (accessed on 17 November 2023).
- Wang, F.; So, K.-F.; Xiao, J.; Wang, H. Organ-organ communication: The liver’s perspective. Theranostics 2021, 11, 3317–3330. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.C.; Cogliano, V.; Guyton, K.; Madia, F.; Straif, K.; Ward, E.M.; Schubauer-Berigan, M.K. Research Recommendations for Selected IARC-Classified Agents: Impact and Lessons Learned. Env. Environ. Health Perspect. 2023, 131, 105001. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yin, X.; Liu, Z.; Wang, J. Non-Alcoholic Fatty Liver Disease (NAFLD) Pathogenesis and Natural Products for Prevention and Treatment. Int. J. Mol. Sci. 2022, 23, 15489. [Google Scholar] [CrossRef] [PubMed]
- Herter-Sprie, G.S.; Kung, A.L.; Wong, K.-K. New cast for a new era: Preclinical cancer drug development revisited. J. Clin. Investig. 2013, 123, 3639–3645. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Saxton, S.H.; Stevens, K.R. 2D and 3D liver models. J. Hepatol. 2023, 78, 873–875. [Google Scholar] [CrossRef]
- Kmieć, Z. Cooperation of liver cells in health and disease. Adv. Anat. Embryol. Cell Biol. 2001, 161, 1–151. [Google Scholar] [CrossRef]
- Tutty, M.A.; Vella, G.; Prina-Mello, A. Pre-clinical 2D and 3D toxicity response to a panel of nanomaterials; comparative assessment of NBM-induced liver toxicity. Drug Deliv. Transl. Res. 2022, 12, 2157–2177. [Google Scholar] [CrossRef]
- Comparison of 2D- and 3D-Culture Models as Drug-Testing Platforms in Breast Cancer. Available online: https://www.spandidos-publications.com/or/33/4/1837 (accessed on 12 November 2023).
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: Recent breakthroughs and future prospects. BioMedical Eng. OnLine 2020, 19, 9. [Google Scholar] [CrossRef]
- Kim, J.; Han, Y.; Jeon, B.G.; Nam, M.S.; Kwon, S.; Heo, Y.J.; Park, M. Development of albumin monitoring system with hepatic hypoxia-on-a-chip. Talanta 2023, 260, 124592. [Google Scholar] [CrossRef]
- Ehrlich, A.; Duche, D.; Ouedraogo, G.; Nahmias, Y. Challenges and Opportunities in the Design of Liver-on-Chip Microdevices. Annu. Rev. Biomed. Eng. 2019, 21, 219–239. [Google Scholar] [CrossRef] [PubMed]
- Zuchowska, A.; Frojdenfal, S.; Trzaskowski, M.; Jastrzebska, E. Advanced three-dimensional in vitro liver models to study the activity of anticancer drugs. Biotechnol. J. 2024, 19, 2400159. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-A.; No, D.Y.; Kang, E.; Ju, J.; Kim, D.-S.; Lee, S.-H. Spheroid-based three-dimensional liver-on-a-chip to investigate hepatocyte–hepatic stellate cell interactions and flow effects. Lab. Chip 2013, 13, 3529–3537. [Google Scholar] [CrossRef]
- Bavli, D.; Prill, S.; Ezra, E.; Levy, G.; Cohen, M.; Vinken, M.; Vanfleteren, J.; Jaeger, M.; Nahmias, Y. Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E2231–E2240. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhao, L.; Zhou, E.-M.; Xu, J.; Shen, S.; Wang, J. On-Chip Construction of Liver Lobule-like Microtissue and Its Application for Adverse Drug Reaction Assay. Anal. Chem. 2016, 88, 1719–1727. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Neuzil, P.; Volk, T.; Manz, A.; Kleber, A. On-chip three-dimensional cell culture in phaseguides improves hepatocyte functions in vitro. Biomicrofluidics 2015, 9, 034113. [Google Scholar] [CrossRef]
- Gong, J.; Tu, W.; Liu, J.; Tian, D. Hepatocytes: A key role in liver inflammation. Front. Immunol. 2023, 13, 1083780. [Google Scholar] [CrossRef]
- Minciuna, I.; Taru, M.G.; Procopet, B.; Stefanescu, H. The Interplay between Liver Sinusoidal Endothelial Cells, Platelets, and Neutrophil Extracellular Traps in the Development and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. J. Clin. Med. 2024, 13, 1406. [Google Scholar] [CrossRef]
- Sufleţel, R.-T.; Melincovici, C.S.; Gheban, B.-A.; Toader, Z.; Mihu, C.M. Hepatic stellate cells—From past till present: Morphology, human markers, human cell lines, behavior in normal and liver pathology. Rom. J. Morphol. Embryol. 2020, 61, 615–642. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-T.; Lin, R.-Z.; Chen, R.-J.; Chin, C.-K.; Gong, S.-E.; Chang, H.-Y.; Peng, H.-L.; Hsu, L.; Yew, T.-R.; Chang, S.-F.; et al. Liver-cell patterning Lab Chip: Mimicking the morphology of liver lobule tissue. Lab. Chip 2013, 13, 3578–3587. [Google Scholar] [CrossRef] [PubMed]
- Engineered Liver-on-a-Chip Platform to Mimic—ProQuest. Available online: https://www.proquest.com/docview/2549023540?sourcetype=Scholarly%20Journals (accessed on 20 July 2024).
- Lee, J.; Choi, B.; No, D.Y.; Lee, G.; Lee, S.-R.; Oh, H.; Lee, S.-H. A 3D alcoholic liver disease model on a chip. Integr. Biol. 2016, 8, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.D.; Wang, Y.T.; Wang, J.R.; Wu, J.L.; Meng, X.S.; Hu, P.; Mu, X.; Liang, Q.L.; Luo, G.A. Design and Fabrication of a Liver-on-a-Chip Platform for Convenient, Highly Efficient, and Safe In Situ Perfusion Culture of 3D Hepatic Spheroids. Lab Chip 2018, 18, 2547–2562. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frojdenfal, S.; Zuchowska, A. Advanced Liver-on-a-Chip Model for Evaluating Drug Metabolism and Hepatotoxicity. Biosensors 2024, 14, 435. https://doi.org/10.3390/bios14090435
Frojdenfal S, Zuchowska A. Advanced Liver-on-a-Chip Model for Evaluating Drug Metabolism and Hepatotoxicity. Biosensors. 2024; 14(9):435. https://doi.org/10.3390/bios14090435
Chicago/Turabian StyleFrojdenfal, Sonia, and Agnieszka Zuchowska. 2024. "Advanced Liver-on-a-Chip Model for Evaluating Drug Metabolism and Hepatotoxicity" Biosensors 14, no. 9: 435. https://doi.org/10.3390/bios14090435
APA StyleFrojdenfal, S., & Zuchowska, A. (2024). Advanced Liver-on-a-Chip Model for Evaluating Drug Metabolism and Hepatotoxicity. Biosensors, 14(9), 435. https://doi.org/10.3390/bios14090435