
A Simple Microfluidic Platform for Long-Term Analysis and Continuous Dual-Imaging Detection of T-Cell Secreted IFN-γ and IL-2 on Antibody-Based Biochip


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Reagents
2.2. Cell Sample
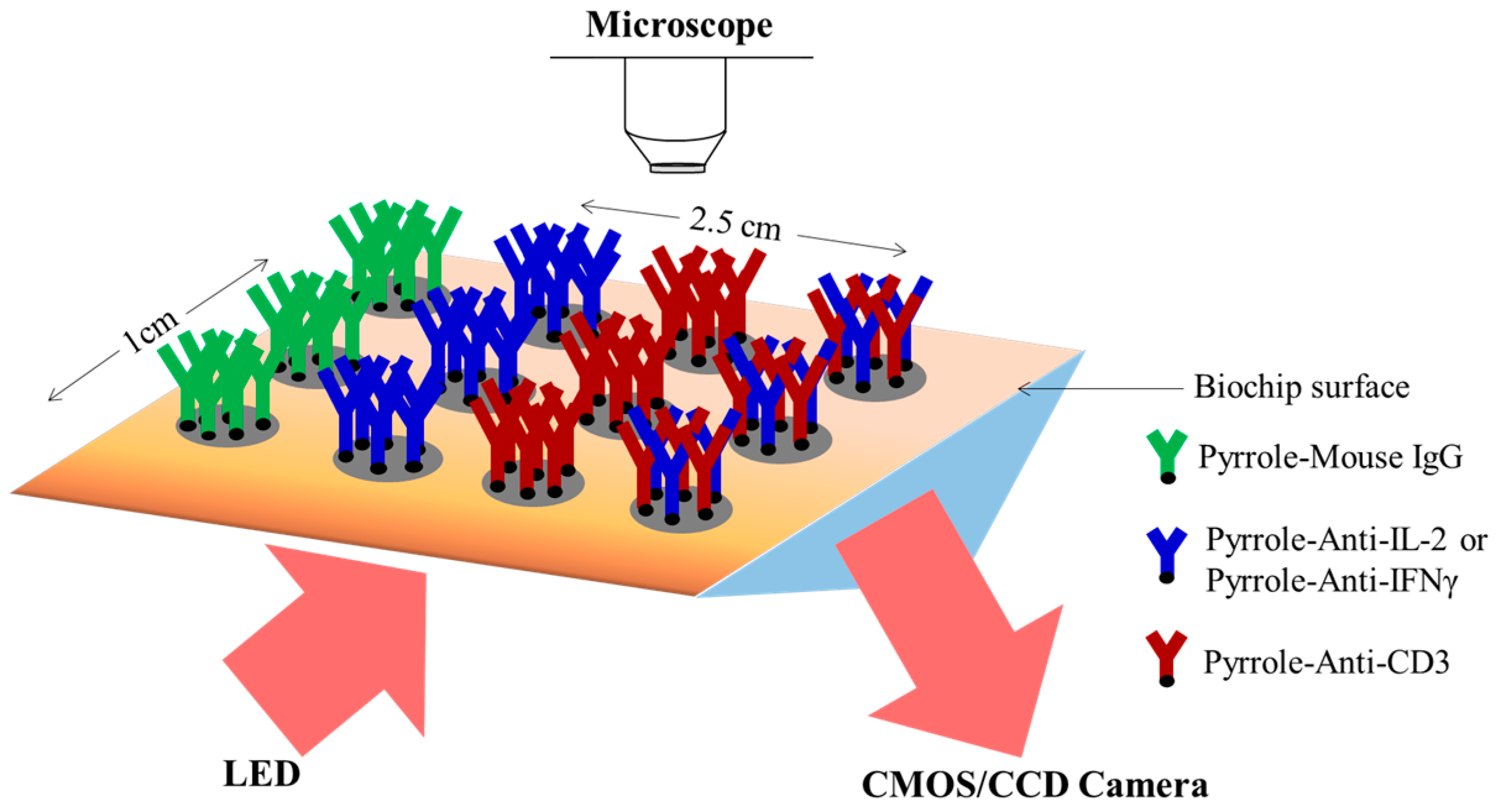
2.3. Biochip Preparation and Surface Treatment
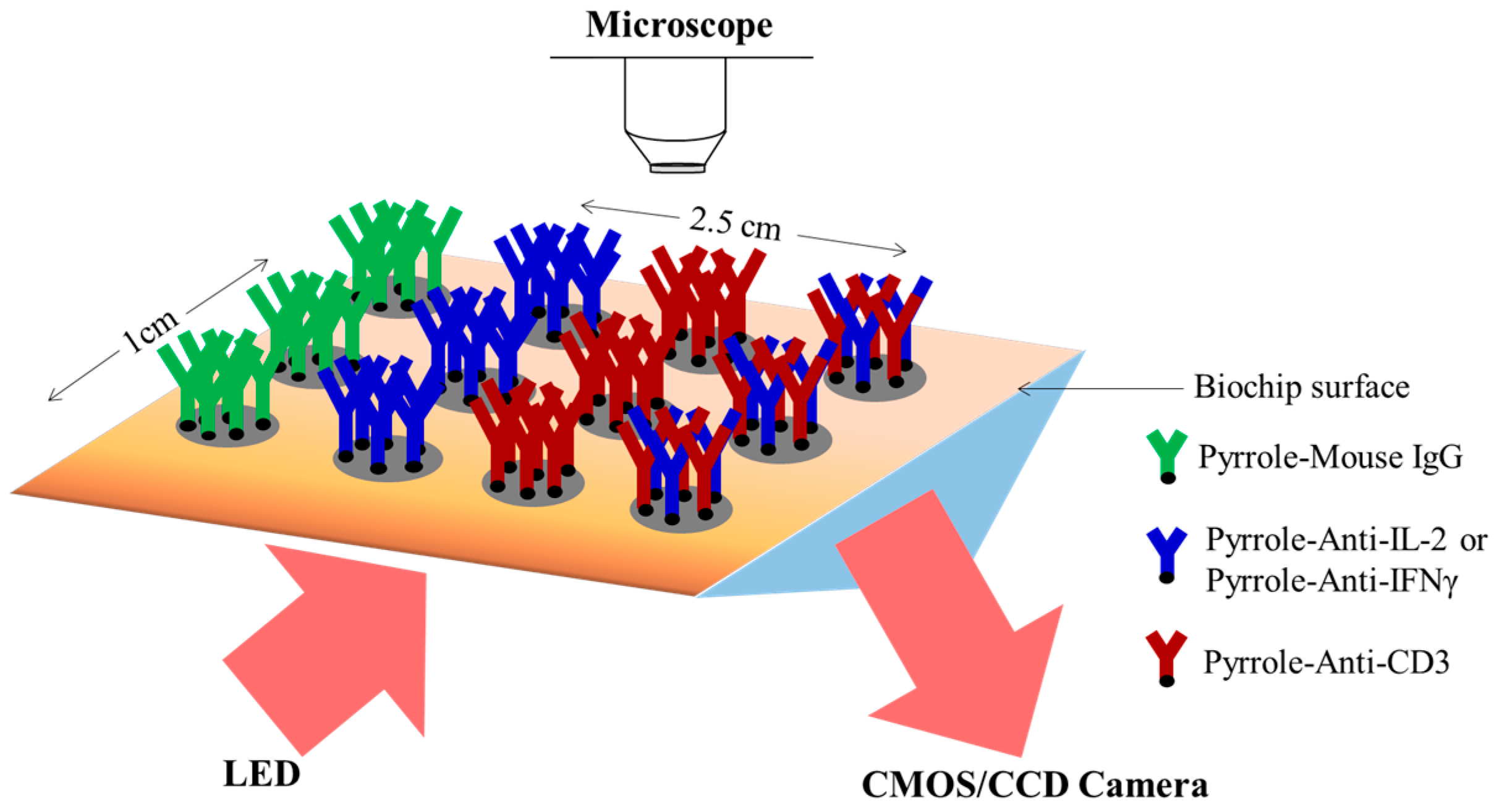
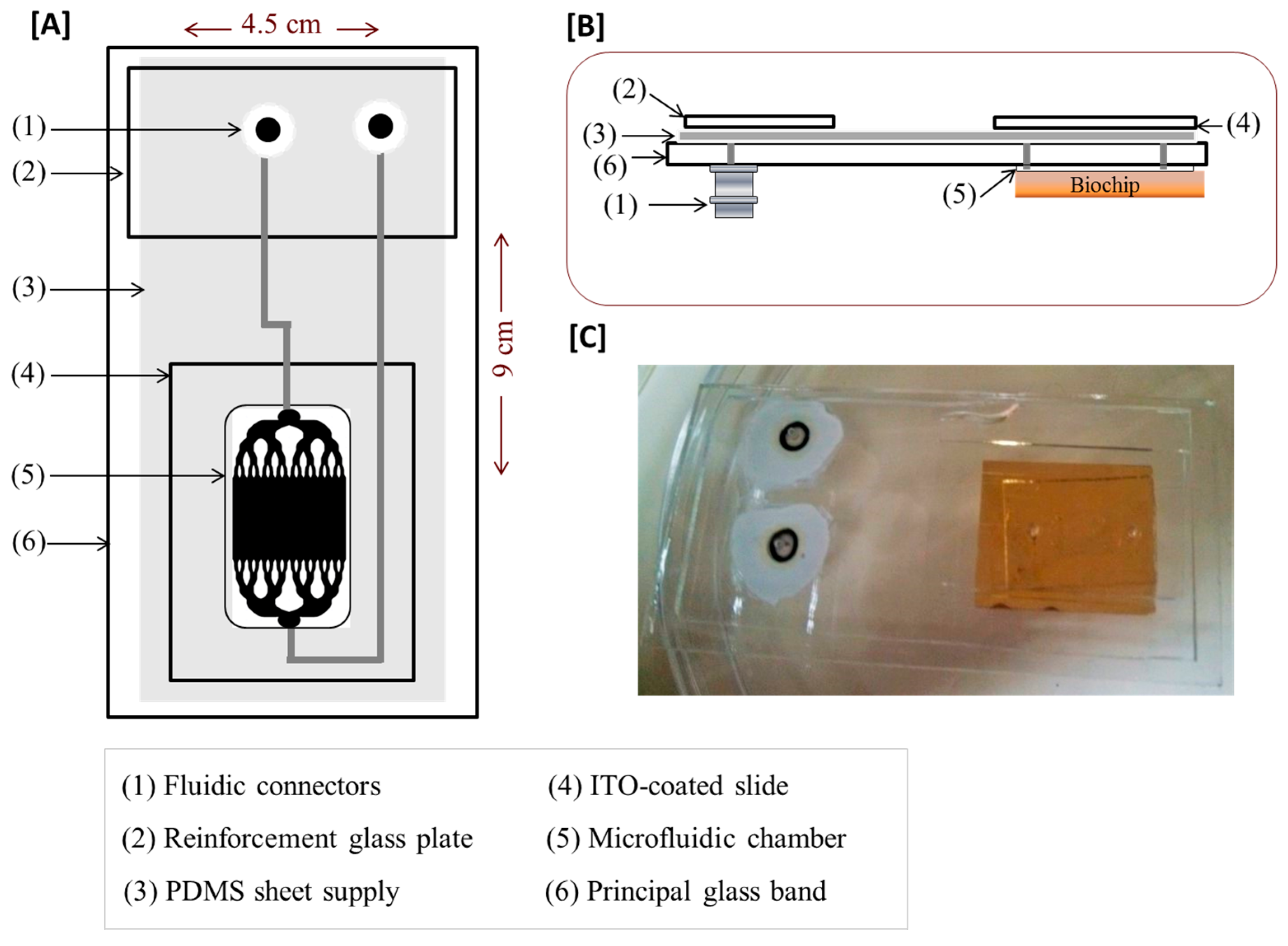
2.4. Design of the Microfluidic Platform

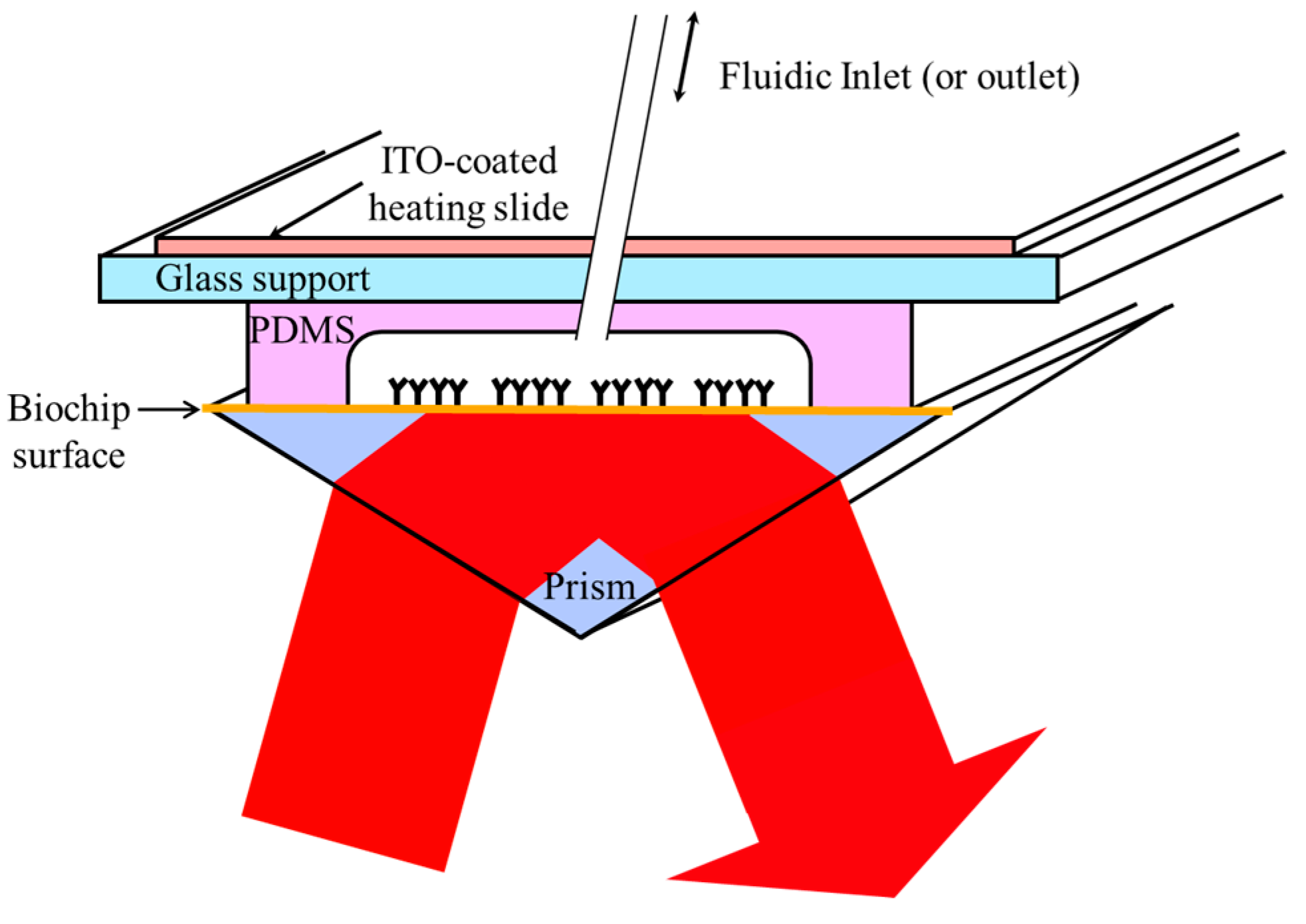
2.5. SPRi Detection Set-Up

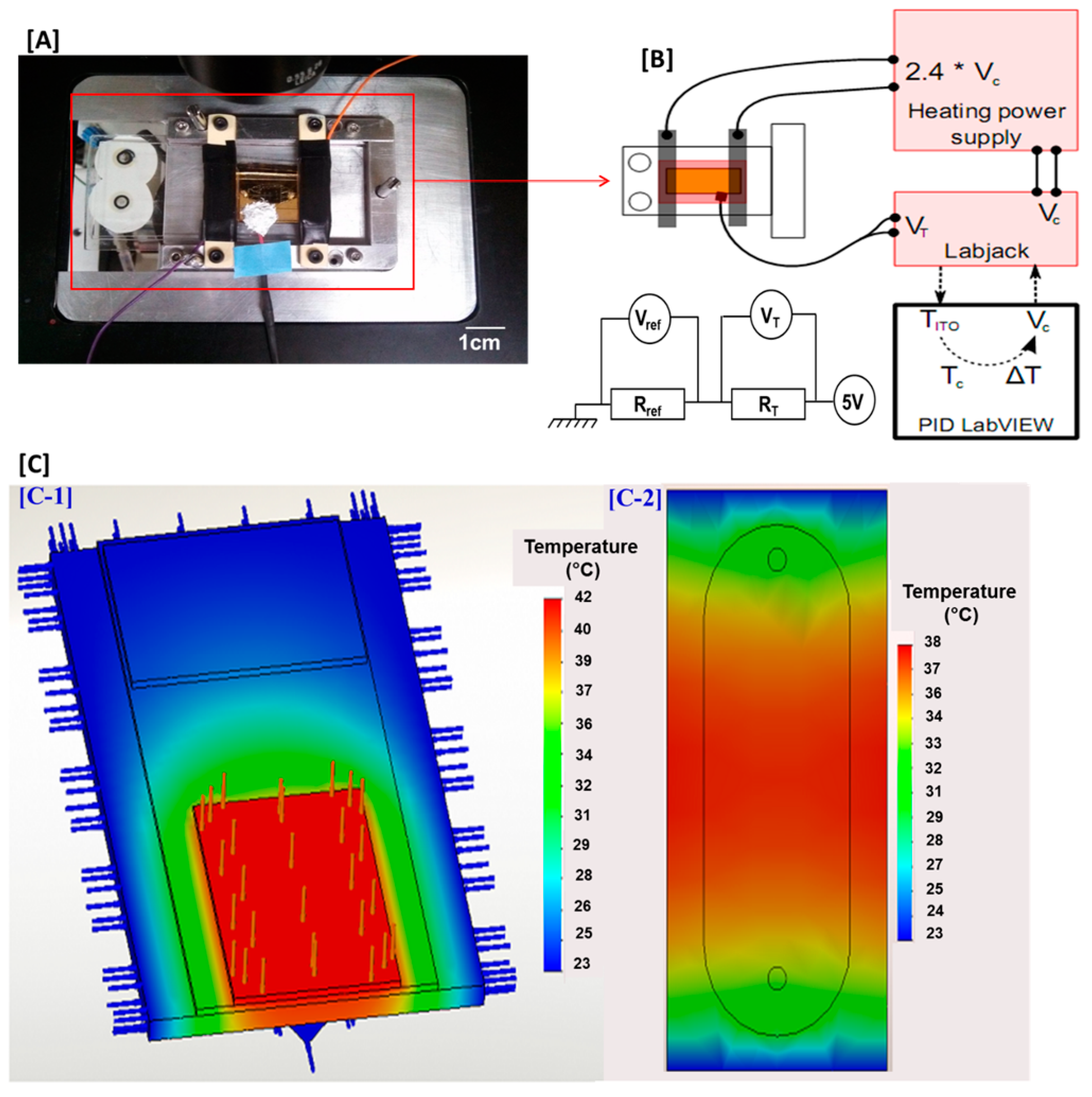
2.6. Heating and Thermoregulation in the Microfluidic Cell

2.7. On-Chip Cell Culture in the Microfluidic Device
2.8. On-Chip Cell Vitality Assay
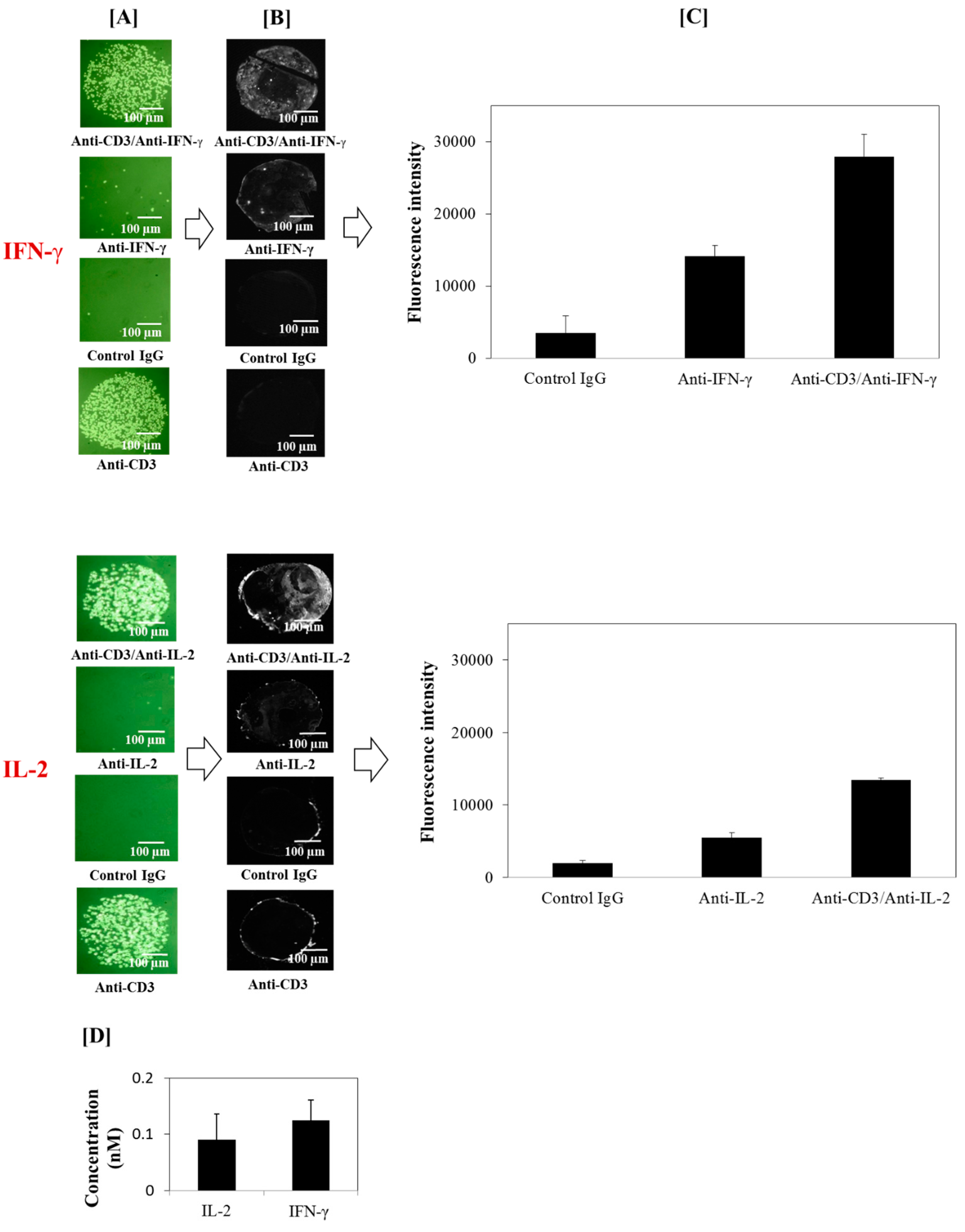
2.9. On-Chip Cytokine Detection
2.10. ELISA Titration of Cytokines
3. Results and Discussion
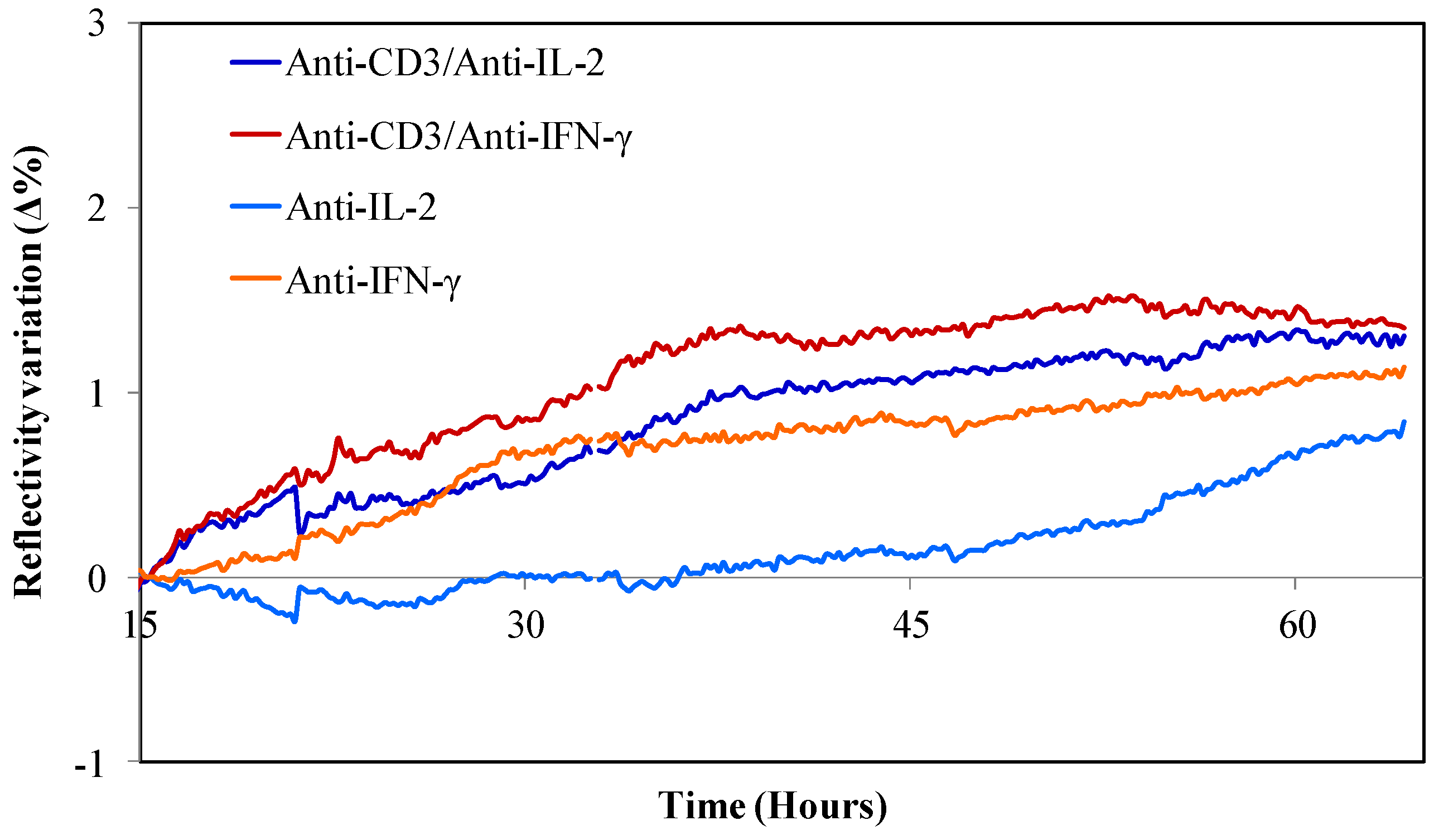
3.1. Specific Capture and Culture of Viable T Cells on the Biochip

3.2. Detection of Captured T Cell Cytokine Secretions


4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Webb, J.T.; Behar, M. Topology, dynamics, and heterogeneity in immune signaling. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J. Cytokines: Past, present, and future. Int. J. Hematol. 2001, 74, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pala, P.; Hussell, T.; Openshaw, P.J. Flow cytometric measurement of intracellular cytokines. J. Immunol. Methods 2000, 243, 107–124. [Google Scholar] [CrossRef]
- Ibrahim, F.S.; van den Engh, G. High-speed cell sorting: Fundamentals and recent advances. Curr. Opin. Biotechnol. 2003, 14, 5–12. [Google Scholar] [CrossRef]
- Desombere, I.; Meuleman, P.; Rigole, H.; Willems, A.; Irsch, J.; Leroux-Roels, G. The interferon gamma secretion assay: A reliable tool to study interferon gamma production at the single cell level. J. Immunol. Methods 2004, 286, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.X.; McElhaney, J.E.; Walston, J.D.; Xie, D.; Fedarko, N.S.; Kuchel, G.A. ELISA and multiplex technologies for cytokine measurement in inflammation and aging research. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Trune, D.R.; Larrain, B.E.; Hausman, F.A.; Kempton, J.B.; MacArthur, C.J. Simultaneous measurement of multiple ear proteins with multiplex ELISA assays. Hear. Res. 2011, 275, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Letsch, A.; Scheibenbogen, C. Quantification and characterization of specific T-cells by antigen-specific cytokine production using ELISPOT assay or intracellular cytokine staining. Methods 2003, 31, 143–149. [Google Scholar] [CrossRef]
- Gazagne, A.; Claret, E.; Wijdenes, J.; Yssel, H.; Bousquet, F.; Levy, E.; Vielh, P.; Scotte, F.; Goupil, T.L.; Fridman, W.H.; et al. A Fluorospot assay to detect single T lymphocytes simultaneously producing multiple cytokines. J. Immunol. Methods 2003, 283, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, A.; Overbergh, L.; Valckx, D.; Decallonne, B.; Bouillon, R.; Mathieu, C. An overview of real-time quantitative PCR: Applications to quantify cytokine gene expression. Methods 2001, 25, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Barten, M.J.; Rahmel, A.; Bocsi, J.; Boldt, A.; Garbade, J.; Dhein, S.; Mohr, F.W.; Gummert, J.F. Cytokine analysis to predict immunosuppression. Cytometry A 2006, 69, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Bagheri, N.; Bradshaw, E.M.; Hafler, D.A.; Lauffenburger, D.A.; Love, J.C. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc. Natl. Acad. Sci. USA 2012, 5, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, Y.; Yamagishi, M.; Shimura, N.; Hijikata, A.; Ohara, O. Toward an understanding of immune cell sociology: Real-time monitoring of cytokine secretion at the single-cell level. IUBMB Life 2013, 65, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Soen, Y.; Stuge, B.T.; Lee, P.P.; Weber, S.J.; Brown, O.P.; Davis, M.M. Marked Differences in Human Melanoma Antigen-Specific T Cell Responsiveness after Vaccination Using a Functional Microarray. PLoS Med. 2005, 2, e265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.D.; Demkowicz, W.E., Jr.; Stern, L.J. HLA-restricted epitope identification and detection of functional T cell responses by using MHC-peptide and costimulatory microarrays. Proc. Natl. Acad. Sci. USA 2005, 102, 3744–3749. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Davis, M.M. Molecular and functional analysis using live cell microarrays. Curr. Opin. Chem. Biol. 2006, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.N.; Lee, J.Y.; Zhu, J.; Stybayeva, G.; Ramanculov, E.; Zern, M.A.; Revzin, A. Multifunctional Protein Microarrays for Cultivation of Cells and Immunodetection of Secreted Cellular Products. Anal. Chem. 2008, 80, 6351–6357. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Stybayeva, G.; Macal, M.; Ramanculov, E.; George, M.D.; Dandekar, S.; Revzin, A. A microdevice for multiplexed detection of T-cell-secreted cytokines. Lab Chip 2008, 8, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Vu, T.; Stybayeva, G.; Pan, T.; Revzin, A. Reconfigurable microfluidics combined with antibody microarrays for enhanced detection of T-cell secreted cytokines. Biomicrofluidics 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, J.; Howland, M.C.; Kwa, T.; Revzin, A. Micropatterned aptasensors for continuous monitoring of cytokine release from human leukocytes. Anal. Chem. 2011, 83, 8286–8292. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kwa, T.; Revzin, A. Simultaneous detection of cell-secreted TNF-α and IFN-γ using micropatterned aptamer-modified electrodes. Biomaterials 2012, 33, 7347–7355. [Google Scholar] [CrossRef] [PubMed]
- Kwa, T.; Zhou, Q.; Gao, Y.; Rahimian, A.; Kwon, L.; Liu, Y.; Revzin, A. Reconfigurable microfluidics with integrated aptasensors for monitoring intercellular communication. Lab Chip 2014, 14, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Cherif, B.; Villiers, L.C.; Paranhos-Baccala, G.; Calemczuk, R.; Marche, N.P.; Livache, T.; Villiers, M.B. Design and application of a microarray for flluorescence and SPR imaging analysis of peptide-antibody interactions. J. Biomed. Nanotechnol. 2006, 2, 1–7. [Google Scholar] [CrossRef]
- Piliarik, M.; Homola, J. SPR Sensor Instrumentation. Springer Serf. Chem. Sens. Biosens. 2006, 4, 95–116. [Google Scholar]
- Stybayeva, G.; Kairova, M.; Ramanculov, E.; Simonian, A.L.; Revzin, A. Detecting interferon-gamma release from human CD4 T-cells using surface plasmon resonance. Colloids Surf. B Biointerfaces 2010, 80, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Milgram, S.; Bombera, R.; Livache, T.; Roupioz, Y. Antibody microarrays for label-free cell-based applications. Methods 2012, 56, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Assenmacher, M.; Löhning, M.; Scheffold, A.; Manz, R.A.; Schmitz, J.; Radbruch, A. Sequential production of IL-2, IFN-gamma and IL-10 by individual staphylococcal enterotoxin B-activated T helper lymphocytes. Eur. J. Immunol. 1998, 28, 1534–1543. [Google Scholar] [CrossRef]
- Suraniti, E.; Sollier, E.; Calemczuk, R.; Livache, T.; Marche, P.N.; Villiers, M.B.; Roupioz, Y. Real-time detection of lymphocytes binding on an antibody chip using SPR imaging. Lab Chip 2007, 7, 1206–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, W.M.; Dharmaraja, S.; Milisavljevic, V.; di Carlo, D. The effects of shear stress on isolated receptor-ligand interactions of Staphylococcus epidermidis and human plasma fibrinogen using molecularly patterned microfluidics. Lab Chip 2011, 11, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Laplatine, L.; Leroy, L.; Calemczuk, R.; Baganizi, D.; Marche, P.N.; Roupioz, Y.; Livache, T. Spatial resolution in prism-based surface plasmon resonance microscopy. Opt. Express 2014, 22, 22771–22785. [Google Scholar] [CrossRef] [PubMed]
- Erickson, D.; Sinton, D.; Li, D. Joule heating and heat transfer in poly(dimethylsiloxane) microfluidic systems. Lab Chip 2003, 3, 141–149. [Google Scholar] [CrossRef] [PubMed]
- De Mello, A.J.; Habgood, M.; Llewellyn, L.N.; Welton, T.; Wootton, R.C.R. Precise temperature control in microfluidic devices using Joule heating of ionic liquids. Lab Chip 2004, 4, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Miralles, V.; Huerre, A.; Malloggi, F.; Jullien, M.C. A Review of Heating and Temperature Control in Microfluidic Systems: Techniques and Applications. Diagnostics 2013, 3, 33–67. [Google Scholar] [CrossRef]
- Altman, A.; Mally, M.I.; Isakov, N. Phorbol ester synergizes with Ca2+ ionophore in activation of protein kinase C (PKC)a and PKCfi isoenzymes in human T cells and in induction of related cellular functions. Immunology 1992, 76, 465–471. [Google Scholar] [PubMed]
- Szmacinski, H.; Toshchakov, V.; Piao, W.; Lakowicz, J.R. Imaging of Protein Secretion from a Single Cell Using Plasmonic Substrates. Bionanoscience 2013, 3, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nath, N.; Reichert, M.W. Parallel Comparison of Sandwich and Direct Label Assay Protocols on Cytokine Detection Protein Arrays. Anal. Chem. 2003, 75, 5274–5281. [Google Scholar] [CrossRef]
- Kaplan, D. Autocrine secretion and the physiological concentration of cytokines. Immunol. Today 1996, 17, 303–304. [Google Scholar] [CrossRef]
- Wang, S.; Ota, S.; Guo, B.; Ryu, J.; Rhodes, C.; Xiong, Y.; Kalim, S.; Zeng, L.; Chen, Y.; Teitell, M.A.; et al. Subcellular resolution mapping of endogenous cytokine secretion by nano-plasmonic-resonator sensor array. Nano Lett. 2011, 11, 3431–3434. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N. Cell Surface Concentrations and Concentration Ranges for Testing In Vitro Autocrine Loops and Small Molecules. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Scarano, S.; Mascini, M.; Turner, A.P.; Minunni, M. Surface plasmon resonance imaging for affinity-based biosensors. Biosens. Bioelectron. 2010, 25, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milgram, S.; Cortes, S.; Villiers, M.B.; Marche, P.; Buhot, A.; Livache, T.; Roupioz, Y. On chip real time monitoring of B-cells hybridoma secretion of immunoglobulin. Biosens. Bioelectron. 2011, 26, 2728–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdeva, N.; Asthana, D. Cytokine quantitation: Technologies and applications. Front. Biosci. 2007, 12, 4682–4695. [Google Scholar] [CrossRef] [PubMed]
- Yanase, Y.; Hiragun, T.; Ishii, K.; Kawaguchi, T.; Yanase, T.; Kawai, M.; Sakamoto, K.; Hide, M. Surface plasmon resonance for cell-based clinical diagnosis. Sensors 2014, 14, 4948–4959. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baganizi, D.R.; Leroy, L.; Laplatine, L.; Fairley, S.J.; Heidmann, S.; Menad, S.; Livache, T.; Marche, P.N.; Roupioz, Y. A Simple Microfluidic Platform for Long-Term Analysis and Continuous Dual-Imaging Detection of T-Cell Secreted IFN-γ and IL-2 on Antibody-Based Biochip. Biosensors 2015, 5, 750-767. https://doi.org/10.3390/bios5040750
Baganizi DR, Leroy L, Laplatine L, Fairley SJ, Heidmann S, Menad S, Livache T, Marche PN, Roupioz Y. A Simple Microfluidic Platform for Long-Term Analysis and Continuous Dual-Imaging Detection of T-Cell Secreted IFN-γ and IL-2 on Antibody-Based Biochip. Biosensors. 2015; 5(4):750-767. https://doi.org/10.3390/bios5040750
Chicago/Turabian StyleBaganizi, Dieudonné R., Loïc Leroy, Loïc Laplatine, Stacie J. Fairley, Samuel Heidmann, Samia Menad, Thierry Livache, Patrice N. Marche, and Yoann Roupioz. 2015. "A Simple Microfluidic Platform for Long-Term Analysis and Continuous Dual-Imaging Detection of T-Cell Secreted IFN-γ and IL-2 on Antibody-Based Biochip" Biosensors 5, no. 4: 750-767. https://doi.org/10.3390/bios5040750