Novel Cecropin-4 Derived Peptides against Methicillin-Resistant Staphylococcus aureus

 ,
, 
Abstract
1. Introduction
2. Results
2.1. Designing of Derived Peptides and Evaluation for Antimicrobial Activity
2.2. Salts and Serum Effect and Synergy with Clinical Antibacterial Agents
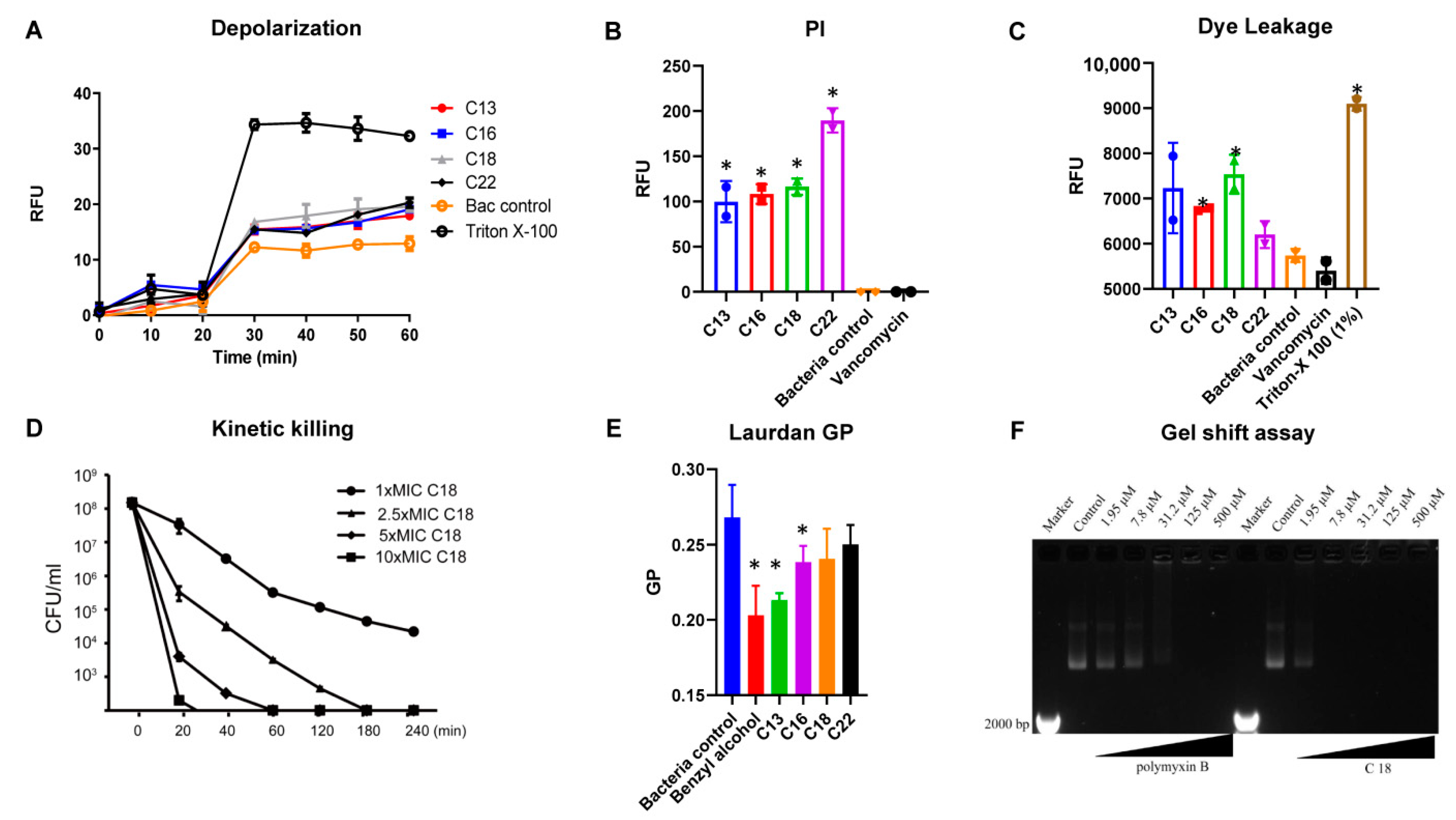
2.3. Mechanism of Action
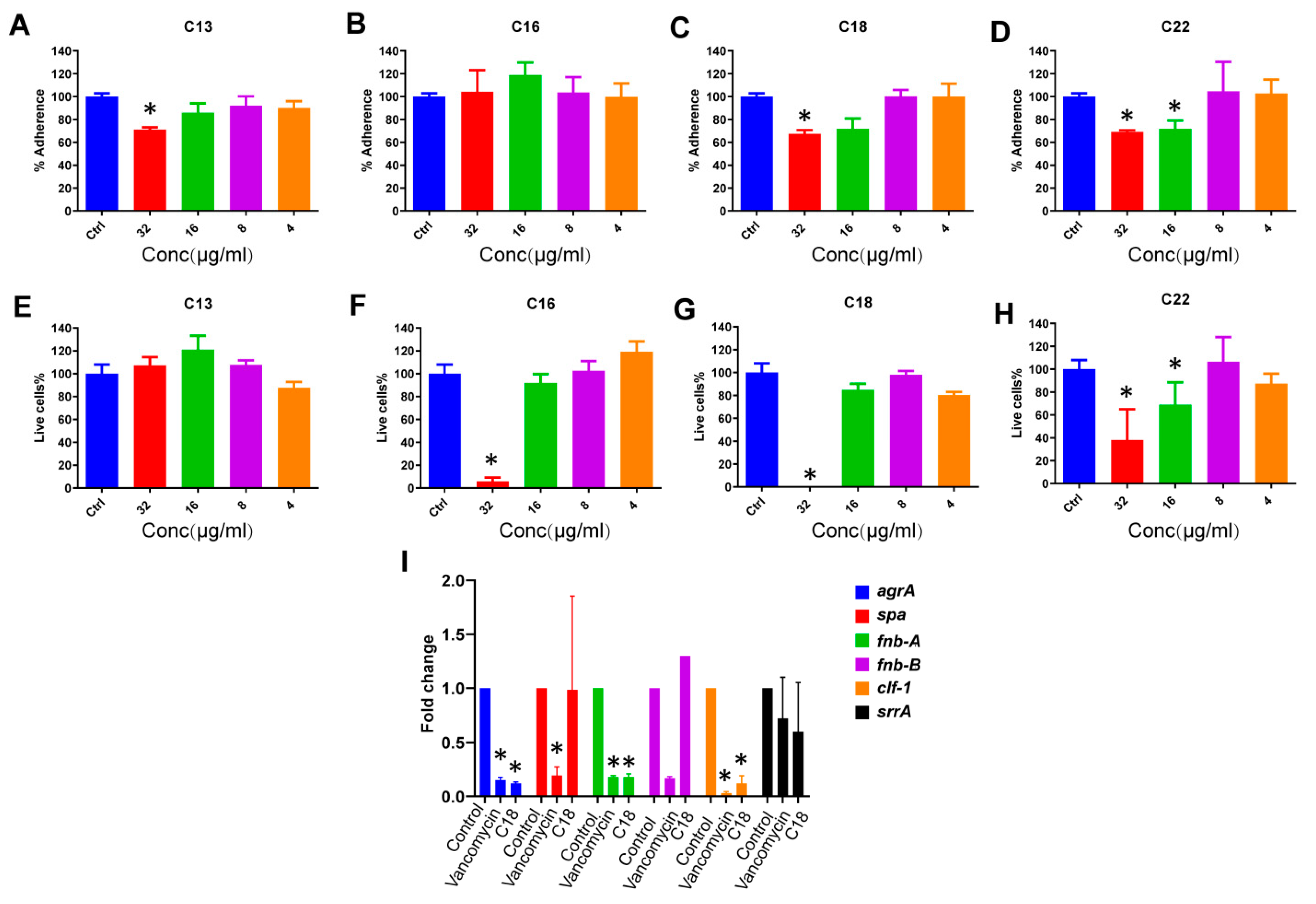
2.4. Anti-Biofilm Effects
2.5. The Effect of C18 on the Regulation of the Virulence Genes in S. aureus
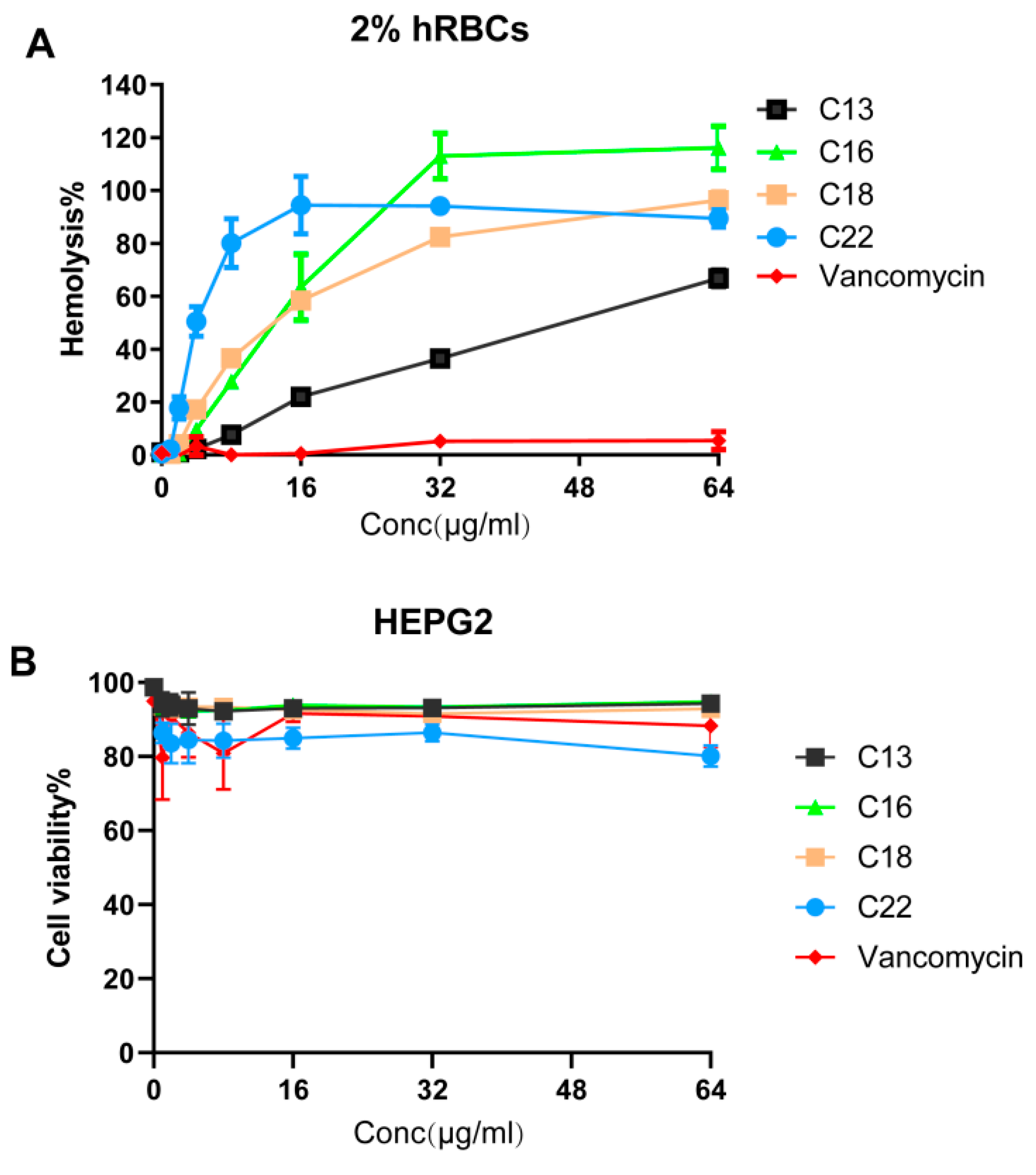
2.6. Hemolytic Activity and Cytotoxicity
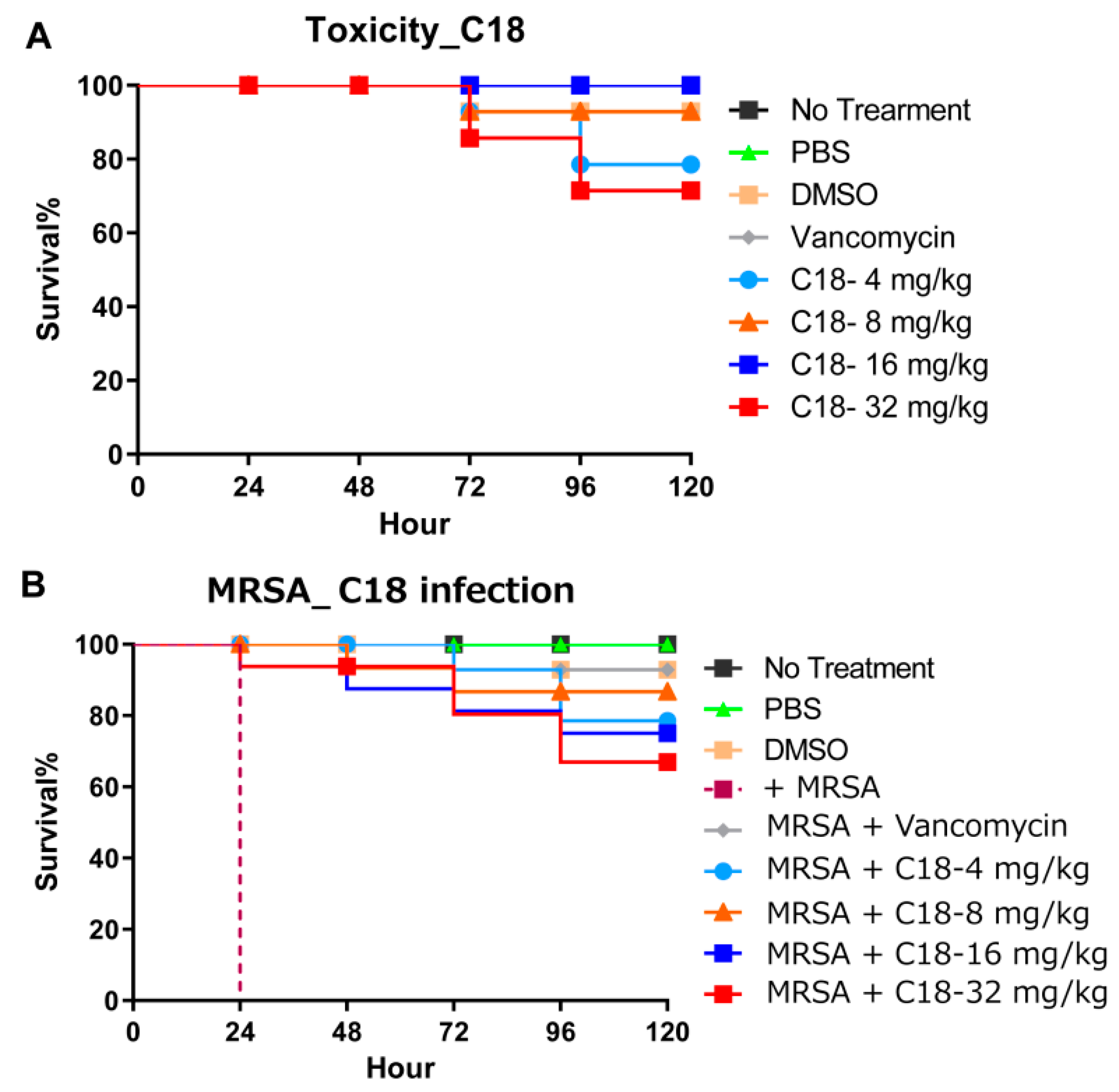
2.7. Galleria Mellonella Assays
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Minimal Inhibitory Concentration (MIC) Assay
4.3. Antimicrobial Activity in the Presence of Salts and Serum
4.4. Synergy with Clinical Antibacterial Agents
4.5. Membrane Depolarization
4.6. Propidium Iodide-Based Membrane Permeability
4.7. Laurdan Based Membrane Fluidity Assay
4.8. S. Aureus Persisters Cell Generation and Time-Kill Assay
4.9. Plasmid Band Shift Assay
4.10. Prevention of S. aureus Static Biofilm Attachment
4.11. Inhibition of S. aureus Biofilm Formation
4.12. Quantitative Polymerase Chain Reaction (qPCR)
4.13. Hemolysis of Human Red Blood Cells (hRBCs)
4.14. Mammalian Cell Cytotoxicity Assays
4.15. Galleria Mellonella In Vivo Assay
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Antimicrobial peptides | AMPs |
| Antimicrobial resistance | AMR |
| Brain-heart infusion | BHI |
| Double distilled water | ddH2O |
| Enterococcus faecium, Staphylococcus aureus, Klebsiella spp., Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp. | ESKAPE |
| Fractional inhibition concentration index | FICI |
| Generalized polarization | GP |
| High-performance liquid chromatography | HPLC |
| Methicillin-resistant Staphylococcus aureus | MRSA |
| Mueller-Hinton broth | MHB |
| Minimal inhibitory concentration | MIC |
| Phosphate buffer saline | PBS |
| Propidium iodide | PI |
| Quantitative polymerase chain reaction | qPCR |
| Sabouraud dextrose broth | SDB |
| tryptic soy broth | TSB |
| Ventilator-associated pneumonia | VAP |
References
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Murray, A.K. The Novel Coronavirus COVID-19 Outbreak: Global Implications for Antimicrobial Resistance. Front. Microbiol. 2020, 11, 1020. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, J.C.; Buehrle, D.J.; Nguyen, M.H. PRO: The COVID-19 pandemic will result in increased antimicrobial resistance rates. JAC Antimicrob. Resist. 2020, 1–3. [Google Scholar] [CrossRef]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections-United States. MMWR Morb. Mortal. Wkly Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef]
- Hidayat, L.K.; Hsu, D.I.; Quist, R.; Shriner, K.A.; Wong-Beringer, A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: Efficacy and toxicity. Arch. Intern. Med. 2006, 166, 2138–2144. [Google Scholar] [CrossRef]
- Yoon, Y.K.; Park, D.W.; Sohn, J.W.; Kim, H.Y.; Kim, Y.S.; Lee, C.S.; Lee, M.S.; Ryu, S.Y.; Jang, H.C.; Choi, Y.J.; et al. Multicenter prospective observational study of the comparative efficacy and safety of vancomycin versus teicoplanin in patients with health care-associated methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2014, 58, 317–324. [Google Scholar] [CrossRef]
- Kato-Hayashi, H.; Niwa, T.; Ohata, K.; Harada, S.; Matsumoto, T.; Kitagawa, J.; Tsurumi, H.; Suzuki, A. Comparative efficacy and safety of vancomycin versus teicoplanin in febrile neutropenic patients receiving hematopoietic stem cell transplantation. J. Clin. Pharm. 2019, 44, 888–894. [Google Scholar] [CrossRef]
- Mishra, B.; Wang, G. The Importance of Amino Acid Composition in Natural AMPs: An Evolutional, Structural, and Functional Perspective. Front. Immunol. 2012, 3, 221. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host defense antimicrobial peptides as antibiotics: Design and application strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef]
- Nagarajan, D.; Roy, N.; Kulkarni, O.; Nanajkar, N.; Datey, A.; Ravichandran, S.; Thakur, C.; Sandeep, T.; Aprameya, I.V.; Sarma, S.P.; et al. Ω76: A designed antimicrobial peptide to combat carbapenem- and tigecycline-resistant Acinetobacter baumannii. Sci. Adv. 2019, 5, eaax1946. [Google Scholar] [CrossRef] [PubMed]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Narayana, J.L.; Mishra, B.; Lushnikova, T.; Wu, Q.; Chhonker, Y.S.; Zhang, Y.; Zarena, D.; Salnikov, E.S.; Dang, X.; Wang, F.; et al. Two distinct amphipathic peptide antibiotics with systemic efficacy. Proc. Natl. Acad. Sci. USA 2020, 117, 19446–19454. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Long, H.; Liu, W.; Wu, Z.; Wang, T.; Zeng, Z.; Guo, G.; Wu, J. Antibacterial mechanism of peptide Cec4 against Acinetobacter baumannii. Infect. Drug Resist. 2019, 12, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wu, Z.; Liu, W.; Long, H.; Zhu, G.; Guo, G.; Wu, J. Antimicrobial functional divergence of the cecropin antibacterial peptide gene family in Musca domestica. Parasites Vectors 2019, 12, 537. [Google Scholar] [CrossRef]
- Yagi-Utsumi, M.; Yamaguchi, Y.; Boonsri, P.; Iguchi, T.; Okemoto, K.; Natori, S.; Kato, K. Stable isotope-assisted NMR characterization of interaction between lipid A and sarcotoxin IA, a cecropin-type antibacterial peptide. Biochem. Biophys. Res. Commun. 2013, 431, 136–140. [Google Scholar] [CrossRef]
- Winkel, J.D.T.; Gray, D.A.; Seistrup, K.H.; Hamoen, L.W.; Strahl, H. Analysis of Antimicrobial-Triggered Membrane Depolarization Using Voltage Sensitive Dyes. Front. Cell Dev. Biol. 2016, 4, 29. [Google Scholar] [CrossRef]
- Rosenberg, M.; Azevedo, N.F.; Ivask, A. Propidium iodide staining underestimates viability of adherent bacterial cells. Sci. Rep. 2019, 9, 6483. [Google Scholar] [CrossRef]
- Mishra, B.; Narayana, J.L.; Lushnikova, T.; Wang, X.; Wang, G. Low cationicity is important for systemic in vivo efficacy of database-derived peptides against drug-resistant Gram-positive pathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 13517–13522. [Google Scholar] [CrossRef]
- Muller, A.; Wenzel, M.; Strahl, H.; Grein, F.; Saaki, T.N.V.; Kohl, B.; Siersma, T.; Bandow, J.E.; Sahl, H.G.; Schneider, T.; et al. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc. Natl. Acad. Sci. USA 2016, 113, E7077–E7086. [Google Scholar] [CrossRef]
- Thomas, P.; Sekhar, A.C.; Upreti, R.; Mujawar, M.M.; Pasha, S.S. Optimization of single plate-serial dilution spotting (SP-SDS) with sample anchoring as an assured method for bacterial and yeast cfu enumeration and single colony isolation from diverse samples. Biotechnol. Rep. 2015, 8, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Jenul, C.; Horswill, A.R. Regulation of Staphylococcus aureus Virulence. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.; Grapputo, A.; Romoli, O.; Sandrelli, F. Insect Cecropins, Antimicrobial Peptides with Potential Therapeutic Applications. Int. J. Mol. Sci. 2019, 20, 5862. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.; Shin, S.Y.; Kang, J.H.; Hahm, K.S.; Kim, K.L.; Kim, Y. NMR structural characterization of cecropin A(1-8)—Magainin 2(1-12) and cecropin A (1-8)—Melittin (1-12) hybrid peptides. J. Pept. Res. 1999, 53, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.B.; Wu, R.J.; Si, D.Y.; Liao, X.D.; Zhang, L.L.; Zhang, R.J. Novel Hybrid Peptide Cecropin A (1-8)-LL37 (17-30) with Potential Antibacterial Activity. Int. J. Mol. Sci. 2016, 17, 983. [Google Scholar] [CrossRef] [PubMed]
- Chicharro, C.; Granata, C.; Lozano, R.; Andreu, D.; Rivas, L. N-terminal fatty acid substitution increases the leishmanicidal activity of CA(1-7)M(2-9), a cecropin-melittin hybrid peptide. Antimicrob. Agents Chemother. 2001, 45, 2441–2449. [Google Scholar] [CrossRef]
- Vylkova, S.; Nayyar, N.; Li, W.; Edgerton, M. Human beta-defensins kill Candida albicans in an energy-dependent and salt-sensitive manner without causing membrane disruption. Antimicrob. Agents Chemother. 2007, 51, 154–161. [Google Scholar] [CrossRef]
- Ciornei, C.D.; Sigurdardottir, T.; Schmidtchen, A.; Bodelsson, M. Antimicrobial and chemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and inhibition by serum of analogs of human cathelicidin LL-37. Antimicrob. Agents Chemother. 2005, 49, 2845–2850. [Google Scholar] [CrossRef]
- Ejim, L.; Farha, M.A.; Falconer, S.B.; Wildenhain, J.; Coombes, B.K.; Tyers, M.; Brown, E.D.; Wright, G.D. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 2011, 7, 348–350. [Google Scholar] [CrossRef]
- Silvestro, L.; Axelsen, P.H. Membrane-induced folding of cecropin A. Biophys. J. 2000, 79, 1465–1477. [Google Scholar] [CrossRef]
- Sato, H.; Feix, J.B. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1245–1256. [Google Scholar] [CrossRef]
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016, 1. [Google Scholar] [CrossRef]
- Heyer, G.; Saba, S.; Adamo, R.; Rush, W.; Soong, G.; Cheung, A.; Prince, A. Staphylococcus aureus agr and sarA functions are required for invasive infection but not inflammatory responses in the lung. Infect. Immun. 2002, 70, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Hauck, C.R.; Ohlsen, K. Sticky connections: Extracellular matrix protein recognition and integrin-mediated cellular invasion by Staphylococcus aureus. Curr. Opin. Microbiol. 2006, 9, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003, 48, 1429–1449. [Google Scholar] [CrossRef]
- Tharmalingam, N.; Khader, R.; Fuchs, B.B.; Mylonakis, E. The Anti-virulence Efficacy of 4-(1,3-Dimethyl-2,3-Dihydro-1H-Benzimidazol-2-yl)Phenol Against Methicillin-Resistant Staphylococcus aureus. Front. Microbiol. 2019, 10, 1557. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Sahu, C.; Jain, V.; Mishra, P.; Prasad, K.N. Clinical and laboratory standards institute versus European committee for antimicrobial susceptibility testing guidelines for interpretation of carbapenem antimicrobial susceptibility results for Escherichia coli in urinary tract infection (UTI). J. Lab. Physicians 2018, 10, 289–293. [Google Scholar] [CrossRef]
- Yan, H.; Hancock, R.E. Synergistic interactions between mammalian antimicrobial defense peptides. Antimicrob. Agents Chemother. 2001, 45, 1558–1560. [Google Scholar] [CrossRef]
- Kim, W.; Zou, G.; Hari, T.P.A.; Wilt, I.K.; Zhu, W.; Galle, N.; Faizi, H.A.; Hendricks, G.L.; Tori, K.; Pan, W.; et al. A selective membrane-targeting repurposed antibiotic with activity against persistent methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2019, 116, 16529–16534. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Conery, A.L.; Rajamuthiah, R.; Fuchs, B.B.; Ausubel, F.M.; Mylonakis, E. Identification of an Antimicrobial Agent Effective against Methicillin-Resistant Staphylococcus aureus Persisters Using a Fluorescence-Based Screening Strategy. PLoS ONE 2015, 10, e0127640. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Lee, A.Y.; Lin, Y.C.; Yu, Y.P.; Huang, W.T.; Wu, S.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Golla, R.M.; Lau, K.; Lushnikova, T.; Wang, G. Anti-Staphylococcal Biofilm Effects of Human Cathelicidin Peptides. ACS Med. Chem. Lett. 2016, 7, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liu, Q.; Kim, W.; Tharmalingam, N.; Fuchs, B.B.; Mylonakis, E. Antimicrobial activity of 1,3,4-oxadiazole derivatives against planktonic cells and biofilm of Staphylococcus aureus. Future Med. Chem. 2018, 10, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Mora-Navarro, C.; Mendez-Vega, J.; Caraballo-Leon, J.; Lee, M.R.; Palecek, S.; Torres-Lugo, M.; Ortiz-Bermudez, P. Hydrophobicity of Antifungal beta-Peptides Is Associated with Their Cytotoxic Effect on In Vitro Human Colon Caco-2 and Liver HepG2 Cells. PLoS ONE 2016, 11, e0149271. [Google Scholar] [CrossRef]
- Kim, W.; Zhu, W.; Hendricks, G.L.; Van Tyne, D.; Steele, A.D.; Keohane, C.E.; Fricke, N.; Conery, A.L.; Shen, S.; Pan, W.; et al. A new class of synthetic retinoid antibiotics effective against bacterial persisters. Nature 2018, 556, 103–107. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Jara, S.; Monga, D.; Eliopoulos, G.M.; Moellering, R.C., Jr.; Mylonakis, E. Galleria mellonella as a model system to study Acinetobacter baumannii pathogenesis and therapeutics. Antimicrob. Agents Chemother. 2009, 53, 2605–2609. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Sequences | E. faecium | S. aureus (ATCC29231) | S. aureus MW2 | K. pneumoniae | A. baumannii | P. aeruginosa | E. aerogenes | C. albicans |
|---|---|---|---|---|---|---|---|---|---|
| G+ | G− | Fungus | |||||||
| C1 | LKKIGKKIWRVGWNTRDATIQ | >128/50.96 | >128/50.96 | >128/50.96 | >128/50.96 | >128/50.96 | >128/50.96 | >128/50.96 | >128/50.96 |
| C2 | LKKIGKKIWRVGWNTR | >128/64.53 | >128/64.53 | >128/64.53 | >128/64.53 | 128/64.53 | >128/64.53 | >128/64.53 | >128/64.53 |
| C3 | LKKIGKKIWRVGWNWR | >128/61.87 | >128/61.87 | 128/61.87 | 64/32.27 | 32/16.13 | 128/61.87 | >128/61.87 | 128/61.87 |
| C4 | LKKIGKKIWRVG | >128/89.77 | >128/89.77 | >128/89.77 | >128/89.77 | >128/89.77 | >128/89.77 | >128/89.77 | >128/89.77 |
| C5 | LKWIGKKIWRVGWNWR | >128/60.19 | 128/60.19 | 64/30.10 | 128/60.19 | 32/15.05 | 128/60.19 | >128/60.19 | 128/60.19 |
| C6 | LKWIGKWIWRVGWNWR | >128/58.59 | >128/58.59 | 64/29.30 | >128/58.59 | 128/58.59 | >128/58.59 | >128/58.59 | >128/58.59 |
| C7 | LKWIGKKIWRVG | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 |
| C8 | LKWIGKWIWRVG | >128/83.01 | >128/83.01 | 64/41.51 | >128/83.01 | >128/83.01 | >128/83.01 | >128/83.01 | >128/83.01 |
| C9 | LWKIGKKIWRVGWNWR | >128/60.19 | 64/30.10 | 64/30.10 | 64/30.10 | 16/7.25 | 64/30.10 | 128/60.19 | 128/60.19 |
| C10 | LWKIGWKIWRVGWNWR | >128/58.59 | 32/14.65 | 32/14.65 | 128/58.59 | 32/14.65 | 64/29.30 | 128/58.59 | 64/29.30 |
| C11 | LWKIGKKIWRVG | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 | >128/86.26 |
| C12 | LWKIGWKIWRVG | >128/83.01 | 64/41.51 | 64/41.51 | 64/41.51 | 32/20.75 | 128/83.01 | 128/83.01 | 128/83.01 |
| C13 | LWKIWKKIWRVGWNWR | >128/56.74 | 16/7.09 | 32/14.19 | 32/14.19 | 32/14.19 | 32/14.19 | 64/28.37 | 32/14.19 |
| C14 | LWKIGKKIWRVWWNWR | >128/56.74 | 16/7.09 | 32/14.19 | 32/14.19 | 32/14.19 | 32/14.19 | 64/28.37 | 32/14.19 |
| C15 | LWKIWKKIWRVWWNWR | 128/53.67 | 32/13.14 | 64/26.84 | 128/53.67 | 128/53.67 | 64/26.84 | 128/53.67 | 128/53.67 |
| C16 | LWKIWKKIWRVWKNWR | >128/55.01 | 8/3.44 | 32/13.75 | 64/27.50 | 64/27.50 | 32/13.75 | 32/13.75 | 32/13.75 |
| C17 | LWKILKKIWRVGWNWR | >128/58.64 | 16/7.33 | 32/14.66 | 64/29.32 | 64/29.32 | 64/29.32 | 128/58.64 | 64/29.32 |
| C18 | LWKIGKKIWRVLWNWR | 128/58.64 | 4/1.83 | 4/1.83 | 16/7.33 | 16/7.33 | 32/14.66 | 16/7.33 | 16/7.33 |
| C19 | LWKILKKIWRVLWNWR | >128/57.17 | 32/14.29 | 64/28.59 | 128/57.17 | 128/57.17 | 128/57.17 | 128/57.17 | 64/28.59 |
| C20 | LWKILKKIWRVLKNWR | >128/58.70 | 32/14.67 | 32/14.67 | 64/29.35 | 128/58.70 | 32/14.67 | 64/29.35 | 32/14.67 |
| C21 | LWKIWWKIWRVWWNWR | >128/52.40 | 128/52.40 | 128/52.40 | >128/52.40 | 128/52.40 | >128/52.40 | >128/52.40 | >128/52.40 |
| C22 | LWKIWWKIWRVWKNWR | >128/53.67 | 8/3.35 | 32/13.42 | 128/53.67 | >128/53.67 | 32/13.42 | 128/53.67 | 64/26.84 |
| C23 | LWKILWKIWRVLWNWR | >128/55.73 | 128/55.73 | 128/55.73 | >128/55.73 | 128/55.73 | >128/55.73 | >128/55.73 | 128/55.73 |
| C24 | LWKILWKIWRVLKNWR | 128/57.2 | 64/28.59 | 128/57.2 | 128/57.2 | >128/57.2 | 128/57.2 | 128/57.2 | >128/57.2 |
| Peptides | MHB | NaCl (150 mM) | CaCl2 (2 mM) | Serum (5%) |
|---|---|---|---|---|
| C13 | 16 | 32 | 16 | 128 |
| C18 | 4 | 4 | 4 | 128 |
| C22 | 32 | 32 | 32 | >128 |
| vancomycin | 2 | 2 | 2 | 2 |
| SL. No. | Antibacterial Agents | FICI |
|---|---|---|
| 1 | daptomycin | 0.313 |
| 2 | vancomycin | 0.625 |
| 3 | gentamicin | 1.25 |
| 4 | oxacillin | 0.75 |
| 5 | ciprofloxacin | 0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, J.; Mishra, B.; Khader, R.; Felix, L.; Mylonakis, E. Novel Cecropin-4 Derived Peptides against Methicillin-Resistant Staphylococcus aureus. Antibiotics 2021, 10, 36. https://doi.org/10.3390/antibiotics10010036
Peng J, Mishra B, Khader R, Felix L, Mylonakis E. Novel Cecropin-4 Derived Peptides against Methicillin-Resistant Staphylococcus aureus. Antibiotics. 2021; 10(1):36. https://doi.org/10.3390/antibiotics10010036
Chicago/Turabian StylePeng, Jian, Biswajit Mishra, Rajamohammed Khader, LewisOscar Felix, and Eleftherios Mylonakis. 2021. "Novel Cecropin-4 Derived Peptides against Methicillin-Resistant Staphylococcus aureus" Antibiotics 10, no. 1: 36. https://doi.org/10.3390/antibiotics10010036
APA StylePeng, J., Mishra, B., Khader, R., Felix, L., & Mylonakis, E. (2021). Novel Cecropin-4 Derived Peptides against Methicillin-Resistant Staphylococcus aureus. Antibiotics, 10(1), 36. https://doi.org/10.3390/antibiotics10010036